Sequenziamento dell'rRNA 16S: una tecnica basata sulla PCR per identificare le specie batteriche
Fonte: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Dipartimento di Scienze Cliniche Lund, Divisione di Medicina delle Infezioni, Centro Biomedico, Università di Lund, 221 00 Lund, Svezia
Il pianeta Terra è un habitat per milioni di specie batteriche, ognuna delle quali ha caratteristiche specifiche. L'identificazione delle specie batteriche è ampiamente utilizzata nell'ecologia microbica per determinare la biodiversità dei campioni ambientali e la microbiologia medica per diagnosticare i pazienti infetti. I batteri possono essere classificati utilizzando metodi microbiologici convenzionali, come la microscopia, la crescita su supporti specifici, test biochimici e sierologici e saggi di sensibilità agli antibiotici. Negli ultimi decenni, i metodi di microbiologia molecolare hanno rivoluzionato l'identificazione batterica. Un metodo popolare è il sequenziamento del gene dell'RNA ribosomiale 16S (rRNA). Questo metodo non è solo più veloce e più accurato rispetto ai metodi convenzionali, ma consente anche l'identificazione di ceppi difficili da coltivare in condizioni di laboratorio. Inoltre, la differenziazione dei ceppi a livello molecolare consente la discriminazione tra batteri fenotipicamente identici (1-4).
L'rRNA 16S si unisce a un complesso di 19 proteine per formare una subunità 30S del ribosoma batterico (5). È codificato dal gene rRNA 16S, che è presente e altamente conservato in tutti i batteri grazie alla sua funzione essenziale nell'assemblaggio dei ribosomi; tuttavia, contiene anche regioni variabili che possono fungere da impronte digitali per particolari specie. Queste caratteristiche hanno reso il gene rRNA 16S un frammento genetico ideale da utilizzare nell'identificazione, nel confronto e nella classificazione filogenetica dei batteri (6).
Il sequenziamento del gene rRNA 16S si basa sulla reazione a catena della polimerasi (PCR) (7-8) seguita dal sequenziamento del DNA (9). La PCR è un metodo di biologia molecolare utilizzato per amplificare specifici frammenti di DNA attraverso una serie di cicli che includono:
i) Denaturazione di un modello di DNA a doppio filamento
ii) Ricottura di primer (oligonucleotidi corti) complementari al template
iii) Estensione dei primer da parte dell'enzima DNA polimerasi, che sintetizza un nuovo filamento di DNA
Una panoramica schematica del metodo è illustrata nella Figura 1.
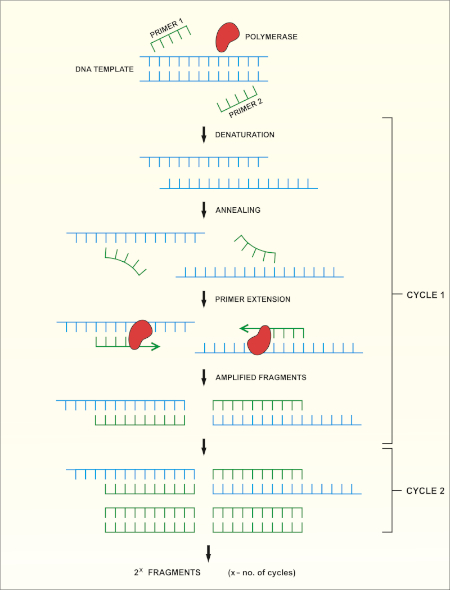
Figura 1: Panoramica schematica della reazione PCR. Fare clic qui per visualizzare una versione più grande di questa figura.
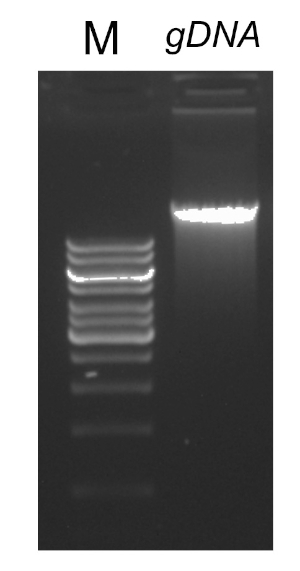
Ci sono diversi fattori che sono importanti per una reazione PCR di successo, uno dei quali è la qualità del modello di DNA. L'isolamento del DNA cromosomico dai batteri può essere eseguito utilizzando protocolli standard o kit commerciali. Particolare attenzione deve essere prestata per ottenere DNA privo di contaminanti che possono inibire la reazione PCR.
Le regioni conservate del gene rRNA 16S consentono la progettazione di coppie di primer universali (una in avanti e una inversa) che possono legarsi e amplificare la regione bersaglio in qualsiasi specie batterica. L'area di destinazione può variare in termini di dimensioni. Mentre alcune coppie di primer possono amplificare la maggior parte del gene rRNA 16S, altre amplificano solo parti di esso. Esempi di primer di uso comune sono mostrati nella Tabella 1 e i loro siti di legame sono illustrati nella Figura 2.
| Nome del primer | Sequenza (5'→3') | Avanti/indietro | Riferimento |
| 8F b) | AGAGTTTGATCCTGGCTCAG | inoltrare | -1 |
| 27F · | AGAGTTTGATCMTGGCTCAG | inoltrare | -10 |
| 515F · | GTGCCAGCMGCCGCGGTAA | inoltrare | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | inverso | -12 |
| 1391R | GACGGGCGGTGTGTRCA | inverso | -11 |
| 1492R | GGTTACCTTGTTACGACTT | inverso | -11 |
Tabella 1: Esempi di oligonucleotidi standard utilizzati nell'amplificazione dei geni rRNA 16S a).
a) Le lunghezze previste del prodotto PCR generato utilizzando le diverse combinazioni di primer possono essere stimate calcolando la distanza tra i siti di legame per il primer avanti e il primer inverso (vedi Figura 2), ad esempio la dimensione del prodotto PCR utilizzando la coppia di primer 8F-1492R è ~ 1500 bp e per la coppia di primer 27F-911R ~ 900 bp.
b) noto anche come fD1

Figura 2: Figura rappresentativa della sequenza di rRNA 16S e dei siti di legame del primer. Le regioni conservate sono colorate in grigio e le regioni variabili sono riempite con linee diagonali. Per consentire la massima risoluzione, il primer 8F e 1492R (nome basato sulla posizione sulla sequenza di rRNA) vengono utilizzati per amplificare l'intera sequenza, consentendo il sequenziamento di diverse regioni variabili del gene. Fare clic qui per visualizzare una versione più grande di questa figura.
Le condizioni cicliche per la PCR(cioè la temperatura e il tempo necessari per denaturare il DNA, ricotto con primer e sintetizzato) dipendono dal tipo di polimerasi utilizzata e dalle proprietà dei primer. Si consiglia di seguire le linee guida del produttore per una particolare polimerasi.
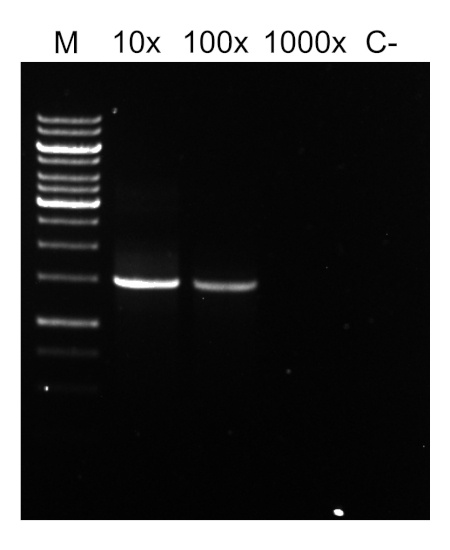
Dopo aver completato il programma PCR, i prodotti vengono analizzati mediante elettroforesi su gel di agarose. Una PCR di successo produce una singola banda di dimensioni previste. Il prodotto deve essere purificato prima del sequenziamento per rimuovere i primer residui, i desossiribonucleotidi, la polimerasi e il tampone che erano presenti nella reazione PCR. I frammenti di DNA purificati vengono solitamente inviati per il sequenziamento ai servizi di sequenziamento commerciali; tuttavia, alcune istituzioni eseguono il sequenziamento del DNA presso le proprie strutture principali.
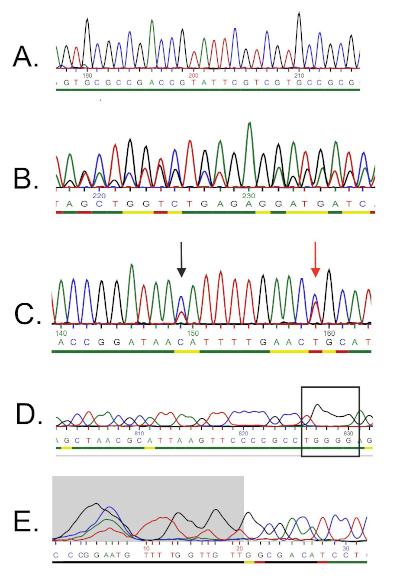
La sequenza di DNA viene generata automaticamente da un cromatogramma del DNA da un computer e deve essere attentamente controllata per verificare la qualità, poiché a volte è necessaria la modifica manuale. Seguendo questo passaggio, la sequenza genica viene confrontata con le sequenze depositate nel database dell'rRNA 16S. Vengono identificate le regioni di somiglianza e vengono consegnate le sequenze più simili.
1. Configurazione
- Durante la manipolazione dei microrganismi, è necessario seguire una buona pratica microbiologica. Tutti i microrganismi, in particolare i campioni sconosciuti, devono essere trattati come potenziali agenti patogeni. Seguire la tecnica asettica per evitare di contaminare i campioni, i ricercatori o il laboratorio. Lavarsi le mani prima e dopo aver maneggiato i batteri, usare guanti e indossare indumenti protettivi.
- Effettuare una valutazione del rischio per il protoc
L'identificazione delle specie batteriche è importante per i diversi ricercatori, così come per quelli nel settore sanitario. Il sequenziamento dell'rRNA 16S è stato inizialmente utilizzato dai ricercatori per determinare le relazioni filogenetiche tra i batteri. Nel tempo, è stato implementato in studi metagenomici per determinare la biodiversità di campioni ambientali e nei laboratori clinici come metodo per identificare potenziali agenti patogeni. Consente un'identificazione rapida e accurata dei batteri presenti...
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
Vai a...
Video da questa raccolta:

Now Playing
Sequenziamento dell'rRNA 16S: una tecnica basata sulla PCR per identificare le specie batteriche
Microbiology
186.6K Visualizzazioni

Creazione di una colonna di Winogradsky: un metodo per arricchire le specie microbiche presenti in un campione di sedimento
Microbiology
127.1K Visualizzazioni

Diluizioni seriali e piastratura: la conta microbica
Microbiology
311.6K Visualizzazioni

Culture di arricchimento: coltura di microbi aerobici e anaerobici su terreni selettivi e differenziali
Microbiology
131.2K Visualizzazioni

Colture pure e piastratura per striscio: isolamento di singole colonie batteriche da un campione misto
Microbiology
164.9K Visualizzazioni

Curve di crescita: generazione di curve di crescita utilizzando le unità formanti colonia e la misurazione della densità ottica
Microbiology
289.8K Visualizzazioni

Test di suscettibilità agli antibiotici: test dell'epsilometro per determinare i valori MIC di due antibiotici e valutare la sinergia antibiotica
Microbiology
93.1K Visualizzazioni

Microscopia e colorazioni: la colorazione di Gram, delle endospore e del capside
Microbiology
361.4K Visualizzazioni

Saggio delle placche: un metodo per determinare il titolo virale in unità formanti placca (UFP)
Microbiology
184.9K Visualizzazioni

Trasformazione di cellule di E. coli tramite l'utilizzo di una procedura basata sul metodo del cloruro di calcio
Microbiology
85.8K Visualizzazioni

Coniugazione: un metodo per trasferire la resistenza all'ampicillina dal donatore al ricevente E. coli
Microbiology
37.8K Visualizzazioni

La trasduzione batterica tramite fagi: un metodo per trasferire la resistenza all'ampicillina da una cellula donatore di E. coli ad una ricevente
Microbiology
28.7K Visualizzazioni
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati