
Sequenciamento de rRNA 16S: uma técnica baseada em PCR para identificar espécies bacterianas
Visão Geral
Fonte: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Departamento de Ciências Clínicas Lund, Divisão de Medicina de Infecção, Centro Biomédico, Universidade de Lund, 221 00 Lund, Suécia
O Planeta Terra é um habitat para milhões de espécies bacterianas, cada uma das quais tem características específicas. A identificação de espécies bacterianas é amplamente utilizada na ecologia microbiana para determinar a biodiversidade de amostras ambientais e microbiologia médica para diagnosticar pacientes infectados. As bactérias podem ser classificadas usando métodos convencionais de microbiologia, como microscopia, crescimento em mídia específica, testes bioquímicos e sorológicos e ensaios de sensibilidade a antibióticos. Nas últimas décadas, os métodos de microbiologia molecular revolucionaram a identificação bacteriana. Um método popular é o sequenciamento genético RNA ribossômico 16S (rRNA). Este método não só é mais rápido e mais preciso do que os métodos convencionais, mas também permite a identificação de cepas difíceis de crescer em condições laboratoriais. Além disso, a diferenciação das cepas no nível molecular permite a discriminação entre bactérias fenotipicamente idênticas (1-4).
16S rRNA une-se a um complexo de 19 proteínas para formar uma subunidade 30S do ribossomo bacteriano (5). É codificado pelo gene 16S rRNA, que está presente e altamente conservado em todas as bactérias devido à sua função essencial no conjunto ribossomo; no entanto, também contém regiões variáveis que podem servir como impressões digitais para determinadas espécies. Essas características tornaram o gene 16S rRNA um fragmento genético ideal a ser usado na identificação, comparação e classificação filogenética de bactérias (6).
O sequenciamento genético rRNA 16S é baseado na reação em cadeia de polimerase (PCR) (7-8) seguida pelo sequenciamento de DNA (9). PCR é um método de biologia molecular usado para amplificar fragmentos específicos de DNA através de uma série de ciclos que incluem:
i) Denaturação de um modelo de DNA duplo encalhado
ii) Ressaração de primers (oligonucleotídeos curtos) que são complementares ao modelo
iii) Extensão dos primers pela enzima dna polimerase, que sintetiza uma nova cadeia de DNA
Uma visão geral esquemática do método é mostrada na Figura 1.
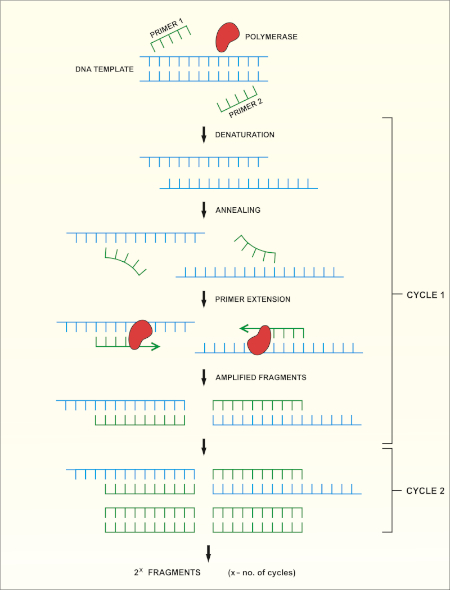
Figura 1: Visão geral esquemática da reação do PCR. Clique aqui para ver uma versão maior desta figura.
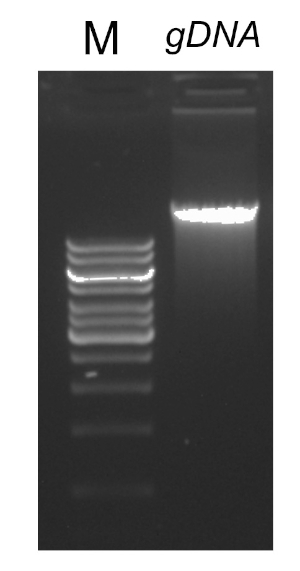
Existem vários fatores que são importantes para uma reação de sucesso do PCR, um dos quais é a qualidade do modelo de DNA. O isolamento do DNA cromossômico de bactérias pode ser realizado usando protocolos padrão ou kits comerciais. Deve-se tomar cuidado especial para obter DNA livre de contaminantes que podem inibir a reação da PCR.
Regiões conservadas do gene rRNA 16S permitem o desenho de pares de primer universais (um para a frente e um inverso) que podem se ligar e amplificar a região alvo em qualquer espécie bacteriana. A região alvo pode variar de tamanho. Enquanto alguns pares de primer podem amplificar a maior parte do gene 16S rRNA, outros amplificam apenas partes dele. Exemplos de primers comumente usados são mostrados na Tabela 1 e seus locais de vinculação são retratados na Figura 2.
| Nome do primer | Sequência (5'→3') | Para a frente/ para trás | Referência |
| 8F b) | AGAGTTTGATCCTGGCTCAG | encaminhar | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | encaminhar | -10 |
| 515F | GTGCCAGCCCCCGGGGTAA | encaminhar | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | inverter | -12 |
| 1391R | GACGGGCGGTGTGTRCA | inverter | -11 |
| 1492R | GGTTACCTTGTTACGACTT | inverter | -11 |
Tabela 1: Exemplos de oligonucleotídeos padrão utilizados na amplificação de genes de rRNA 16S a).
a) Os comprimentos esperados do produto PCR gerados usando as diferentes combinações de primer podem ser estimados calculando-se a distância entre os locais de ligação para o primer para a frente e para o primer (ver Figura 2), por exemplo, o tamanho do produto PCR usando o par de primer 8F-1492R é de ~1500 bp, e para o par de primer 27F-911R ~900 bp.
b) também conhecido como fD1

Figura 2: Figura representativa da sequência de rRNA 16S e dos sites de vinculação de primer. As regiões conservadas são coloridas em cinza e regiões variáveis são preenchidas com linhas diagonais. Para permitir a maior resolução, o primer 8F e 1492R (nome baseado na localização na sequência de rRNA) são usados para amplificar toda a sequência, permitindo o sequenciamento de várias regiões variáveis do gene. Clique aqui para ver uma versão maior desta figura.
As condições de ciclismo para PCR (ou seja, a temperatura e o tempo necessários para que o DNA seja desnaturado, enrosco com primers e sintetizado) dependem do tipo de polimerase que é usado e das propriedades dos primers. Recomenda-se seguir as diretrizes do fabricante para uma determinada polimerase.
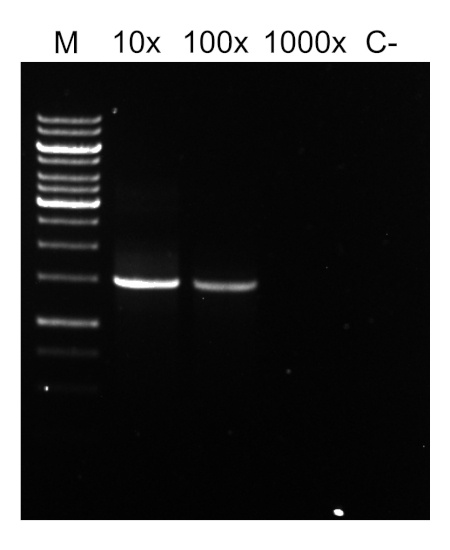
Após a conclusão do programa PCR, os produtos são analisados por eletroforese de gel agarose. Um PCR bem sucedido rende uma única faixa de tamanho esperado. O produto deve ser purificado antes do sequenciamento para remover primers residuais, desoxiribonucleotídeos, polimerase e tampão que estavam presentes na reação pcr. Os fragmentos de DNA purificados são geralmente enviados para sequenciamento para serviços de sequenciamento comercial; no entanto, algumas instituições realizam sequenciamento de DNA em suas próprias instalações principais.
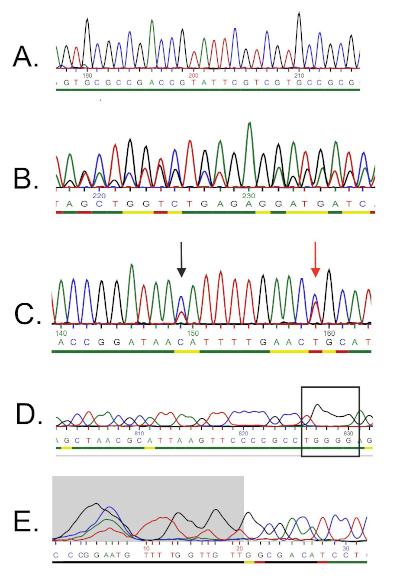
A sequência de DNA é gerada automaticamente a partir de um cromatgrama de DNA por um computador e deve ser cuidadosamente verificada para a qualidade, pois a edição manual às vezes é necessária. Após esta etapa, a sequência genética é comparada com sequências depositadas no banco de dados de rRNA 16S. As regiões de semelhança são identificadas, e as sequências mais semelhantes são entregues.
Procedimento
1. Configurar
- Ao manusear microrganismos, é necessário seguir uma boa prática microbiológica. Todos os microrganismos, especialmente amostras desconhecidas, devem ser tratados como patógenos potenciais. Siga a técnica asséptica para evitar contaminar as amostras, pesquisadores ou o laboratório. Lave as mãos antes e depois de manusear bactérias, use luvas e use roupas de proteção.
- Realizar uma avaliação de risco para o protocolo experimental para o isolamento genômico de DNA e purifica
Aplicação e Resumo
Identificar espécies bacterianas é importante para diferentes pesquisadores, bem como para aqueles em saúde. O sequenciamento de rRNA 16S foi inicialmente utilizado por pesquisadores para determinar as relações filogenéticas entre bactérias. Com o tempo, foi implementado em estudos metagenômicos para determinar a biodiversidade de amostras ambientais e em laboratórios clínicos como método para identificar potenciais patógenos. Permite uma identificação rápida e precisa das bactérias presentes em amostras ...
Referências
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
Pular para...
Vídeos desta coleção:

Now Playing
Sequenciamento de rRNA 16S: uma técnica baseada em PCR para identificar espécies bacterianas
Microbiology
189.6K Visualizações

Criando uma coluna de Winogradsky: um método para enriquecer as espécies microbianas em uma amostra de sedimento
Microbiology
129.9K Visualizações

Diluições em série e plaqueamento: enumeração microbiana
Microbiology
317.0K Visualizações

Culturas de enriquecimento: cultivo de micróbios aeróbicos e anaeróbicos em meios seletivos e diferenciais
Microbiology
132.2K Visualizações

Culturas puras e semeadura por esgotamento: isolamento de colônias bacterianas únicas de uma amostra mista
Microbiology
166.4K Visualizações

Curvas de crescimento: gerando curvas de crescimento usando unidades formadoras de colônias e medições de densidade óptica
Microbiology
297.8K Visualizações

Teste de sucetibilidade a antibióticos: testes de epsilômetro para determinar valores de MIC de dois antibióticos e avaliar a sinergia de antibióticos
Microbiology
93.9K Visualizações

Microscopia e Coloração: Coloração de Gram, Cápsula e Endósporo
Microbiology
364.0K Visualizações

Ensaio de placa: um método para determinar o título viral como unidades formadoras de placa (PFU)
Microbiology
186.5K Visualizações

Transformação de células de E. coli usando um protocolo adaptado de cloreto de cálcio
Microbiology
87.1K Visualizações

Conjugação: um método para transferir a resistência à ampicilina da E. coli doadora para a receptora
Microbiology
38.4K Visualizações

Transdução fágica: um método para transferir a resistência à ampicilina da E. coli doadora para a receptora
Microbiology
29.1K Visualizações
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados