
16S rRNA 시퀀싱: 박테리아 종 식별을위한 PCR 기반 기술
Overview
출처: 에와 부코스카-패니반드1,틸드 앤더슨1,롤프 루드1
1 임상 과학 룬드학과, 감염 의학 학과, 생물 의학 센터, 룬드 대학, 221 00 룬드, 스웨덴
행성 지구는 세균 종의 수백만에 대한 서식지입니다, 각각의 특정 특성을 가지고. 세균성 종의 식별은 감염된 환자를 진단하기 위해 환경 샘플 및 의료 미생물학의 생물 다양성을 결정하기 위해 미생물 생태학에서 널리 사용됩니다. 박테리아는 현미경 검사법과 같은 기존의 미생물학 방법, 특정 매체에 대한 성장, 생화학 및 혈청 술 검사 및 항생제 민감성 분석을 사용하여 분류될 수 있습니다. 최근 수십 년 동안 분자 미생물학 방법은 세균 식별에 혁명을 일으켰습니다. 16S 리보소말 RNA(rRNA) 유전자 염기서열분석이 인기 있는 방법이다. 이 방법은 기존의 방법보다 빠르고 정확할 뿐만 아니라 실험실 조건에서 성장하기 어려운 균주를 식별할 수 있습니다. 더욱이, 분자 수준에서 균주의 분화는 표현형 동일 박테리아 (1-4)의 차별을 가능하게합니다.
16S rRNA는 세균성 리보솜(5)의 30S 하위 단위를 형성하기 위해 19개의 단백질의 복합체와 결합한다. 그것은 16S rRNA 유전자에 의해 인코딩된다, 이는 존재하고 리보솜 조립에 그것의 필수적인 기능때문에 모든 박테리아에 높게 보존; 그러나 특정 종에 대한 지문 역할을 할 수있는 가변 영역도 포함되어 있습니다. 이러한 특징은 16S rRNA 유전자를 박테리아의 식별, 비교 및 계통유전학 적 분류에 사용되는 이상적인 유전 적 단편으로 만들었습니다 (6).
16S rRNA 유전자 염기서열분석은 중합효소 연쇄 반응(PCR) (7-8)를 기반으로 DNA 염기서열 분석(9)을 선행한다. PCR은 다음과 같은 일련의 주기를 통해 DNA의 특정 단편을 증폭시키는 데 사용되는 분자 생물학 방법입니다.
i) 이중 좌초 된 DNA 템플릿의 거부
ii) 템플릿에 보완프라이머 (짧은 올리고 뉴클레오티드)의 안다골
iii) 새로운 DNA 가닥을 합성하는 DNA 폴리머라제 효소에 의한 프라이머 확장
메서드의 회로도 개요는 도 1에표시됩니다.
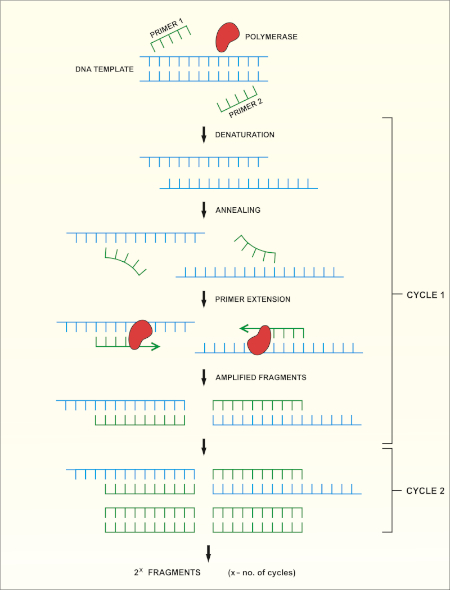
그림 1: PCR 반응의 회로도 개요. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
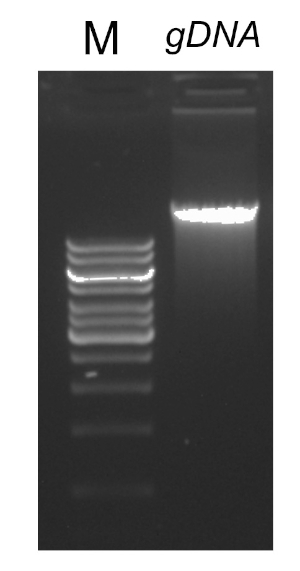
성공적인 PCR 반응에 중요한 몇 가지 요인이 있으며, 그 중 하나는 DNA 템플릿의 품질입니다. 박테리아로부터 염색체 DNA의 격리는 표준 프로토콜 또는 상업적 키트를 사용하여 수행될 수 있다. PCR 반응을 억제할 수 있는 오염물질이 없는 DNA를 얻기 위해 특별한 주의를 기울여야 한다.
16S rRNA 유전자의 보존된 영역은 임의의 세균종에서 표적 부위에 결합하고 증폭될 수 있는 범용 프라이머 쌍(1개의 전진 및 1개의 역)의 설계를 허용한다. 대상 영역의 크기가 다를 수 있습니다. 몇몇 프라이머 쌍은 16S rRNA 유전자의 대부분을 증폭할 수 있는 동안, 그 외는 그것의 단지 부분을 증폭합니다. 일반적으로 사용되는 프라이머의 예는 표 1에 표시되며 바인딩 사이트는 그림 2에표시됩니다.
| 프라이머 이름 | 시퀀스(5'→3') | 전진/역방향 | 참조 |
| 8F b) | 아가그트가트CCTGGCTCAG | 전달 | -1 |
| 27F | AGAGTGATCMTGGCTCAG | 전달 | -10 |
| 515F | GTGCCAGCMGCCGCGGGTAA | 전달 | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | 후진 | -12 |
| 1391R | 가CGGGCGGTGTGTRCA | 후진 | -11 |
| 1492R | GGTTGTTACGACTT | 후진 | -11 |
표 1: 16S rRNA 유전자a)의증폭에 사용되는 표준 올리고뉴클레오티드의 예.
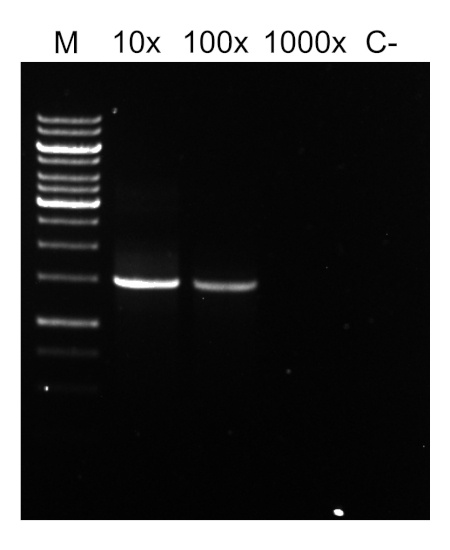
a) 상이한 프라이머 조합을 사용하여 생성된 PCR 제품의 예상 길이는 전방 및 역프라이머(그림 2 참조)에 대한 결합 부위 사이의 거리를 계산하여 추정할 수 있으며, 예를 들어 프라이머 쌍 8F-1492R을 사용하여 PCR 제품의 크기는 ~1500bp, 프라이머 쌍 27F-91R~90bp.90 bp.
b) fD1이라고도 함

그림 2: 16S rRNA 서열 및 프라이머 결합 부위의 대표적인 수치. 보존된 영역은 회색으로 표시되고 가변 영역은 대각선으로 채워져 있습니다. 가장 높은 해상도를 허용하기 위해, 프라이머 8F 및 1492R(rRNA 서열에 근거한 이름)는 전체 서열을 증폭시키는 데 사용되어 유전자의 여러 가변 영역의 시퀀싱을 허용한다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
PCR에 대한 사이클링조건(즉, DNA가 변성되고, 프라이머로 어닐링되고, 합성되는 데 필요한 온도 및 시간)은 사용되는 폴리머라제의 종류와 프라이머의 특성에 의존한다. 특정 폴리머라제에 대한 제조업체의 지침을 따르는 것이 좋습니다.
PCR 프로그램이 완료되면, 제품은 아가로즈 젤 전기포광에 의해 분석된다. 성공적인 PCR은 예상 크기의 단일 밴드를 생성합니다. 제품은 PCR 반응에 존재했던 잔류 프라이머, 데옥시리보뉴클레오티드, 폴리머라제 및 완충물을 제거하기 위해 시퀀싱 전에 정제되어야 한다. 정제 된 DNA 단편은 일반적으로 상업적 시퀀싱 서비스에 시퀀싱을 위해 전송됩니다. 그러나 일부 기관은 자신의 핵심 시설에서 DNA 시퀀싱을 수행합니다.
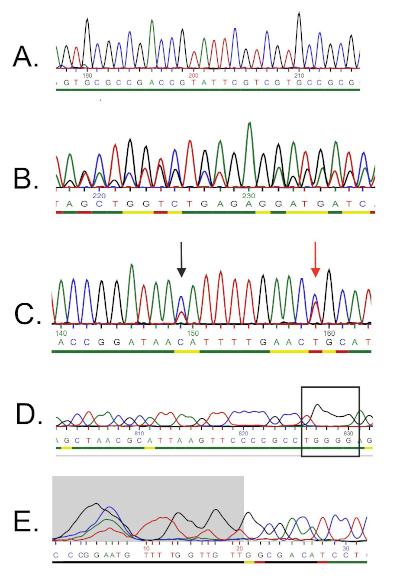
DNA 서열은 컴퓨터에 의해 DNA 크로마토그램에서 자동으로 생성되며 수동 편집이 때로는 필요하기 때문에 품질을 주의 깊게 검사해야합니다. 이 단계에 이어, 유전자 서열은 16S rRNA 데이터베이스에 증착된 서열과 비교된다. 유사성 영역이 식별되고 가장 유사한 시퀀스가 전달됩니다.
Procedure
1. 설정
- 미생물을 취급하는 동안, 좋은 미생물 사례를 따르는 것이 필요합니다. 모든 미생물, 특히 알려지지 않은 샘플은 잠재적인 병원균으로 취급되어야 합니다. 샘플, 연구원 또는 실험실을 오염하지 않도록 무균 기술을 따르십시오. 박테리아를 취급하기 전과 후에 손을 씻고 장갑을 착용하고 보호복을 착용하십시오.
- 게놈 DNA 격리 및 PCR 제품 정제를 위한 실험 프로토콜에 대한 위험 평가를 수행한다. 일부 시약은 유해할 수 있습니다!
- 순수한 배양은 16S rRNA 시퀀싱에 필수적입니다. 게놈 DNA의 격리를 진행하기 전에, 시작 물질이 완전히 순수한지 확인하십시오. 이것은 개별 식민지를 격리하기 위하여 줄무늬 도금에 의해 행적할 수 있습니다. 이들은 더 개별적으로 접시에 줄무늬, 또는 국물에 줄무늬, 필요한 경우.
- 필요한 실험실 장비:
- PCR용 열 사이클러. 열 사이클러의 기능은 세트 프로그램에 따라 온도를 높이고
Application and Summary
세균성 종을 확인하는 것은 다른 연구원을 위해, 뿐만 아니라 헬스케어에 있는 사람들을 위해 중요합니다. 16S rRNA 시퀀싱은 처음에 박테리아 사이의 물리 유전학 관계를 결정하기 위해 연구원에 의해 사용되었다. 시간이 지남에 따라, 그것은 잠재적인 병원균을 식별하는 방법으로 환경 샘플및 임상 실험실의 생물 다양성을 결정하기 위해 메타게노믹 연구에서 구현되었습니다. 그것은 임상 견본에...
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
Tags
건너뛰기...
이 컬렉션의 비디오:

Now Playing
16S rRNA 시퀀싱: 박테리아 종 식별을위한 PCR 기반 기술
Microbiology
189.6K Views

위노그라드스키 칼럼 생성: 퇴적물 검체에서 미생물 종을 풍부하게하는 방법
Microbiology
129.9K Views

연속 희석 및 플레이팅: 미생물 나열
Microbiology
317.0K Views

농축 배양: 선택 및 차동 매체에서 호기성 및 혐기성 미생물 배양
Microbiology
132.2K Views

순수 배양 및 줄무늬 평판배양: 혼합 검체에서 단일 박테리아 군집 분리
Microbiology
166.4K Views

성장 곡선: 군집 형성 단위 및 광학 밀도 측정을 사용하여 성장 곡선 생성
Microbiology
297.8K Views

항생 물질 감수성 시험: 두 항생제의 MIC 값을 결정하고 항생 시너지를 평가하기 위한 Epsilometer 검사
Microbiology
93.9K Views

현미경 및 염색: 그램, 캡슐 및 내생포자 염색
Microbiology
364.0K Views

플라크 분석: 플라크 형성 단위(PFU)로서 바이러스 역가를 결정하는 방법
Microbiology
186.5K Views

적응 염화칼슘 절차를 이용한 E.coli 세포의 형질변환
Microbiology
87.1K Views

접합: 공여 E.coli에서 수용 E.coli로 암피실린 내성을 전달하는 방법
Microbiology
38.4K Views

파지 형질도입: 공여 E.coli에서 수용 E.coli로 암피실린 내성을 전달하는 방법
Microbiology
29.1K Views
Copyright © 2025 MyJoVE Corporation. 판권 소유