16S rRNA测序:基于PCR的技术,用于识别细菌物种
Overview
资料来源:埃瓦·布科夫斯卡-法尼班德1号,蒂尔德·安德斯森1号,罗尔夫·卢德1号
1临床科学隆德系,感染医学系,生物医学中心,隆德大学,221 00 隆德,瑞典
地球是数百万种细菌的栖息地,每种细菌物种都有其特殊特征。细菌物种的鉴定在微生物生态学中广泛用于确定环境样本的生物多样性和医学微生物学,以诊断受感染的患者。细菌可以使用传统的微生物学方法进行分类,如显微镜、特定介质的生长、生化和血清学测试以及抗生素敏感性检测。近几十年来,分子微生物学方法彻底改变了细菌鉴定。一个流行的方法是16S核糖体RNA(rRNA)基因测序。该方法不仅比传统方法更快、更准确,而且能够识别在实验室条件下难以生长的菌株。此外,在分子水平上对菌株进行分化,使表性相同的细菌(1-4)之间具有鉴别力。
16S rRNA 与 19 种蛋白质的复合物结合,形成细菌核糖体 (5) 的 30S 亚单位。它由16S rRNA基因编码,由于其在核糖体组装中的基本功能,在所有细菌中都存在并高度保存;然而,它也包含可变区域,可以作为特定物种的指纹。这些特征使16S rRNA基因成为理想的基因片段,可用于细菌的鉴定、比较和遗传分类(6)。
16S rRNA基因测序基于聚合酶链反应(PCR)(7-8),然后是DNA测序(9)。PCR 是一种分子生物学方法,用于通过一系列周期来扩增 DNA 的特定片段,其中包括:
i) 双绞合DNA模板的变性
ii) 与模板互补的引物(短寡核苷酸)的退火
iii) DNA聚合酶的引物延伸,合成新的DNA链
方法的原理图概述如图1所示。
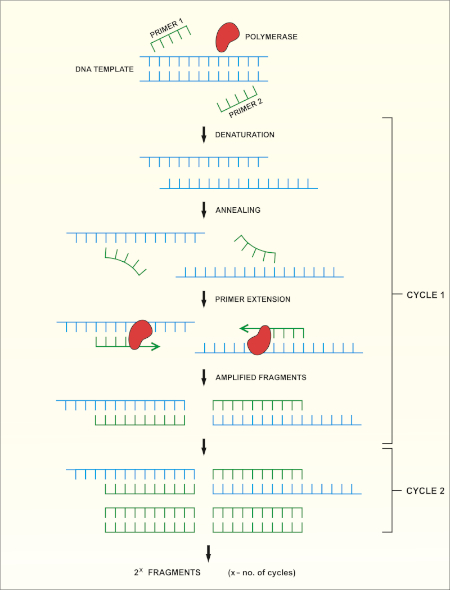
图 1:PCR 反应的原理概述。请点击此处查看此图的较大版本。
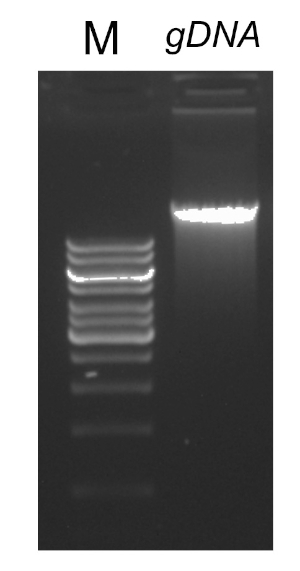
有几个因素对成功的PCR反应很重要,其中之一是DNA模板的质量。使用标准协议或商业试剂盒从细菌中分离染色体DNA。应特别注意获得不含污染物的DNA,这些污染物可以抑制PCR反应。
16S rRNA基因的保存区域允许设计通用引物对(一个正向和一个反向),可以结合和放大任何细菌物种的目标区域。目标区域的大小可能有所不同。虽然一些引热剂对可以扩增大多数16S rRNA基因,但其他仅扩增部分。常用引种示例如图1所示,其绑定位点如图2所示。
| 引种名称 | 序列 (5'~3') | 前进/后退 | 参考 |
| 8F b) | 阿加加特加特格格格格格格格股份公司 | 向前 | -1 |
| 27F | 阿加加特·加·加金茨格格格股份公司 | 向前 | -10 |
| 515F | 格格格格·CMGCCGGGTA | 向前 | -11 |
| 911R | 海合会加特克特科特加特加 | 反向 | -12 |
| 1391R | GACGGGGTGTGTRCA | 反向 | -11 |
| 1492R | GGTTACCTGTTT | 反向 | -11 |
表 1:用于扩增16S rRNA基因a)的标准寡核苷酸的例子。
a)通过计算正向和反向底漆的装订位点之间的距离(见图2),可以估计使用不同引漆组合生成的PCR产品的预期长度,例如PCR的大小使用底漆对 8F-1492R 的产品为 ±1500 bp,对于底漆对 27F-911R =900 bp。
b)也称为 fD1

图 2:16S rRNA序列和引结位点的代表性图。保留区域为灰色,可变区域用对角线填充。为了达到最高分辨率,引基8F和1492R(基于rRNA序列位置的名称)用于放大整个序列,从而允许对基因的几个可变区域进行测序。请点击此处查看此图的较大版本。
PCR 的循环条件(即脱氧核糖核酸变性、引物退火和合成所需的温度和时间)取决于所使用的聚合酶类型和引物的特性。建议遵循制造商针对特定聚合酶的指南。
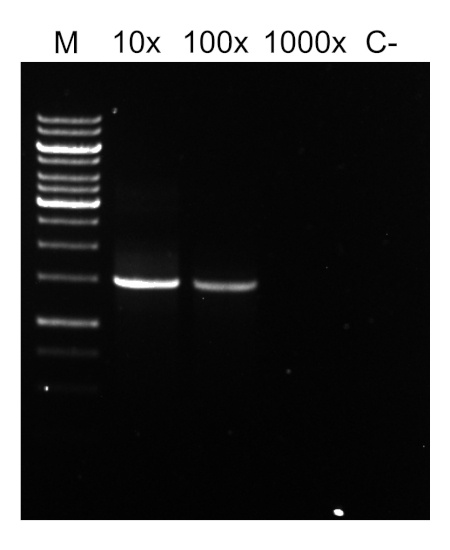
PCR程序完成后,通过胶质电泳对产品进行分析。成功的 PCR 产生一个预期大小的单个波段。在测序之前,必须对产品进行纯化,以去除PCR反应中存在的残留底漆、脱氧核苷酸、聚合酶和缓冲液。纯化的DNA片段通常被发送到商业测序服务;然而,一些机构在他们自己的核心设施进行DNA测序。
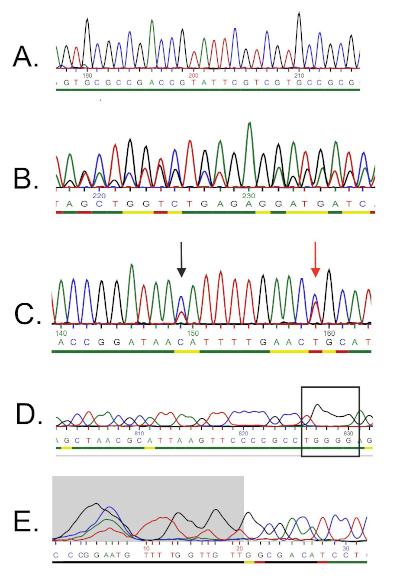
DNA 序列由计算机从 DNA 色谱图自动生成,必须仔细检查质量,因为有时需要手动编辑。按照此步骤,将基因序列与沉积在16S rRNA数据库中的序列进行比较。识别相似区域,并传递最相似的序列。
Procedure
1. 设置
- 在处理微生物时,需要遵循良好的微生物实践。所有微生物,特别是未知样品,都应作为潜在的病原体处理。遵循无菌技术,避免污染样品、研究人员或实验室。处理细菌前后洗手,使用手套,并穿防护服。
- 对基因组DNA分离和PCR产品纯化的实验方案进行风险评估。有些试剂可能是有害的!
- 纯培养对16S rRNA测序至关重要。在继续分离基因组DNA之前,确保起始材料是完全纯净的。这可以通过条纹电镀来隔离单个菌落。如果需要,这些可以单独在盘子上或肉汤中进一步生长。
- 需要实验室设备:
- 用于 PCR 的热循环器。热循环器的功能是根据设定的程序提高和降低温度。创建程序时,系统将要求您输入每个 PCR 步骤的温度和时间值以及循环总数。
- 阿加玫瑰凝胶电泳系统。它用于根据DNA片段的大小和电荷分离DNA片段。在此协议中,阿加糖凝胶电泳将用于可视化分离基因组DNA和PCR产品的质量。
2. 议定书
Application and Summary
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.
Tags
跳至...
此集合中的视频:

Now Playing
16S rRNA测序:基于PCR的技术,用于识别细菌物种
Microbiology
187.9K Views

创建维诺格拉茨基柱:在沉积物样本中丰富微生物物种的方法
Microbiology
128.2K Views

连续稀释和电镀:微生物枚举
Microbiology
313.9K Views

富集培养:在选择性和微分介质上培养有氧和厌氧微生物
Microbiology
131.7K Views

纯培养和条纹电镀:从混合样品中分离单一细菌菌落
Microbiology
165.7K Views

生长曲线:使用菌落成形单位和光学密度测量生成生长曲线
Microbiology
293.0K Views

抗生素敏感性测试:Epsilo计测试,以确定两种抗生素的MIC值,并评估抗生素协同效应
Microbiology
93.4K Views

显微镜和染色:革兰克、胶囊和内孔染色
Microbiology
362.4K Views

斑块测定:确定病毒性皮石为斑块形成单位(PFU)的方法
Microbiology
185.6K Views

使用自适应氯化钙程序对大肠杆菌细胞的转化
Microbiology
86.3K Views

结合:将阿霉素耐药性从捐赠者转移到受体大肠杆菌的方法
Microbiology
38.1K Views

噬菌体转导:将氨基青霉素耐药性从捐赠者转移到受体大肠杆菌的方法
Microbiology
28.9K Views
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。