
Method Article
Edição de genoma mediado por CRISPR-Cas9 no Filamentous Ascomycete Huntiella omanensis
Neste Artigo
Resumo
O sistema de edição de genomas CRISPR-Cas9 é um editor de genomas fácil de usar que tem sido usado em espécies modelo e não-modelo. Aqui apresentamos uma versão baseada em proteínas deste sistema que foi usada para introduzir um codon stop prematuro em um gene de acasalamento de um fungo ascomiceto não-modelado.
Resumo
O sistema de edição de genomas CRISPR-Cas9 é uma ferramenta molecular que pode ser usada para introduzir mudanças precisas nos genomas de espécies modelo e não-modelo. Esta tecnologia pode ser usada para uma variedade de abordagens de edição de genomas, desde nocautes genéticos e knockins até mudanças mais específicas, como a introdução de alguns nucleotídeos em um local alvo. A edição de genomas pode ser usada para uma infinidade de aplicações, incluindo a caracterização funcional parcial dos genes, a produção de organismos transgênicos e o desenvolvimento de ferramentas de diagnóstico. Em comparação com as estratégias de edição de genes anteriormente disponíveis, o sistema CRISPR-Cas9 tem se mostrado fácil de estabelecer em novas espécies e possui alta eficiência e especificidade. A principal razão para isso é que a ferramenta de edição usa uma molécula de RNA para atingir o gene ou sequência de interesse, tornando o design da molécula alvo simples, dado que as regras padrão de pareamento de base podem ser exploradas. Semelhante a outros sistemas de edição de genomas, os métodos baseados em CRISPR-Cas9 também requerem protocolos de transformação eficientes e eficazes, bem como acesso a dados de sequência de boa qualidade para o projeto das moléculas de RNA e DNA alvo. Desde a introdução deste sistema, em 2013, tem sido usado para criar geneticamente uma variedade de espécies modelo, incluindo Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster e Mus musculus. Posteriormente, pesquisadores que trabalham com espécies não-modelo têm aproveitado o sistema e utilizado para o estudo de genes envolvidos em processos tão diversos quanto o metabolismo secundário em fungos, crescimento de nematoides e resistência a doenças nas plantas, entre muitos outros. Este protocolo detalhado abaixo descreve o uso do protocolo de edição do genoma CRISPR-Cas9 para a truncação de um gene envolvido no ciclo sexual de Huntiella omanensis, um fungo ascomíceo filamentoso pertencente à família Ceratocystidaceae.
Introdução
A crescente disponibilidade de genomas e transcritos de alta qualidade, totalmente montados, melhorou muito a capacidade de estudar uma ampla variedade de processos biológicos em uma matriz de organismos1. Isso é verdade tanto para espécies modelo quanto para espécies não-modelo, muitas das quais podem oferecer uma compreensão mais diversificada dos processos biológicos. Esses tipos de dados podem ser usados para a descoberta genética, a identificação de redes de transcrição e comparações de genoma e transcriptome inteiros, cada uma delas com seu próprio conjunto de aplicações. No entanto, enquanto os genes estão sendo previstos, anotados e putativamente ligados a diferentes vias funcionais a uma taxa nunca antes vista, a caracterização funcional desses genes permanece para trás, limitada pelos kits de ferramentas moleculares disponíveis para muitas espécies. Este é particularmente o caso das espécies não-modelo, onde os dados genômicos são relativamente fáceis de gerar, mas onde a caracterização molecular adicional tem sido quase impossível1,,2.
A caracterização parcial das funções de genes específicos importantes para a biologia das espécies fúngicas pode ser alcançada por experimentos de nocaute ou knockin seguidos de análise fenotípica das cepas mutantes3. Esses dois sistemas dependem inteiramente da disponibilidade de protocolos de engenharia genética, incluindo, no mínimo, um sistema de transformação e um sistema de edição genética. Existem uma série de diferentes sistemas de transformação que foram desenvolvidos em uma variedade de fungos filamentosos4. Sistemas físicos como os que dependem da biolística e eletroporação foram desenvolvidos em Trichoderma harzianum5 e Aspergillus niger6 ,respectivamente. Sistemas que utilizam produtos químicos como cloreto de cálcio ou acetato de lítio foram desenvolvidos em Neurospora crassa7. Por fim, sistemas biológicos que dependem do uso de tumefaciens agrobacterium para transformação têm sido utilizados com sucesso no Ceratocystis albifundus8.
Em contraste com a disponibilidade de diferentes protocolos de transformação, os sistemas de edição de genomas são menos abundantes. Muitos dos experimentos tradicionais de caracterização funcional realizados em fungos filamentosos utilizaram uma construção de nocaute de marcador dividido na forma de um marcador selecionável ladeado por regiões de homologia à região-alvo ou gene no genoma3. O método conta com a reparação de DNA direcionada à homologia (RH), que instala recombinação homologiosa entre a construção do nocaute e a região de interesse. Este evento de recombinação resulta na substituição do gene de interesse pela sequência do marcador selecionável. Infelizmente, embora isso tenha sido bem sucedido em muitas espécies, incluindo Cercospora nicotianae10, Aspergillus fumigatus11 e Grosmannia clavigera12, as taxas de recombinação homologos são altamente variáveis entre diferentes espécies fúngicas, tornando este um protocolo ineficiente e às vezes inutilizável em determinadas espécies3.
Outros sistemas de edição de genomas, incluindo aqueles que fazem uso de núcleos de dedo de zinco (ZFNs) e núcleos de efeitos (TALENs) semelhantes a ativação de transcrição (TALENs) representaram uma grande melhoria nos sistemas mais antigos, particularmente dadas suas habilidades para fazer mudanças específicas e direcionadas13. Tanto zfns quanto TALENs são compostos por uma proteína nuclease e uma proteína capaz de reconhecer sequências específicas de nucleotídeos13. Após o reconhecimento, a nuclease induz uma quebra de DNA duplamente encalhada que pode facilitar a introdução de mutações específicas. Para provocar mudanças no genoma, a região proteica que reconhece a sequência de nucleotídeos precisa ser projetada especificamente para cada experimento. Devido a essa dependência de interações proteína-nucleicas de ácido para orientar a edição, projetar e produzir as moléculas de alvo para cada experimento de nocaute ou knockin é difícil e trabalhosa intensiva14,15. Ilustrativos desses desafios, pouquíssimos fungos filamentosos foram submetidos à edição de genomas usando esses sistemas. Um exemplo é o sistema baseado em TALENs que foi desenvolvido no fungo da explosão de arroz, Magnaporthe oryzae16.
Sem dúvida, a maior revolução para o campo da edição de genomas foi a descoberta e o desenvolvimento subsequente do sistema CRISPR-Cas9, um editor de genomas que permite o decote direcionado de uma sequência de interesse por uma endonuclease que é guiada por uma molécula de RNA. Esta foi uma grande melhoria nos editores de genomas previamente desenvolvidos que dependiam de interações proteína-nucleicos ácidos como a principal vantagem do sistema CRISPR-Cas9 é que ele conta com uma molécula de RNA para atingir a região de interesse. Isso significa que o sistema depende de uma interação RNA-DNA e, portanto, as regras padrão de pareamento base podem ser exploradas ao projetar cada experimento15.
O sistema CRISPR-Cas9 conforme detalhado aqui é composto por três componentes principais: um único guia RNA (sgRNA), a enzima Cas9 e um DNA doador (dDNA)17. O sgRNA é composto por uma região de 20 nucleotídeos chamada protoespaço, bem como uma região mais longa chamada andaime18. A região do protoespaço é usada para guiar o sistema de edição para a região alvo e, portanto, é redesenhada para cada experimento. O andaime é a região do RNA que se liga fisicamente à enzima Cas9 para formar a ribonucleoproteína (RNP) e é, portanto, idêntica independentemente da região a ser alvo. A enzima Cas9 facilita fisicamente o decote do DNA alvo, utilizando o protoespaço como guia para identificar essa região19. O último componente, o dDNA, é opcional e seu uso depende do experimento particular20. O dDNA abriga a sequência que deve ser especificamente inserida na região sendo cortada pela enzima Cas9, e é, portanto, ideal para experimentos de knockin genético onde um gene está sendo introduzido no genoma ou para experimentos de nocaute genético onde um gene de resistência a antibióticos ou outro marcador selecionável está sendo introduzido para substituir o gene de interesse. O dDNA também pode ser projetado de forma a introduzir novas sequências no genoma. Por exemplo, conforme detalhado abaixo, é possível introduzir um codon stop in-frame em uma determinada região no gene de interesse quando uma truncação genética é necessária21. Outras aplicações incluem a mutação de regiões específicas do gene, como um domínio funcional22, ou a introdução de uma sequência de marcação23.
Um grande benefício do uso do sistema CRISPR-Cas9 é sua versatilidade24. Um exemplo dessa adaptabilidade é que a enzima Cas9 pode ser introduzida na célula hospedeira em uma de suas três formas: DNA, RNA ou proteína, dependendo do sistema de transformação particular que está sendo utilizado. Quando introduzido na forma de DNA, o gene cas9 é frequentemente incluído em um plasmídeo juntamente com um marcador selecionável, um para expressar o sgRNA e, se necessário, um codificando a sequência dDNA25. A principal vantagem deste sistema é que apenas uma única construção precisa ser transformada na célula e a transformação bem sucedida garante que todos os componentes necessários para a edição de genomas mediados pelo CRISRP-Cas9 estejam presentes. No entanto, este método conta com a disponibilidade de um sistema de expressão para as espécies hospedeiras. Para cas9 induzir com sucesso danos de DNA, ele precisa ser expresso em níveis elevados e, portanto, é necessário um promotor adequado e potencialmente específico. Para espécies não-modelo onde tais promotores ainda não foram desenvolvidos, isso pode ser um fator desfante e, portanto, a capacidade de introduzir Cas9 em RNA ou forma proteica pode ser uma opção mais atraente. A introdução do RNA na célula traz seus próprios desafios, particularmente no que o RNA é instável e pode não sobreviver ao processo de transformação. Além disso, quando introduzida na forma DEM ou RNA, a sequência genética Cas9 pode precisar ser otimizada para uso no sistema hospedeiroespecífico 17. Por exemplo, o gene cas9 de Streptococcus pyogenes pode não funcionar em uma célula hospedeira de mamíferos e um gene cas9 que foi otimizado para uso em uma célula mamífera pode não funcionar em uma célula vegetal. Todos esses desafios podem ser superados usando a forma proteica do Cas9, que, juntamente com o sgRNA, pode ser montado em um RNP e transformado na célula hospedeira26,27. Este sistema não conta com nenhum sistema de expressão endógena ou otimização de codon e deve, portanto, funcionar na maioria das espécies não-modelo. A desvantagem do sistema à base de proteínas é que ele não é compatível com sistemas de transformação baseados em DNA, como a transferência mediada pelo Agrobacterium. Assim, para que o método baseado em proteínas funcione, um protocolo de transformação como aqueles que dependem de protoplastos ou biolísticas precisa estar disponível. Este sistema baseado em RNP tem sido usado com sucesso nos fungos filamentosos, Fusarium oxysporum26 e Mucor circinelloides27.
Huntiella omanensis, um membro da família Ceratocystidaceae, é um fungo cosmopolita frequentemente encontrado em plantas lenhosas recém-feridas28. Enquanto dados de genoma e transcriptome de alta qualidade estão disponíveis para esta espécie28,,29,,30, nenhum protocolo de transformação ou edição de genomas foram desenvolvidos. Até o momento, a pesquisa sobre H. omanensis se concentrou nos componentes genéticos subjacentes de seu ciclo sexual29,31. Este fungo exibe um ciclo sexual heterotálico típico, com reprodução sexual ocorrendo exclusivamente entre isolados dos tipos de acasalamento MAT1-1 e MAT1-231. Em contraste, os isolados MAT1-2 da Huntiella moniliformis intimamente relacionadas são capazes de reprodução sexual independente e completam um ciclo sexual na ausência de um parceiro MAT1-131. Acredita-se que essa diferença nas capacidades sexuais seja, pelo menos em parte, devido a uma grande diferença no gene de acasalamento, MAT1-2-7, onde h. omanensis abriga uma cópia completa e intacta, enquanto o gene é severamente truncado em H. moniliformis29,31. Para caracterizar ainda mais o papel desse gene na reprodução sexual, o gene MAT1-2-7 de H. omanensis foi truncado para imitar a truncação vista em H. moniliformis21.
O protocolo abaixo detalha a transformação da H. omanensis e a truncação do gene MAT1-2-7 usando uma versão baseada em proteínas do sistema de edição de genomas CRISPR-Cas9. Este protocolo foi desenvolvido depois que as abordagens de substituição de genes baseados em recombinação homólogo e edição de genoma CRISPR-Cas9 baseada em plasmídeos não tiveram sucesso.
Protocolo
1. Design e síntese do sgRNA
- Para identificar possíveis regiões protoespaciais que farão parte do sgRNA, pesquise manualmente através do gene de interesse para trigêmeos de 5' NGG 3', usando a função de pesquisa em qualquer programa que esteja sendo usado. Anote esses trigêmeos como sequências PAM.
NOTA: Vários programas de software estão disponíveis que pesquisam e anotam as sequências potenciais de PAM e protoespaço.- Selecione o upstream de 20 bps de cada uma das sequências PAM identificadas e anote essas sequências como potenciais protoespaciadores.
- Para decidir sobre um único protoespaço, execute as seguintes etapas de filtragem para descartar sequências de baixa qualidade.
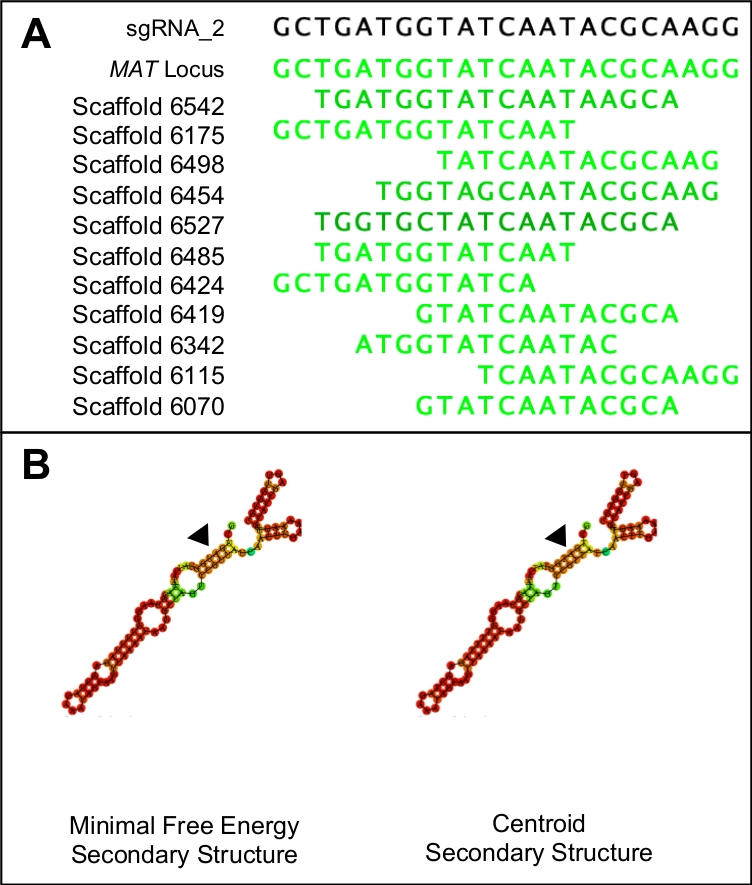
- Para testar a especificidade dos potenciais protoespaciares, combine a sequência PAM e o protoespaço em uma única sequência e use-a como uma consulta BLASTn contra todo o genoma. Descarte quaisquer protoespaciares que mostrem semelhança com qualquer região do genoma que não seja a região alvo(Figura 1A).
- Para garantir que a molécula de RNA se dobre na estrutura 3D correta para se ligar à enzima Cas9, crie uma sequência combinada, incluindo o protoespaço e a sequência de andaimes específicas da enzima Cas9.
- Carregue cada uma das sequências combinadas de andaimes protoespaço-andaimes em uma ferramenta de previsão de estrutura secundária RNA(Tabela de Materiais).
- Analise os resultados comparando a energia mínima livre e as estruturas secundárias centroides.
NOTA: Os candidatos ideais protoespaçoares terão energia mínima mínima idêntica e estruturas secundárias centroides. Ambas as estruturas secundárias devem ser compostas por três laços de haste, interrompidos por cinco estruturas de anéis. As estruturas também devem apresentar altas probabilidades de ligação (indicadas em vermelho) em toda a estrutura, exceto para a região que representa o protoespaço(Figura 1B). Os valores energéticos reais não são relevantes.
- Escolha um candidato final daqueles que passaram as etapas de filtragem acima selecionando o candidato mais próximo da região específica que está sendo alvo.
- Para sintetizar o sgRNA como uma única molécula de RNA, use um kit de síntese de sgRNA(Tabela de Materials) compatível com a enzima Cas9 específica que será usada (por exemplo, Cas9 de Streptococcus pyogenes).
NOTA: As seguintes etapas podem depender do kit de síntese sgRNA utilizado. Caso seja utilizado um kit diferente, siga as instruções do fabricante. Alternativamente, o sgRNA pode ser encomendado pré-sintetizado. Ao trabalhar com RNA, use reagentes sem nuclease e descartáveis.- Se o protoespaço de 20 bp escolhido na etapa 1.3 acima não abrigar um G no final de 5', adicione um G a esta região.
- Adicione a sequência de promotores T7 ao final de 5' da sequência de destino. Esta sequência é padrão e é de 5' TTCTAATACGACTCACTATAG 3'.
- Adicione uma sequência de 14 nt ao final de 3' da sequência de destino. Esta sequência é específica do kit e é de 5' GTTTTAGAGCTAGA 3' para o kit usado aqui.
- Ordene o fragmento resultante 5' TTCTAATACGACTCACTATAG(N)20 GTTTTAGAGCTAGA, com (N)20 representando o protoespaço selecionado presintigido(Tabela de Materiais).
- De acordo com o protocolo do fabricante (Tabela de Materiais), combine os seguintes reagentes à temperatura ambiente: 2 μL de água, 10 μL de tampão de reação, 5 μL de sequência de protoespaço sintetizado, 1 μL de 0,1 M DTT e 2 μL de enzima transcriptase.
- Incubar esta solução a 37 °C por 30 min e transferir para o gelo.
- Adicione 30 μL de água e 2 μL de DNase I, misture e incubar a 37 °C por mais 15 minutos.
- Visualize o sgRNA resultante em um gel de 2% de agarose.
2. Testando a capacidade in vitro de decote do sgRNA
NOTA: Esta etapa é opcional, mas é recomendada.
- Projetilhas de design que amplificarão um fragmento que abriga a sequência do local que o sgRNA escolhido irá atingir e amplificar a região usando uma polimerase de DNA padrão.
NOTA: Se possível, desenhei os primers de tal forma que o decote no local alvo produzirá dois fragmentos de tamanhos muito diferentes que podem ser facilmente distinguidos um do outro em um gel de agarose padrão. - Monte a ribonucleoproteína sgRNA-Cas9 combinada (RNP) incubando uma solução composta por 30 nM sgRNA como sintetizada acima, proteína Cas9 de 30 nM, tampão de reação de 10x e 10 μL de água a 25 °C por 10 min.
- Teste a capacidade de decote do sgRNA adicionando o produto PCR da região alvo na solução RNP a uma concentração final de 3 nM.
- Incubar a solução a 37 °C por 15 min.
- Adicione 3 μg de proteinase K e 2 μg de RNase à solução para parar a reação de decote e incubar à temperatura ambiente por 10 minutos.
- Visualize os fragmentos de DNA resultantes em um gel de 2% de agarose. O sgRNA é adequado para experimentação in vivo se duas faixas do tamanho esperado forem observadas no gel.
3. Design e síntese do dDNA
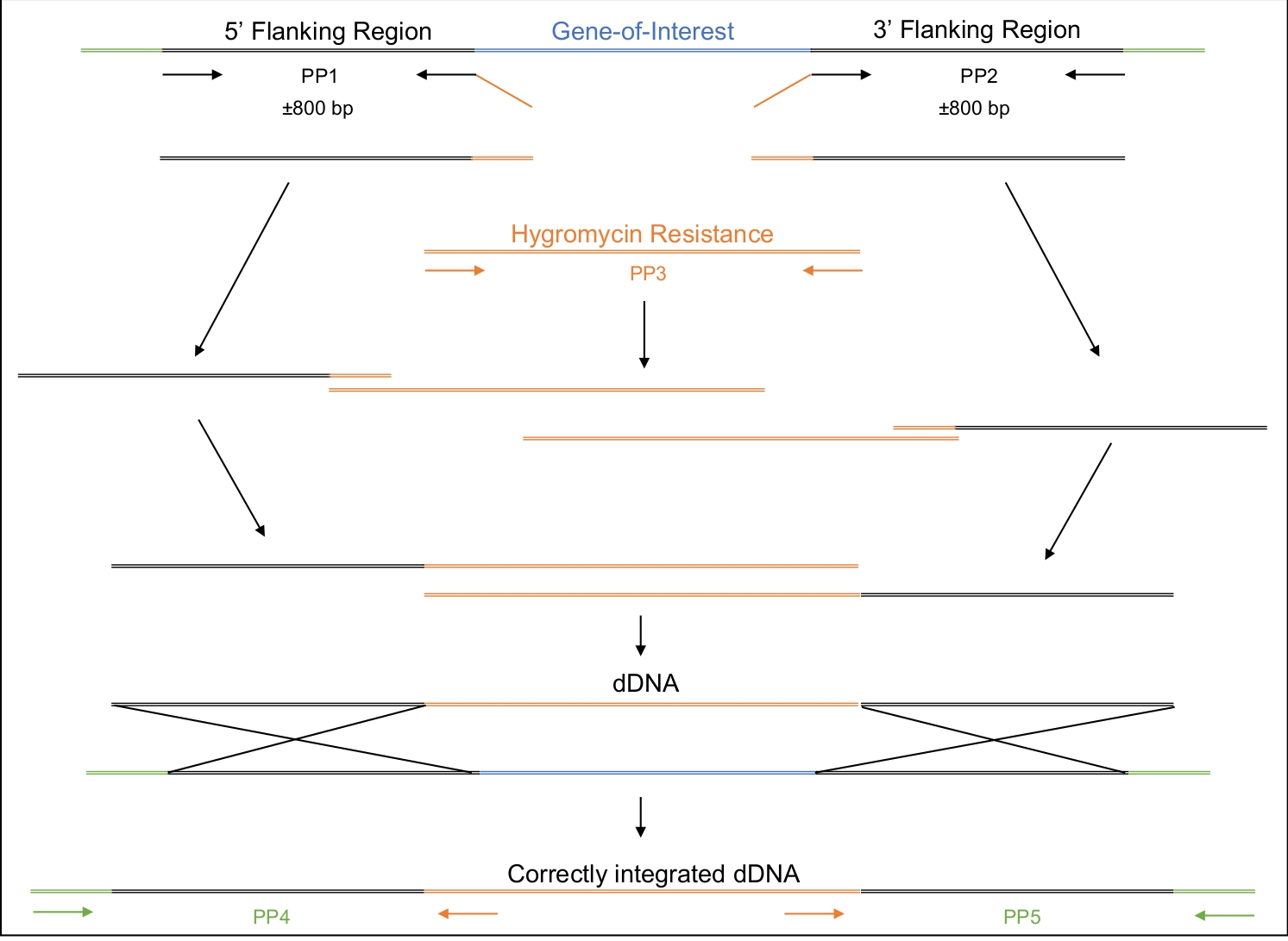
- Projetar o dDNA a ser composto por três regiões: 5' e 3' regiões com complementaridade à região genômica sendo alvo e uma região média abrigando um marcador selecionável(Tabela de Materiais, Figura 2).
NOTA: Outras sequências específicas também podem ser adicionadas a este marcador selecionável. Para este experimento em particular, uma sequência de stop codon (5' TGA 3') foi adicionada pouco antes do marcador selecionável, introduzindo assim um codon stop in-frame no gene. O dDNA pode ser ordenado pré-sintthesized. Alternativamente, o dDNA pode ser amplificado e montado usando uma abordagem PCR de forma passo a passo, sobreposta conforme detalhado abaixo.- Projete primers para amplificar aproximadamente 800 bp das regiões de flanqueamento de 5' e 3'. Adicione 20 nt de sequência que é complementar à sequência do marcador selecionável ao primer reverso da região de 5' e primer dianteiro da região de 3' e primer dianteiro da região de 3'.
- Primers de design que amplificam o marcador selecionável, garantindo que o produto amplificado abriga o gene de resistência, bem como um promotor conhecido por trabalhar nas espécies de interesse.
- Utilizando uma polimerase de DNA de alta fidelidade(Tabela de Materiais),amplifique as três regiões de DDNA. Amplifique as regiões de 5' e 3' do gDNA do organismo que está sendo editado. Amplie o marcador selecionável de uma fonte relevante.
- Em uma única reação, combine a região amplificada de 5' com o marcador selecionável e, usando uma polimerase de DNA de longo alcance e alta fidelidade, amplie toda a região.
- Em uma segunda reação única, combine a região amplificada de 3' com o marcador selecionável e, usando uma polimerase de DNA de longo alcance e alta fidelidade, amplie toda a região.
- Finalmente, combine os dois produtos PCR anteriores em uma única reação e amplie toda a sequência dDNA com uma polimerase de DNA de longo alcance e alta fidelidade.
- Visualize o fragmento de DNA em um gel de 1% de agarose. No caso de dois ou mais fragmentos serem produzidos, purifique o fragmento de tamanho correto do gel usando um kit de purificação de gel.
4. Extração de protoplastos
- Para produzir conidia, inocular 200 mL de caldo fresco de extrato de malte de 2% (MEB) em um frasco de 500 mL com um bloco de ágar coberto de 1 cm x 1 cm de mycelia.
NOTA: Nem todos os fungos são capazes de produzir conidia. Nesse caso, a micélio também pode ser usada. Isso normalmente exigirá maiores concentrações de enzimas ainda mais no protocolo.- Incubar a cultura líquida em uma incubadora de agitação a 25 °C com agitação a 120 rpm por 24 - 48 h.
NOTA: Este tempo de incubação e temperatura foram otimizados para H. omanensis. Isso precisará ser otimizado para outras espécies. - Para colher a conidia; filtrar a cultura líquida através de uma camada de pano de laboratório estéril (por exemplo, Miracloth), transfira a suspensão coniifical em tubos de centrífugas de 50 mL e centrífuga a 3.220 x g a 4 °C por 10 min. Descarte o supernaspeso.
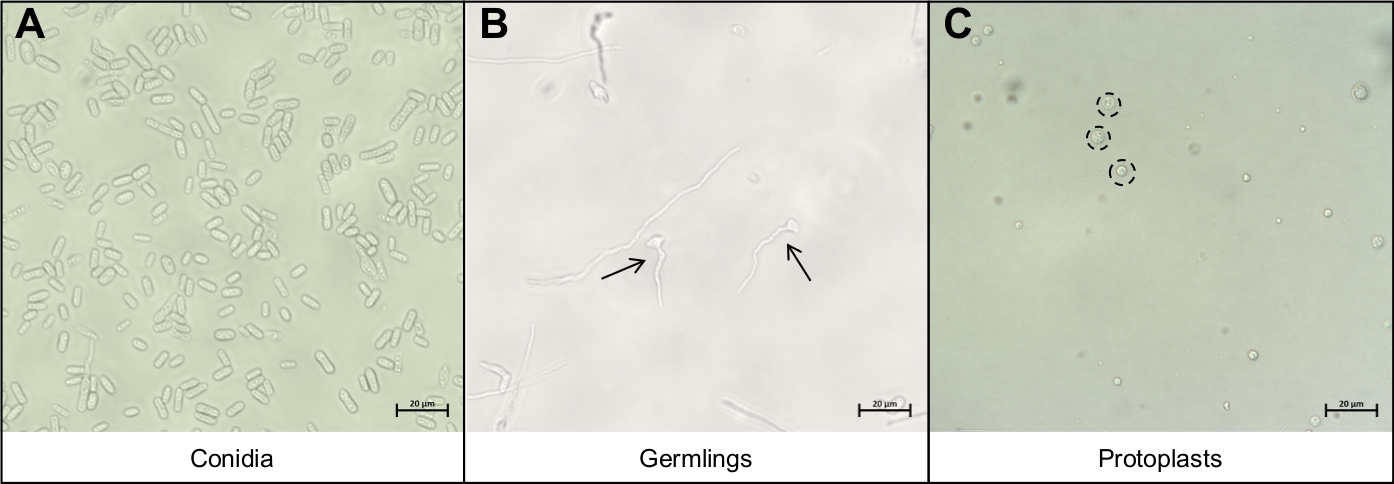
- Resuspenda a conidia em 5 mL de água e pipeta 10 μL da solução conidia em um slide de microscópio e cubra com um deslizamento de tampa. Visualize usando um microscópio composto sob ampliação de 40x para garantir que apenas conidia tenha sido recuperada(Figura 3A).
- Em um frasco de 500 mL, inocular 200 mL de MEB fresco de 1% com o volume total de conidia resuspended.
- Incubar a cultura líquida em uma incubadora de agitação a 25 °C com agitação a 120 rpm por até 12 h.
NOTA: Este tempo de incubação foi otimizado para H. omanensis. Isso precisará ser otimizado para outras espécies. - Para colher os germes, transfira a cultura líquida para tubos de centrífugas de 50 mL e centrífuga a 3.220 x g a 4 °C por 10 min. Descarte o supernaspeso.
- Resuspend os germlings em até 10 mL de sorbitol de 1 M.
- Pipeta 10 μL da solução germinante em um slide de microscópio e tampa com um deslizamento de tampa. Visualize usando um microscópio composto sob ampliação de 40x para garantir que apenas germes tenham sido recuperados(Figura 3B).
NOTA: O protocolo pode ser pausado aqui. Armazene germlings em 1 M sorbitol a -80 °C.
- Incubar a cultura líquida em uma incubadora de agitação a 25 °C com agitação a 120 rpm por 24 - 48 h.
- Para lise as paredes celulares dos germes jovens e solte os protoplastos, adicione 1 mL da suspensão germinal a 9 mL de enzima lysing em várias concentrações em um frasco estéril de 50 mL.
NOTA: Diferentes concentrações de enzimas e tempos de incubação são usados e podem ser encontrados na Tabela 1. As enzimas e concentrações também podem variar dependendo do fungo e precisarão ser otimizadas para cada espécie.- Incubar a solução de enzimas de esporo em uma incubadora de agitação a 25 °C com agitação a 80 rpm por 2 a 3 h.
- Filtre a solução de protoplasto através de uma camada de pano de laboratório estéril e colete os protoplastos por centrifugação a 1.810 x g a 4 °C por 10 min. Descarte o supernaspeso.
NOTA: Protoplastos são células sem paredes celulares e, portanto, são muito sensíveis à interrupção mecânica. Certifique-se de manuseá-los com cuidado, especialmente quando estiver em pipeta. - Resuspenque cuidadosamente a pelota de protoplasto em 200 μL de tampão STC(Tabela de Materiais).
- Pipeta 10 μL da solução protoplastia em um slide de microscópio e tampa com um deslizamento de tampa. Visualize usando um microscópio composto sob ampliação de 40x para garantir que apenas protoplastos tenham sido recuperados(Figura 3C).
- Usando um hemócito, conte e calcule o número de protoplastos gerados nas etapas acima. Diluir a solução protoplasta em alíquotas contendo aproximadamente 5 x 106 protoplastos.
NOTA: O protocolo pode ser pausado aqui. Armazene protoplastos no buffer STC a -80 °C.
5. Transformação assistida por Protoplast e PEG e recuperação transformadora
- Para iniciar a transformação, combine aproximadamente 5 x 106 protoplastos com um único volume da solução RNP e aproximadamente 6 μg do fragmento dDNA.
NOTA: Os protoplastos são muito sensíveis à interrupção mecânica. Certifique-se de manuseá-los com cuidado, especialmente quando estiver em pipeta.- Usando uma pipeta, escorra lentamente 1 mL de uma solução recém-preparada de 30% PTC na solução de protoplasto e incubar a solução à temperatura ambiente por 20 minutos.
NOTA: Este passo é um passo sensível e muito importante. Certifique-se de usar a solução PTC recém-preparada e soltar a solução sobre as células o mais lentamente e uniformemente possível, criando uma camada hidrofóbica em toda a superfície celular. - Adicione 5 mL de meio de controle osmótico (OCM) à solução protoplastia e pipeta lentamente e suavemente para garantir que a solução esteja completamente misturada.
- Incubar a solução de protoplasto em uma incubadora de agitação a 25 °C com agitação a 80 rpm durante a noite.
- Usando uma pipeta, escorra lentamente 1 mL de uma solução recém-preparada de 30% PTC na solução de protoplasto e incubar a solução à temperatura ambiente por 20 minutos.
- Para selecionar os isolados transformados, divida a solução em 5 placas de cultura vazias de 60 mm.
- Adicione 10 mL de ágar OCM suplementado com 30 μg/mL de higincina B a cada placa de cultura e gire lentamente cada placa para misturar bem.
- Deixe a primeira camada de ágar definir antes de adicionar 10 mL de ágar médio de controle osmótico suplementado com 40 μg/mL de higincina B.
- Permita que a segunda camada de ágar defina e incuba as culturas a 25 °C até que isolados únicos possam ser vistos crescendo através de ambas as camadas de ágar.
- Para recuperar isolados transformados com sucesso, transfira os isolados individuais capazes de crescer através da camada ágar suplementada com 40 μg/mL de higincina B para placas de extrato de malte fresco (MEA) complementadas com 50 μg/mL de higmicina B (MEA-50).
- Incubar as culturas frescas a 25 °C por 5 dias, verificando diariamente o crescimento. Culturas capazes de crescimento sustentado nesta mídia foram transformadas com sucesso e podem ser usadas para estudos mais aprofundados.
6. Confirmação da integração e estabilidade do dDNA
- Para confirmar que o dDNA foi integrado ao genoma na região alvo, projetam primers que flanqueiam os locais de inserção previstos de 5' e 3'(Figura 2).
- Execute dois PCRs usando estes dois conjuntos de primer e uma polimerase de DNA de alta fidelidade. Se ambos os PCRs produzirem amplicons de tamanho e sequência esperados, o dDNA foi integrado com sucesso na região alvo. Em seguida, avalie a mancha mutante para integração estável do dDNA.
- A fim de confirmar que o dDNA foi firmemente integrado ao genoma e será mantido durante o crescimento vegetativo, realize um teste de transferência de mídia.
- Transfira um bloco de ágar coberto de micélio de um isolado mutante em médio MEA-50 para meio MEA não upplementado. Incubar a 25 °C durante 3 dias.
- Transfira um bloco de ágar coberto de mycelia do isolado que cresce no meio MEA para o mea-50 médio. Incubar a 25 °C durante 3 dias.
- Repita este processo, transferindo a micelia de crescimento ativo de meio suplementado para não-aturado por pelo menos quatro rodadas.
NOTA: Se o isolado é capaz de crescimento sustentado no mea-50 médio após muitas transferências, o dDNA foi perfeitamente integrado ao genoma e pode ser mantido através do crescimento vegetativo. A mancha mutante pode ser avaliada para a presença de apenas uma única cópia do dDNA integrado.
- Para confirmar que o dDNA foi integrado ao genoma em um único local, realize uma análise de manchas do Sul.
- Digerir um total de 30 μg de gDNA de cada cepa mutante usando enzimas de restrição HindIII e EcoRI de acordo com os protocolos do fabricante.
NOTA: Embora a escolha da enzima de restrição seja do pesquisador, certifique-se de que o local de reconhecimento da enzima de restrição não esteja presente na sequência dDNA. - Separe o gDNA digerido em um gel de 0,75% de agarose e transfira o DNA para uma membrana de nylon usando procedimentos padrão32.
- Sujeitar a membrana à hibridização usando uma sonda visando a sequência dDNA.
- Projete primers para amplificar uma região curta (300 bp) do dDNA.
- Usando estes primers, sintetize a sonda usando uma mistura de rotulagem PCR DIG.
- Use a sonda recém-sintetizada para hibridização, tratamento e visualização de membranas utilizando procedimentos padrão32. Se apenas uma única banda for vista em cada pista, o dDNA está presente em apenas um único local no genoma. A cepa mutante agora pode ser usada para análise fenotípica e experimentos de caracterização funcional.
- Digerir um total de 30 μg de gDNA de cada cepa mutante usando enzimas de restrição HindIII e EcoRI de acordo com os protocolos do fabricante.
7. Análise fenotípica das cepas mutantes
- Realizar experimentos de acasalamento para determinar se a interrupção do gene MAT teve um efeito sobre as capacidades sexuais do fungo que está sendo estudado.
NOTA: Este passo depende do gene e espécies em particular que estão sendo estudados. Neste caso, acredita-se que o gene alvo esteja envolvido na reprodução sexual e, portanto, foram realizados testes de acasalamento. Se o gene foi pensado, por exemplo, para estar envolvido na reprodução assexual, então algo como produção conidial poderia ser medido.- A fim de testar as capacidades heterotalólicas da cepa mutante, co-inocular um meio MEA fresco com uma cepa mutante, bem como uma cepa de acasalamento oposto. No caso de H. omanensis,mantenha as tampas das placas fechadas, mas não seladas e incubadas à temperatura ambiente por 7 dias. Avaliar visualmente para a produção de estruturas sexuais.
- Para testar as capacidades homotalólicas da cepa mutante, inoculará o meio MEA fresco com uma cepa mutante. No caso de H. omanensis,mantenha as tampas das placas fechadas, mas não seladas e incubadas à temperatura ambiente por 7 dias. Avaliar visualmente para a produção de estruturas sexuais.
- Realizar experimentos de taxa de crescimento para determinar se a interrupção do gene MAT teve um efeito na taxa de crescimento do fungo que está sendo estudado.
- Crie plugues de ágar cobertos de micélios da borda crescente das culturas mutantes e selvagens, inserindo a parte de trás de uma grande ponta de pipeta estéril no ágar.
- Inocular meio MEA fresco com estes plugues de ágar. Certifique-se de que pelo menos três réplicas por cada tipo de cultura sejam feitas.
- Após 3 dias de crescimento a 20 °C, meça o crescimento em dois diâmetros perpendiculares.
- Compare os dados do tipo selvagem e das cepas mutantes.
Resultados
O protocolo descrito acima facilitou a introdução de um códon de parada prematura em um gene de acasalamento do ascomiceto não-modelo, H. omanensis. Este processo utilizou uma versão do sistema de edição de genomas CRISPR-Cas9 e, como tal, um dos passos mais importantes deste protocolo é o design e a síntese de um sgRNA de alta qualidade. A Figura 1 mostra como essa molécula foi projetada de tal forma que a A) tem como alvo especificamente o gene de interesse e mostra pouca semelhança com outras regiões do genoma e B) dobra corretamente para se ligar à proteína Cas9. O sgRNA também deve ser capaz de efetivamente cortar a região alvo. A capacidade do sgRNA de segmentar e permitir o decote da região alvo foi realizada in vitro,produzindo dois produtos do tamanho esperado.
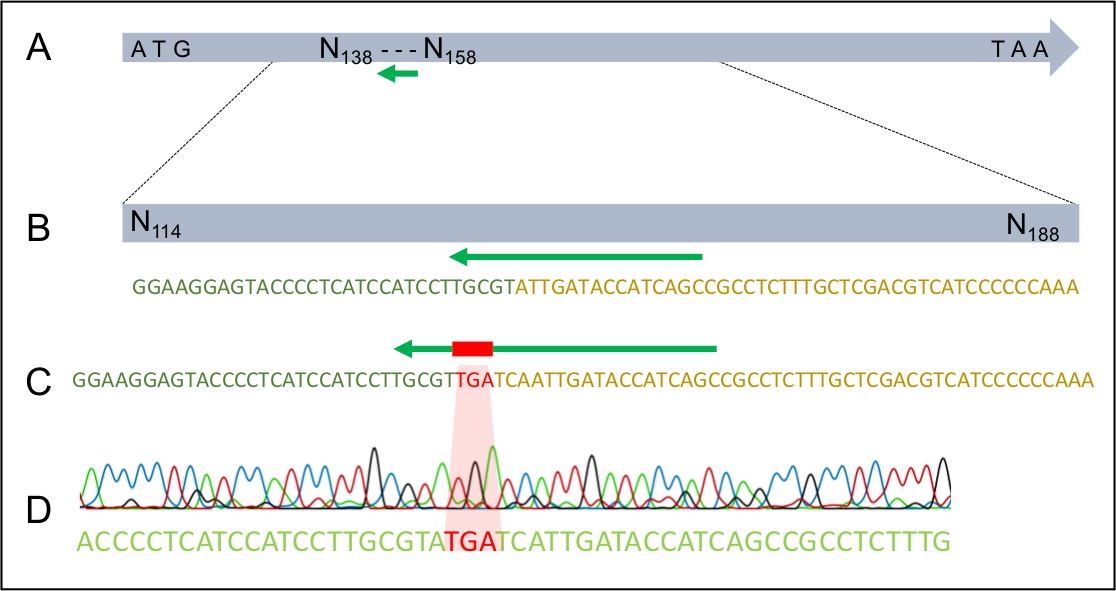
Uma vez que a transformação bem sucedida tenha ocorrido, é importante garantir que o dDNA tenha se integrado ao genoma apenas uma vez e no lugar esperado. A Figura 2 ilustra o design dos primers PCR que visam os sites de inserção, que podem ser usados para rastrear os potenciais transformadores para o local de integração correto. Ao projetar primers que flanqueiam os locais de inserção de 5' e 3', a amplificação só é possível se o dDNA estiver inserido na região correta. A Figura 4 ilustra que o códon de parada prematura foi introduzido no gene MAT1-2-7 no quadro de leitura correto, garantindo que o gene seria truncado de forma semelhante à de H. moniliformis. Além disso, a análise da mancha sulista mostrou que a construção dDNA foi integrada apenas em um único local do genoma.
O sucesso do protocolo foi confirmado após a análise fenotípica das cepas mutantes. No caso do experimento de interrupção do MAT1-2-7, duas cepas mutantes independentes foram desenvolvidas. Em ambos os isolados, a taxa de crescimento radial vegetativo foi significativamente reduzida, sugerindo um efeito pleiotrópico do novo gene de acasalamento(Figura 5). Além disso, os isolados mutantes eram incapazes de completar um ciclo sexual, produzindo apenas estruturas sexuais imaturas que não produziam esporos sexuais(Figura 5). Isso contrastava com os isolados do tipo selvagem, que completaram todo o ciclo sexual dentro de poucos dias de incubação(Figura 5).

Figura 1: Escolhendo um candidato sgRNA adequado.
(A) Um sgRNA adequado só terá semelhança com a região alvo do genoma (neste caso indicado pela sequência de lócus mat). (B) Um sgRNA adequado terá energia mínima mínima idêntica e estruturas secundárias centroides, com os três laços de haste e cinco anéis no laço de passo primário. Além disso, a maioria da estrutura terá altas probabilidades de ligação (indicadas em laranja escuro e vermelho), enquanto probabilidades de ligação mais baixas devem ser vistas na região do protoespaço (indicada pelos triângulos negros). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Design, amplificação e montagem do dDNA.
Os pares de primeira e segunda primer (PP1 e PP2) são usados para amplificar aproximadamente 800 bp upstream (5') e 800 bp downstream (3') do gene de interesse. A cartilha inversa do PP1 e a cartilha dianteira do PP2 incluem regiões de homologia ao de resistência à hisgromicina. O terceiro par de primer amplifica todo o de resistência à hisgromicina. De forma stepwise, os vários amplicons são montados até que todo o DDNA, composto pela região de 5', o de resistência à higinina e região de 3', seja montado. Quando transformado na célula, o dDNA deve recombinar na região onde a enzima Cas9 terá sido direcionada para cortar, substituindo assim o gene de interesse pelo de resistência à hisgromicina. PP4 e PP5 podem ser usados para determinar se o dDNA foi corretamente inserido no genoma no local apropriado. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Os diferentes tipos de células importantes durante o protocolo de extração de protoplasto.
(A) Conidia são usados como material inicial para o protocolo. Estas conidia são permitidas a germinar e crescer até serem(B)germes jovens. A fase ideal de crescimento dos jovens germes é indicada pelas duas setas negras. Outros fios miceliais vistos em(B) são muito maduros para degradação e não devem ser usados. A etapa final do protocolo é a liberação dos protoplastos redondos (C),indicados pelos círculos pretos pontilhados. Essas células não têm mais paredes celulares e, portanto, são muito sensíveis à interrupção mecânica. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: A integração bem sucedida do TGA parar codon no gene MAT1-2-7 de H. omanensis.
(A) O gene H. omanensis MAT1-2-7, com o local alvo sgRNA indicado pelo arqueiro verde. (B) Um esquema ampliado do local alvo sgRNA dentro do gene H. omanensis MAT1-2-7. (C) Um esquema ampliado de uma região do dDNA mostrando o códon de parada ladeado por braços homólogos ao gene MAT1-2-7 de H. omanensis. (D) Cromatograma sequenciamento de Sanger indicando a integração bem sucedida do códon stop no gene MAT1-2-7. Modificado a partir de Wilson et al. 202021. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: As diferenças fenotípicas entre (A) isolados selvagens e (B) isolados mutantes.
As três primeiras imagens em cada painel mostram as diferenças nas capacidades sexuais dos dois tipos isolados. Enquanto o tipo selvagem isola forma ascomata madura durante a reprodução sexual, completa com a exugação de esporos das pontas dos pescoços ascomatal, os isolados mutantes formam apenas estruturas sexuais imaturas que não produzem esporos sexuais. A quarta imagem em cada painel mostra a diferença na taxa de crescimento e morfologia dos dois tipos isolados. Enquanto o isolado do tipo selvagem cresce muito mais rápido e com mais micélio aéreo, o mutante mostra mais lento e está submerso dentro do ágar. Modificado a partir de Wilson et al. 202021. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Reação | Concentração de enzimas | Tempo de degradação |
| Um | 1.250 mg/mL | 180 min. |
| B | 1.875 mg/mL | 180 min. |
| C | 2.500 mg/mL | 150 min. |
| D | 3.750 mg/mL | 150 min. |
| E | 4.375 mg/mL | 120 min. |
| F | 5.000 mg/mL | 120 min. |
Tabela 1: Degradação da solução germosa/mycelia com enzimas de lise de Trichoderma harzianum. As diferentes concentrações de enzimas correspondem a diferentes períodos de incubação, com concentrações mais baixas que requerem incubações mais longas.
Discussão
O protocolo para a transformação bem sucedida de H. omanensis e edição do gene MAT1-2-7 foi demonstrado pela introdução de um codon de parada prematuro no quadro, juntamente com um gene para resistência à higmicina B21. Isso foi conseguido usando uma versão baseada em proteínas do sistema de edição de genomas CRISPR-Cas9. O experimento envolveu a transcrição in vitro do sgRNA, montagem baseada em PCR do dDNA e a co-transformação desses dois ácidos nucleicos com uma enzima Cas9 comercialmente disponível em protoplastos extraídos de H. omanensis
Ao contrário de outros protocolos que dependem da disponibilidade de muitas outras ferramentas moleculares, o protocolo descrito acima pode ser usado com sucesso em espécies para as quais a caixa de ferramentas moleculares ainda é bastante limitada21. O protocolo conta apenas com um sistema de transformação estabelecido e a disponibilidade de dados de NGS, preferencialmente toda a sequência de genomas. Embora um sistema de transformação eficaz possa ter alguma otimização em uma espécie para a qual isso não está disponível, existem muitos protocolos diferentes disponíveis para uma variedade de espécies. Além disso, os dados do genoma estão se tornando cada vez mais disponíveis até mesmo para as espécies mais obscuras e está se tornando mais fácil de gerar de novo se ele ainda não existe.
Dada a duração do protocolo, existem muitas etapas em que as modificações podem ser introduzidas e onde a solução de problemas pode ser necessária. Isso é particularmente verdadeiro para os passos que são considerados espécies específicas. Por exemplo, existem muitas etapas de incubação neste protocolo que precisam ser conduzidas a temperaturas específicas e por períodos específicos de tempo, a fim de gerar tipos de células importantes para o experimento. Essas etapas exigiriam, assim, otimização específica das espécies. Sempre que possível, micrografos das células particulares ou fases de crescimento foram fornecidos para auxiliar na transferência deste protocolo para uma espécie diferente (Figure 1). O tipo e concentração de enzimas utilizadas para degradar as paredes celulares das células fúngicas para liberar os protoplastos também será específico para as espécies de fungos que estão sendo estudadas. Neste protocolo, apenas uma fonte de enzimas de lise é usada, enquanto diferentes combinações de enzimas são necessárias para a extração de protoplastos em espécies como fusarium verticillioides33. Esta etapa depende inteiramente da produção química da parede celular e, portanto, precisará ser otimizada em uma espécie para base de espécies.
Este método é particularmente significativo para aqueles que estudam espécies não-modelo, pois não há dependência de um sistema de expressão. Um método popular de estabelecer o sistema de edição de genomas CRISPR-Cas9 é expressar a proteína Cas9, o sgRNA, bem como o dDNA de um ou dois plasmídeos que são transformados nas células de escolha. Neste caso, o Cas9 precisa ser expresso por um promotor capaz de altos níveis de expressão no organismo em particular que está sendo estudado. Promotores gerais foram desenvolvidos para uso em fungos filamentosos e, embora não sejam compatíveis em todas as espécies, eles permitem uma expressão de baixo nível e podem ser usados com sucesso para expressar, por exemplo, genes de resistência a antibióticos. Esses promotores, no entanto, muitas vezes não permitem altos níveis de expressão e, portanto, não podem ser usados para expressar a proteína Cas9. O uso de uma versão baseada em proteínas do sistema de edição de genomas CRISPR-Cas9 supera essa limitação e permite que o sgRNA e o dDNA sejam co-transformados na célula com uma enzima Cas9 já produzida.
O desenvolvimento deste sistema baseado em proteínas para uso em H. omanensis veio depois de muitas tentativas mal sucedidas de edição de genomas usando tanto a abordagem clássica de marcador split como o sistema CRISPR-Cas9 baseado em plasmídeos. Embora as eficiências diferem de espécies para espécies, a abordagem de marcador dividido tem sido usada com sucesso com 100% de eficiência em espécies tão diversas quanto alternaria alternada34,35e C. nicotianae36. Em contrapartida, a eficiência desse sistema em H. omanensis foi zero, apesar de mais de 80 eventos independentes de transformação e integração. Da mesma forma, o sistema CRISPR-Cas9 baseado em plasmídeo tem sido usado com sucesso com alta eficiência em Trichoderma reesei (>93%)17 e Penicillium chrysogenum (até 100%)37. Isto é, novamente, em contraste com a utilidade deste sistema em H. omanensis. A expressão suficiente da proteína Cas9 não era alcançável em H. omanensis, apesar de tentar uma série de promotores potenciais, incluindo dois promotores específicos de espécies previstos a partir de genes de limpeza. Assim, este sistema não poderia ser usado em tudo. Usando a versão baseada em proteínas do sistema CRISPR-Cas9, no entanto, produziu muitos transformadores independentes, dois dos quais abrigavam o dDNA integrado no local correto. Além disso, este experimento foi tentado apenas uma vez e foi bem sucedido, ilustrando ainda mais a facilidade com que este sistema pode ser usado.
As aplicações futuras deste protocolo incluem sua otimização e uso em outras espécies do Ceratocystidaceae. Já existe uma riqueza de dados de NGS disponíveis para essas espécies30,,38,,39 e estudos sobre sua especificidade de hospedeiro40, taxa de crescimento e virulência41 foram realizados. Esses estudos podem ser reforçados pela caracterização funcional dos genes que se acredita estarem envolvidos nesses processos, pesquisas que agora se tornarão possíveis devido à disponibilidade de um protocolo de transformação e edição de genomas.
Em conclusão, uma investigação minuciosa sobre os genes subjacentes a importantes processos biológicos em espécies não-modelo está se tornando mais acessível graças à disponibilidade de protocolos de edição de genomas fáceis de usar que não dependem da existência de extensos recursos biológicos e kits de ferramentas moleculares. Estudar espécies não-modelo está se tornando mais fácil e permitirá a descoberta de novos caminhos e desvios interessantes dos processos biológicos padrão que foram elucidados em espécies modelo.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Este projeto contou com o apoio da Universidade de Pretória, do Departamento de Ciência e Tecnologia (DST)/Fundação Nacional de Pesquisa (NRF) Centro de Excelência em Biotecnologia em Saúde da Árvore (CTHB). O projeto foi apoiado adicionalmente pela cadeira DST/NRF SARChI do Prof BD Wingfield em Gêmica Fúngica (número de subvenção: 98353) bem como pela bolsa de doutorado NRF do Dr. AM Wilson (108548). Os bolsistas reconhecem que opiniões, conclusões e conclusões ou recomendações expressas neste trabalho são dos pesquisadores e que os órgãos financiadores não aceitam qualquer responsabilidade a esse respeito.
Materiais
| Name | Company | Catalog Number | Comments |
| EcoRI-HF | New England Biolabs, Ipswich, USA | R3101S | |
| EnGen Spy Cas9 NLS protein | New England Biolabs, Ipswich, USA | M0646T | Used to assemble the RNP |
| Eppendorf 5810 R centrifuge | Eppendorf, Hamberg, Germany | ||
| FastStart Taq DNA Polymerase | Sigma, St Louis, USA | 12032902001 | Standard DNA polyermase |
| GeneJET Gel Extraction Kit | ThermoFisher Scientific, Waltham, USA | K0691 | |
| HindIII-HF | New England Biolabs, Ipswich, USA | R3104S | |
| HiScribeTM T7 Quick High Yield RNA synthesis kit | New England Biolabs, Ipswich, USA | E2050S | |
| Hygromycin B from Streptomyces hygroscopicus | Sigma, St Louis, USA | 10843555001 | |
| Infors HT Ecotron Shaking Incubator | Infors AG, Bottmingen, Switzerland | ||
| LongAmp Taq DNA Polymerase | New England Biolabs, Ipswich, USA | M0323S | Long-range, high-fidelity DNA polymerase |
| Malt extract agar, 2% (MEA) | 20 g ME and 20 g agar in 1 l ddH20 | ||
| Malt extract | Sigma, St Louis, USA | 70167-500G | |
| Agar | Sigma, St Louis, USA | A5306 | |
| Malt Extract broth, 1% (MEB) | Sigma, St Louis, USA | 70167-500G | 2 g ME in 200 ml ddH20 |
| Malt Extract broth, 2% (MEB) | Sigma, St Louis, USA | 70167-500G | 4 g ME in 200 ml ddH20 |
| Miracloth | Merck Millipore, New Jersey, USA | 475855 | |
| Nylon membrane (positively charged) | Sigma, St Louis, USA | 11209299001 | |
| Osmotic control medium (OCM) | 0.3% yeast extract, 20% sucrose, 0.3% casein hydrolysate | ||
| Casein Hydrolysate | Sigma, St Louis, USA | 22090 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Yeast extract | Sigma, St Louis, USA | Y1625 | |
| Osmotic control medium (OCM) agar | Osmotic control medium (OCM) + 1% agar | ||
| Agar | Sigma, St Louis, USA | A5306 | |
| PCR DIG Labeling Mix | Sigma, St Louis, USA | 11585550910 | |
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific, Waltham, USA | F-530XL | High fidelity DNA polymerase |
| Plasmid pcb1004 | N/A | N/A | From: Carroll et al., 1994 |
| Presynthesized sgRNA | Inqaba Biotec, Pretoria, South Africa | Ordered as an synthesized dsDNA with specified sequence | |
| Proteinase K | Sigma, St Louis, USA | P2308 | |
| PTC Solution | 30% polyethylene glycol 8000 in STC buffer from above | ||
| Polyethylene glycol 8000 | Sigma, St Louis, USA | 1546605 | |
| RNase A | ThermoFisher Scientific, Waltham, USA | 12091021 | |
| RNAfold Webserver | Institute for Theoretical Chemistry, University of Vienna | N/A | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| RNAstructure | Mathews Lab | N/A | https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html |
| Sorbitol, 1 M | Sigma, St Louis, USA | 1617000 | 182.17g sorbitol in 1 l ddH20 |
| STC Buffer | 20% sucrose, 50 mM Tris-HCl pH 8.00 and 50 mM CaCl2 | ||
| Calcium chloride | Sigma, St Louis, USA | 429759 | |
| Tris-HCl pH 8.00 | Sigma, St Louis, USA | 10812846001 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Trichoderma harzianum lysing enzymes | Sigma, St Louis, USA | L1412 | |
| Zeiss Axioskop 2 Plus Ergonomic Trinocular Microscope | Zeiss, Oberkochen, Germany |
Referências
- Ekblom, R., Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 107, 1-15 (2011).
- Russell, J. J., et al. Non-model model organisms. BMC Biology. 15 (55), 1-31 (2017).
- Kück, U., Hoff, B. New tools for the genetic manipulation of filamentous fungi. Applied Microbiology and Biotechnology. 86, 51-62 (2010).
- Li, D., Tang, Y., Lin, J., Cai, W. Methods for genetic transformation of filamentous fungi. Microbial Cell Factories. 16 (168), 1-13 (2017).
- Lorito, M., Hayes, C. K., Di Pietro, A., Harman, G. E. Biolistic transformation of Trichoderma harzianum and Gliocladium virens using plasmid and genomic DNA. Current Biotechnology. 24, 349-356 (1993).
- Taylor, P., et al. Transformation of intact Aspergillus niger by electroporation. Bioscience, Biotechnology, and Biochemistry. 58 (12), 2224-2227 (2014).
- Dhawale, S. S., Paietta, J. V., Marzluf, G. A. A new, rapid and efficient transformation procedure for Neurospora. Current Genetics. 8, 77-79 (1984).
- Sayari, M., Van Der Nest, M. A., Steenkamp, E. T., Adegeye, O. O., Marincowitz, S. Agrobacterium-mediated transformation of Ceratocystis albifundus. Microbiological Research. 226, 55-64 (2019).
- Meyer, V. Genetic engineering of filamentous fungi- Progress, obstacles and future trends. Biotechnology Advances. 26, 177-185 (2008).
- You, B. J., Lee, M. H., Chung, K. R. Gene-specific disruption in the filamentous fungus Cercospora nicotianae using a split-marker approach. Archives of Microbiology. 191, 615-622 (2009).
- Gravelat, F. N., Askew, D. S., Sheppard, D. C. Targeted gene deletion in Aspergillus fumigatus using the hygromycin-resistance split-marker approach. Host-Fungus Interactions. 845, 119-130 (2012).
- Wang, Y., Diguistini, S., Bohlmann, J., Breuil, C. Agrobacterium-meditated gene disruption using split-marker in Grosmannia clavigera, a mountain pine beetle associated pathogen. Current Genetics. 56, 297-307 (2010).
- Wood, A. J., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333, 307 (2011).
- Mahfouz, M. M., Piatek, A., Neal, C. Genome engineering via TALENs and CRISPR/Cas9 systems: Challenges and perspectives. Plant Biotechnology. 12, 1006-1014 (2014).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85 (1), 227-264 (2016).
- Arazoe, T., et al. Tailor-made TALEN system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnology and Bioengineering. 112 (7), 1335-1342 (2015).
- Liu, R., Chen, L., Jiang, Y., Zhou, Z., Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discovery. 1, 1-11 (2015).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-822 (2012).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences. 109 (39), 2579-2586 (2012).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Cell. 8 (11), 2281-2308 (2013).
- Wilson, A. M., Wilken, P. M., Van Der Nest, M. A., Wing, M. J., Wing, B. D. The novel Huntiella omanensis mating gene, MAT1-2-7, is essential for ascomatal maturation. Fungal Genetics and Biology. 137, 103335 (2020).
- Miao, J., et al. Characterization of an N-terminal non-core domain of RAG1 gene disrupted Syrian Hamster model generated by CRISPR Cas9. Viruses. 10 (243), 10050243 (2018).
- Roberts, B., et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Molecular Biology of the Cell. 28 (21), 2854-2874 (2016).
- Schneider, S., Kirchner, M., Kirchner, M., Schneider, S. CRISPR-Cas: From the bacterial adaptive immune system to a versatile tool for genome engineering. Angewandte Chemie International Edition. 54 (46), 13508-13514 (2015).
- Nødvig, C. S., Nielsen, J. B., Kogle, M. E., Mortensen, U. H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 10 (7), 1-18 (2015).
- Wang, Q., Cobine, P. A., Coleman, J. J. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genetics and Biology. 117, 21-29 (2018).
- Nagy, G., et al. Development of a plasmid free CRISPR-Cas9 system for the genetic modification of Mucor circinelloides. Scientific Reports. 7 (16800), 1-10 (2017).
- Al-Subhi, A. M., Al-Adawi, A. O., Van Wyk, M., Deadman, M. L., Wingfield, M. J. Ceratocystis omanensis, a new species from diseased mango trees in Oman. Mycological Research. 110 (2), 237-245 (2006).
- Wilson, A. M., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Pheromone expression reveals putative mechanism of unisexuality in a saprobic ascomycete fungus. PLoS ONE. 13 (3), 0192517 (2018).
- van der Nest, M. A., et al. Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus. 5 (2), 472-485 (2014).
- Wilson, A. M., Godlonton, T., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Unisexual reproduction in Huntiella moniliformis. Fungal Genetics and Biology. 80, 1-9 (2015).
- Sambrook, J., Green, M. . Molecular cloning: A laboratory manual. , (2012).
- Ramamoorthy, V., Govindaraj, L., Dhanasekaran, M., Vetrivel, S., Kumar, K. K., Ebenezar, E. Combination of driselase and lysing enzyme in one molar potassium chloride is effective for the production of protoplasts from germinated conidia of Fusarium verticillioides. Journal of Microbiological Methods. , (2015).
- Lin, C., Yang, S. L., Wang, N., Chung, K. The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata functions independently or cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic processes. Fungal Genetics and Biology. 47 (4), 381-391 (2010).
- Lin, C., Chung, K. Specialized and shared functions of the histidine kinase- and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, a filamentous fungal pathogen of citrus. Fungal Genetics and Biology. 47 (10), 818-827 (2010).
- Choquer, M., et al. The CTB1 gene encoding a fungal polyketide synthase is required for cercosporin biosynthesis and fungal virulence of Cercospora nicotianae. Molecular Plant-Microbe Interactions. 18 (5), 468-476 (2005).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- van der Nest, M. A. M. A., et al. Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans and Ceratocystis moniliformis. IMA Fungus. 5 (1), 135-140 (2014).
- Wingfield, B. D., et al. Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus. 7 (2), 317-323 (2016).
- Fourie, A., Van Der Nest, M. A., De Vos, L., Wingfield, M. J., Wingfield, B. D., Barnes, I. QTL mapping of mycelial growth and aggressiveness to distinct hosts in Ceratocystis pathogens. Fungal Genetics and Biology. 131, 103242 (2019).
- Lee, D. H., Roux, J., Wingfield, B. D., Wingfield, M. J. Variation in growth rates and aggressiveness of naturally occurring self-fertile and self-sterile isolates of the wilt pathogen Ceratocystis albifundus. Plant Pathology. 64 (5), 1103-1109 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados