
Method Article
CRISPR-Cas9-Mediated Genome Editing in the Filamentous Ascomycete Huntiella omanensis
In This Article
Summary
The CRISPR-Cas9 genome editing system is an easy-to-use genome editor that has been used in model and non-model species. Here we present a protein-based version of this system that was used to introduce a premature stop codon into a mating gene of a non-model filamentous ascomycete fungus.
Abstract
The CRISPR-Cas9 genome editing system is a molecular tool that can be used to introduce precise changes into the genomes of model and non-model species alike. This technology can be used for a variety of genome editing approaches, from gene knockouts and knockins to more specific changes like the introduction of a few nucleotides at a targeted location. Genome editing can be used for a multitude of applications, including the partial functional characterization of genes, the production of transgenic organisms and the development of diagnostic tools. Compared to previously available gene editing strategies, the CRISPR-Cas9 system has been shown to be easy to establish in new species and boasts high efficiency and specificity. The primary reason for this is that the editing tool uses an RNA molecule to target the gene or sequence of interest, making target molecule design straightforward, given that standard base pairing rules can be exploited. Similar to other genome editing systems, CRISPR-Cas9-based methods also require efficient and effective transformation protocols as well as access to good quality sequence data for the design of the targeting RNA and DNA molecules. Since the introduction of this system in 2013, it has been used to genetically engineer a variety of model species, including Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster and Mus musculus. Subsequently, researchers working on non-model species have taken advantage of the system and used it for the study of genes involved in processes as diverse as secondary metabolism in fungi, nematode growth and disease resistance in plants, among many others. This protocol detailed below describes the use of the CRISPR-Cas9 genome editing protocol for the truncation of a gene involved in the sexual cycle of Huntiella omanensis, a filamentous ascomycete fungus belonging to the Ceratocystidaceae family.
Introduction
The increasing availability of high quality, fully assembled genomes and transcriptomes has greatly improved the ability to study a wide variety of biological processes in an array of organisms1. This is true of both model species as well as non-model species, many of which may offer a more diverse understanding of biological processes. These kinds of data can be used for gene discovery, the identification of transcription networks and both whole genome and transcriptome comparisons, each of which come with their own set of applications. However, while genes are being predicted, annotated and putatively linked to different functional pathways at a never-before-seen rate, the functional characterization of these genes remains behind, limited by the molecular toolkits available for many species. This is particularly the case for the non-model species, where genomic data is relatively easy to generate but where further molecular characterization has been near impossible1,2.
Partial characterization of the functions of specific genes important to the biology of fungal species can be achieved by either knockout or knockin experiments followed by phenotypic analysis of the mutant strains3. These two systems rely entirely on the availability of genetic engineering protocols, including, at minimum, a transformation system and a genetic editing system. There are a number of different transformation systems that have been developed in a variety of filamentous fungi4. Physical systems like those that rely on biolistics and electroporation have been developed in Trichoderma harzianum5 and Aspergillus niger6, respectively. Systems that utilize chemicals such as calcium chloride or lithium acetate have been developed in Neurospora crassa7. Lastly, biological systems that rely on the use of Agrobacterium tumefaciens for transformation have been successfully used in Ceratocystis albifundus8.
In contrast to the availability of different transformation protocols, genome editing systems are less abundant. Many of the traditional functional characterization experiments conducted in filamentous fungi utilized a split marker knockout construct in the form of a selectable marker flanked by regions of homology to the target region or gene in the genome3. The method relies on homology directed (HR) DNA repair, which facilities homologous recombination between the knockout construct and the region of interest. This recombination event results in the replacement of the gene of interest with the sequence of the selectable marker. Unfortunately, while this was successful in many species including Cercospora nicotianae10, Aspergillus fumigatus11 and Grosmannia clavigera12, the rates of homologous recombination are highly variable across different fungal species, making this an inefficient and sometimes unusable protocol in certain species3.
Other genome editing systems, including those that make use of zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) represented a great improvement on the older systems, particularly given their abilities to make specific and targeted changes13. Both ZFNs and TALENs are comprised of a nuclease protein and a protein that is capable of recognizing specific nucleotide sequences13. Upon recognition, the nuclease induces a double stranded DNA break that can facilitate the introduction of specific mutations. In order to bring about genome changes, the protein region that recognizes the nucleotide sequence needs to be specifically designed for each experiment. Because of this reliance on protein-nucleic acid interactions to guide the editing, designing and producing the targeting molecules for each knockout or knockin experiment is difficult and labor intensive14,15. Illustrative of these challenges, very few filamentous fungi have been subjected to genome editing using these systems. One example is the TALENs-based system that was developed in the rice blast fungus, Magnaporthe oryzae16.
Arguably the greatest revolution to the field of genome editing was the discovery and subsequent development of the CRISPR-Cas9 system- a genome editor that allows for the targeted cleavage of a sequence of interest by an endonuclease that is guided by an RNA molecule. This was a huge improvement on the previously developed genome editors that relied on protein-nucleic acid interactions as the major advantage of the CRISPR-Cas9 system is that it relies on an RNA molecule to target the region of interest. This means that the system relies on an RNA-DNA interaction and thus standard base pairing rules can be exploited when designing each experiment15.
The CRISPR-Cas9 system as detailed here is comprised of three major components: a single guide RNA (sgRNA), the Cas9 enzyme and a donor DNA (dDNA)17. The sgRNA is composed of a 20 nucleotide region called the protospacer as well as a longer region called the scaffold18. The protospacer region is used to guide the editing system to the target region and is thus redesigned for each experiment. The scaffold is the region of RNA that physically binds to the Cas9 enzyme to form the ribonucleoprotein (RNP) and is thus identical regardless of the region to be targeted. The Cas9 enzyme physically facilitates the cleavage of the target DNA, using the protospacer as a guide to identify this region19. The last component, the dDNA, is optional and its use depends on the particular experiment20. The dDNA harbors the sequence that should specifically be inserted into the region being cut by the Cas9 enzyme, and is thus ideal for gene knockin experiments where a gene is being introduced into the genome or for gene knockout experiments where an antibiotic resistance gene or other selectable marker is being introduced to replace the gene of interest. The dDNA can also be designed in such a way as to introduce novel sequences into the genome. For example, as detailed below, it is possible to introduce an in-frame stop codon into a particular region in the gene of interest when a gene truncation is required21. Other applications include the mutation of specific regions of the gene, such as a functional domain22, or the introduction of a tagging sequence23.
A major benefit of using the CRISPR-Cas9 system is its versatility24. One example of this adaptability is that the Cas9 enzyme can be introduced into the host cell in one of its three forms- DNA, RNA or protein- depending on the particular transformation system being used. When introduced in DNA form, the cas9 gene is often included on a plasmid along with a selectable marker, a cassette to express the sgRNA and, if necessary, a cassette encoding the dDNA sequence25. The primary advantage of this system is that only a single construct needs to be transformed into the cell and successful transformation ensures that all the necessary components for CRISRP-Cas9-mediated genome editing are present. However, this method relies on the availability of an expression system for the host species. For Cas9 to successfully induce DNA damage, it needs to be expressed at high levels, and thus, a suitable and potentially specific-specific promoter is required. For non-model species where such promoters have not yet been developed, this may be a detracting factor and thus the ability to introduce Cas9 in RNA or protein form may be a more attractive option. The introduction of RNA into the cell brings its own challenges- particularly in that RNA is unstable and may not survive the transformation process. Furthermore, when introduced in either DNA or RNA form, the Cas9 gene sequence may need to be codon-optimized for use in the particular host system17. For example, the cas9 gene from Streptococcus pyogenes may not work in a mammalian host cell and a cas9 gene that has been codon-optimized for use in a mammalian cell may not work in a plant cell. All of these challenges can be overcome using the protein form of Cas9, which, together with the sgRNA, can be assembled into an RNP and transformed into the host cell26,27. This system does not rely on any endogenous expression system or codon optimization and should thus work in the majority of non-model species. The disadvantage of the protein-based system is that it is not compatible with DNA-based transformation systems like Agrobacterium-mediated transfer. Thus, for the protein-based method to work, a transformation protocol such as those that rely on protoplasts or biolistics needs to be available. This RNP-based system has been successfully used in the filamentous fungi, Fusarium oxysporum26 and Mucor circinelloides27.
Huntiella omanensis, a member of the Ceratocystidaceae family, is a cosmopolitan fungus often found on freshly wounded woody plants28. While high quality genome and transcriptome data are available for this species28,29,30, no transformation or genome editing protocols have been developed. To date, research on H. omanensis has focused on the underlying genetic components of its sexual cycle29,31. This fungus exhibits a typical heterothallic sexual cycle, with sexual reproduction occurring exclusively between isolates of the MAT1-1 and MAT1-2 mating types31. In contrast, MAT1-2 isolates of the closely related Huntiella moniliformis are capable of independent sexual reproduction and complete a sexual cycle in the absence of a MAT1-1 partner31. This difference in sexual capabilities is thought to be, at least partly, due to a major difference in the mating gene, MAT1-2-7, where H. omanensis harbors a full length and intact copy, while the gene is severely truncated in H. moniliformis29,31. To further characterize the role of this gene in sexual reproduction, the MAT1-2-7 gene of H. omanensis was truncated to mimic the truncation seen in H. moniliformis21.
The protocol below details the transformation of H. omanensis and the truncation of the MAT1-2-7 gene using a protein-based version of the CRISPR-Cas9 genome editing system. This protocol was developed after the approaches of homologous recombination-based gene replacement and plasmid-based CRISPR-Cas9 genome editing were unsuccessful.
Protocol
1. Design and synthesis of the sgRNA
- To identify potential protospacer regions that will form part of the sgRNA, manually search through the gene-of-interest for 5’ NGG 3’ triplets, using the search function in whichever program is being used. Annotate these triplets as PAM sequences.
NOTE: Various software programs are available that search for and annotate the potential PAM and protospacer sequences.- Select the 20 bp upstream of each of the identified PAM sequences and annotate these sequences as potential protospacers.
- To decide on a single protospacer, perform the following filtering steps to discard low quality sequences.
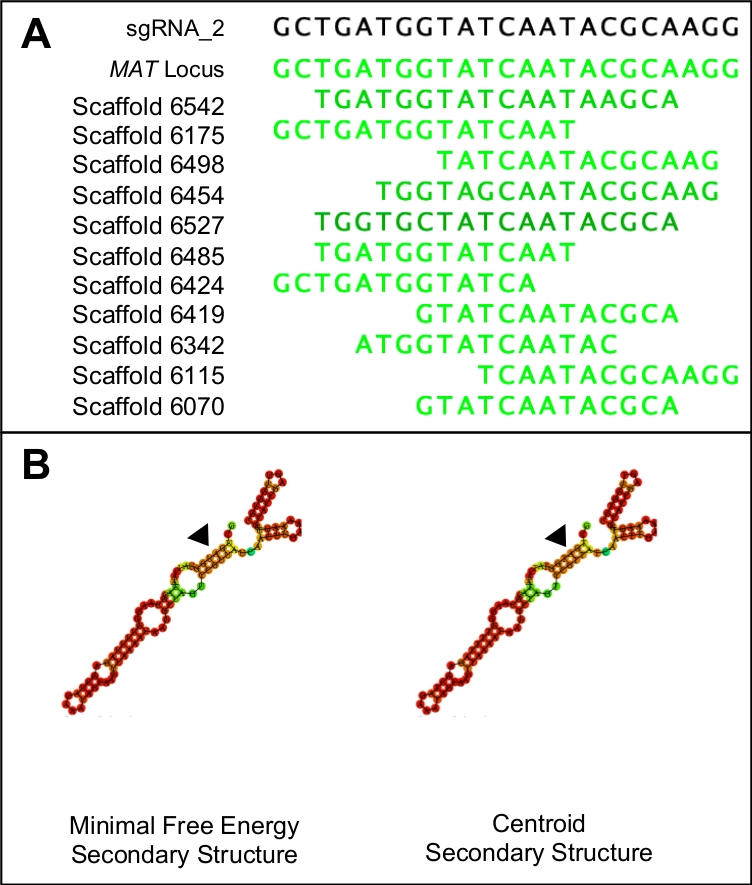
- To test for the specificity of the potential protospacers, combine the PAM sequence and protospacer into a single sequence and use it as a BLASTn query against the entire genome. Discard any protospacers that show similarity to any region in the genome other than the target region (Figure 1A).
- To ensure that the RNA molecule will fold into the correct 3D structure for binding to the Cas9 enzyme, create a combined sequence including the protospacer and the scaffold sequence specific to the Cas9 enzyme.
- Upload each of the combined protospacer-scaffold sequences into an RNA secondary structure prediction tool (Table of Materials).
- Analyze the results by comparing the minimal free energy and centroid secondary structures.
NOTE: Ideal protospacer candidates will have identical minimal free energy and centroid secondary structures. Both secondary structures should be comprised of three stem loops, interrupted by five ring structures. The structures should also exhibit high binding probabilities (indicated in red) throughout the entire structure, except for the region representing the protospacer (Figure 1B). The actual energy values are not relevant.
- Choose a final candidate from those that passed the above filtering steps by selecting the candidate that is closest to the specific region being targeted.
- To synthesize the sgRNA as a single RNA molecule, use an sgRNA synthesis kit (Table of Materials) compatible with the particular Cas9 enzyme that will be used (e.g., Cas9 from Streptococcus pyogenes).
NOTE: The following steps may be dependent on the sgRNA synthesis kit used. In the event that a different kit is used, follow the manufacturer’s instructions. Alternatively, the sgRNA can be ordered pre-synthesized. When working with RNA, use nuclease-free reagents and disposables.- If the 20 bp protospacer chosen in step 1.3 above does not harbor a G at the 5’ end, add a G to this region.
- Add the T7 promoter sequence to the 5’ end of the target sequence. This sequence is standard and is 5’ TTCTAATACGACTCACTATAG 3’.
- Add a 14 nt overlap sequence to the 3’ end of the target sequence. This sequence is kit-specific and is 5’ GTTTTAGAGCTAGA 3’ for the kit used here.
- Order the resulting fragment 5’ TTCTAATACGACTCACTATAG(N)20 GTTTTAGAGCTAGA, with (N)20 representing the selected protospacer presynthesized (Table of Materials).
- In accordance with the manufacturer’s protocol (Table of Materials), combine the following reagents at room temperature: 2 µL of water, 10 µL of reaction buffer, 5 µL of synthesized protospacer sequence, 1 µL of 0.1 M DTT and 2 µL of transcriptase enzyme.
- Incubate this solution at 37 °C for 30 min and transfer to ice.
- Add 30 µL of water and 2 µL of DNase I, mix and incubate at 37 °C for a further 15 min.
- Visualize the resulting sgRNA on a 2% agarose gel.
2. Testing the in vitro Cleavage Ability of the sgRNA
NOTE: This step is optional but is recommended.
- Design primers that will amplify a fragment harboring the sequence of the site that the chosen sgRNA will target and amplify the region using a standard DNA polymerase.
NOTE: If possible, design the primers in such a way that cleavage at the target site will produce two fragments of very different sizes that can easily be distinguished from one another on a standard agarose gel. - Assemble the combined sgRNA-Cas9 ribonucleoprotein (RNP) by incubating a solution comprised of 30 nM sgRNA as synthesized above, 30 nM Cas9 protein, 10x reaction buffer and 10 µL of water at 25 °C for 10 min.
- Test the cleavage ability of the sgRNA by adding the PCR product of the target region into the RNP solution to a final concentration of 3 nM.
- Incubate the solution at 37 °C for 15 min.
- Add 3 µg of proteinase K and 2 µg of RNase to the solution to stop the cleavage reaction and incubate at room temperature for 10 min.
- Visualize the resulting DNA fragments on a 2% agarose gel. The sgRNA is suitable for in vivo experimentation if two bands of the expected size are observed on the gel.
3. Design and synthesis of the dDNA
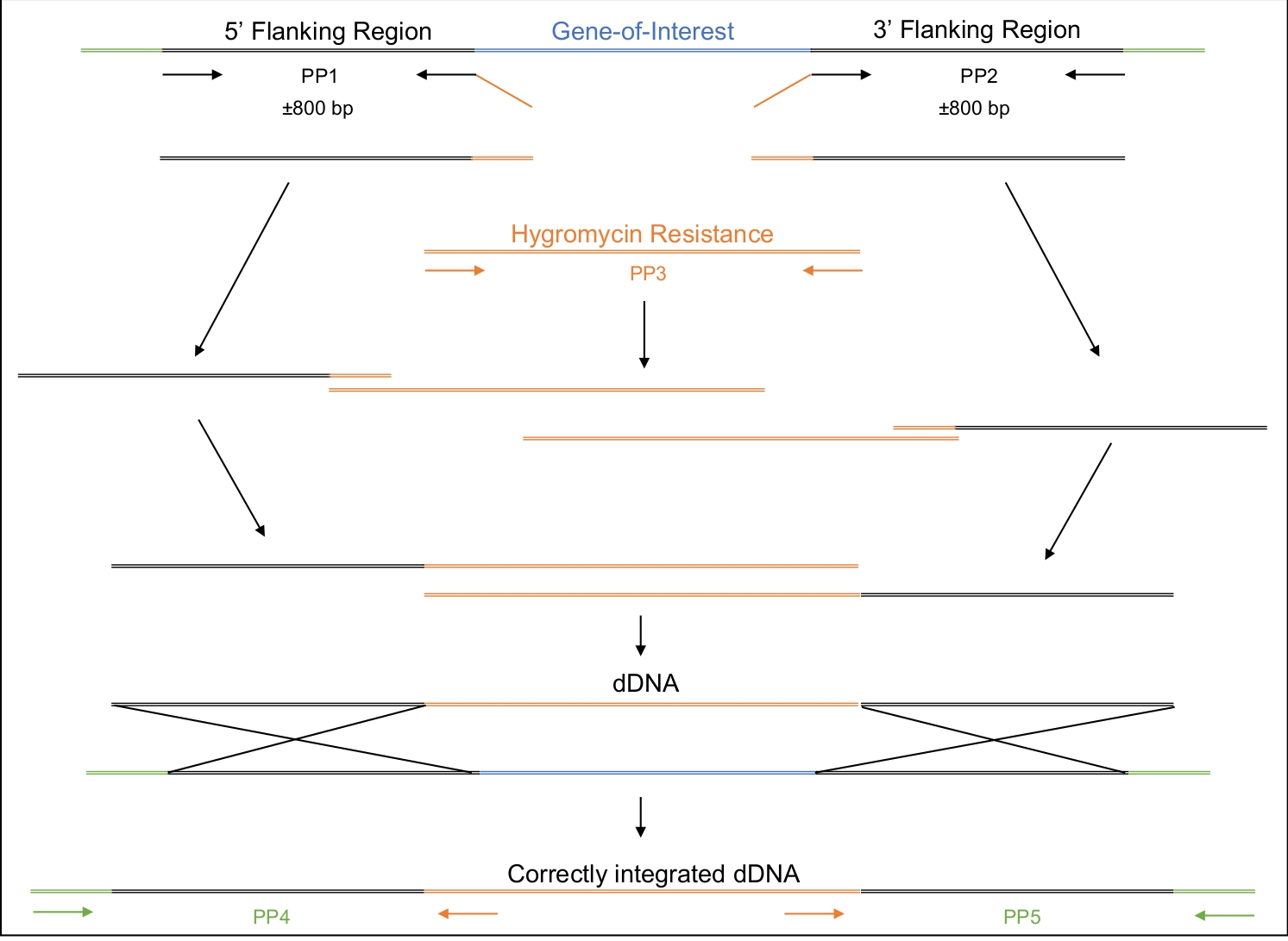
- Design the dDNA to be comprised of three regions- 5’ and 3’ regions with complementarity to the genomic region being targeted and a middle region harboring a selectable marker (Table of Materials, Figure 2).
NOTE: Other specific sequences can also be added to this selectable marker. For this particular experiment, a stop codon sequence (5’ TGA 3’) was added just before the selectable marker, thereby introducing an in-frame stop codon into the gene. The dDNA can be ordered presynthesized. Alternatively, the dDNA can be amplified and assembled using a stepwise, overlap PCR approach as detailed below.- Design primers to amplify approximately 800 bp of the 5’ and 3’ flanking regions. Add 20 nt of sequence that is complementary to the selectable marker’s sequence to the 5’ region’s reverse primer and 3’ region’s forward primer.
- Design primers that amplify the selectable marker, ensuring that the amplified product harbors the resistance gene as well as a promoter known to work in the species of interest.
- Using a high-fidelity DNA polymerase (Table of Materials), amplify the three dDNA regions. Amplify the 5’ and 3’ regions from the gDNA of the organism being edited. Amplify the selectable marker from a relevant source.
- In a single reaction, combine the amplified 5’ region with the selectable marker and, using a long-range, high-fidelity DNA polymerase, amplify the entire region.
- In a second single reaction, combine the amplified 3’ region with the selectable marker and, using a long-range, high-fidelity DNA polymerase, amplify the entire region.
- Finally, combine the two preceding PCR products into a single reaction and amplify the entire dDNA sequence with a long-range, high-fidelity DNA polymerase.
- Visualize the DNA fragment on a 1% agarose gel. In the event that two or more fragments are produced, purify the correctly sized fragment from the gel using a gel purification kit.
4. Extraction of protoplasts
- To produce conidia, inoculate 200 mL of fresh 2% malt extract broth (MEB) in a 500 mL flask with a 1 cm x 1 cm mycelia-covered agar block.
NOTE: Not all fungi are able to produce conidia. In that case, mycelia can also be used. This will typically require higher concentrations of lysing enzyme further in the protocol.- Incubate the liquid culture in a shaking incubator at 25 °C with shaking at 120 rpm for 24 - 48 h.
NOTE: This incubation time and temperature has been optimized for H. omanensis. This will need to be optimized for other species. - To harvest the conidia; filter the liquid culture through a layer of sterile laboratory cloth (e.g., Miracloth), transfer the conidial suspension into 50 mL centrifuge tubes and centrifuge at 3,220 x g at 4 °C for 10 min. Discard the supernatant.
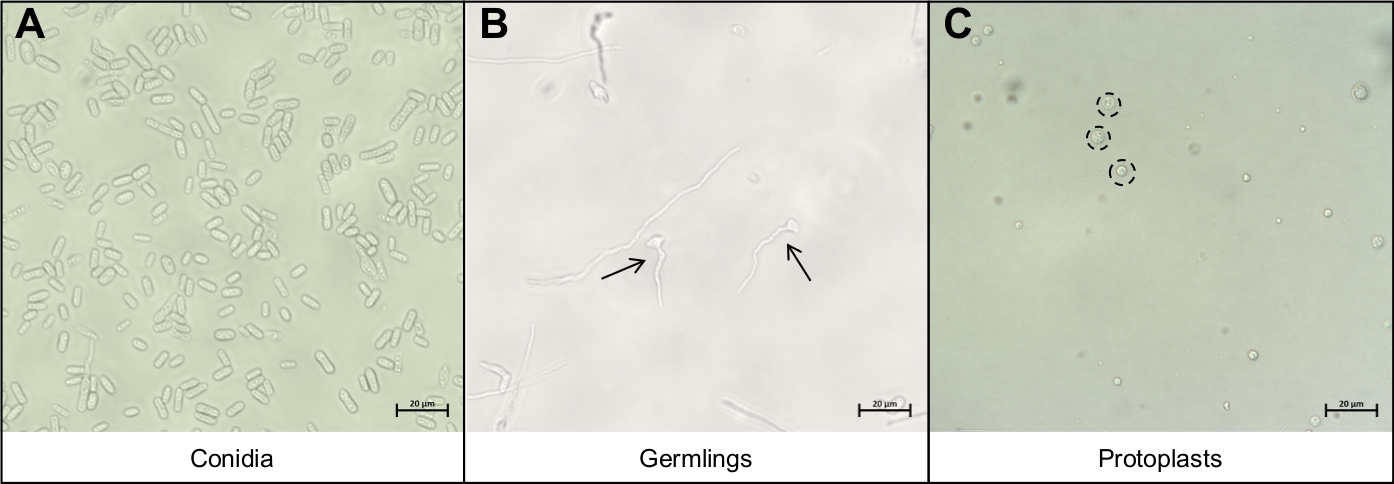
- Resuspend the conidia in 5 mL of water and pipette 10 µL of the conidia solution onto a microscope slide and cover with a coverslip. Visualize using a compound microscope under 40x magnification to ensure only conidia have been recovered (Figure 3A).
- In a 500 mL flask, inoculate 200 mL of fresh 1% MEB with the total volume of resuspended conidia.
- Incubate the liquid culture in a shaking incubator at 25 °C with shaking at 120 rpm for up to 12 h.
NOTE: This incubation time has been optimized for H. omanensis. This will need to be optimized for other species. - To harvest the germlings, transfer the liquid culture into 50 mL centrifuge tubes and centrifuge at 3,220 x g at 4 °C for 10 min. Discard the supernatant.
- Resuspend the germlings in up to 10 mL of 1 M sorbitol.
- Pipette 10 µL of the germling solution onto a microscope slide and cover with a coverslip. Visualize using a compound microscope under 40x magnification to ensure only germlings have been recovered (Figure 3B).
NOTE: The protocol can be paused here. Store germlings in 1 M sorbitol at -80 °C.
- Incubate the liquid culture in a shaking incubator at 25 °C with shaking at 120 rpm for 24 - 48 h.
- To lyse the cell walls of the young germlings and release the protoplasts, add 1 mL of the germling suspension to 9 mL of lysing enzyme at various concentrations in a sterile 50 mL flask.
NOTE: Different enzyme concentrations and incubation times are used and can be found in Table 1. The enzymes and concentrations are also likely to vary depending on the fungus and will need to be optimized for each species.- Incubate the spore-enzyme solution in a shaking incubator at 25 °C with shaking at 80 rpm for 2 to 3 h.
- Filter the protoplast solution through a layer of sterile laboratory cloth and collect the protoplasts by centrifugation at 1,810 x g at 4 °C for 10 min. Discard the supernatant.
NOTE: Protoplasts are cells without cell walls and are thus very sensitive to mechanical disruption. Be sure to handle them carefully, particularly when pipetting. - Carefully resuspend the protoplast pellet in 200 µL of STC buffer (Table of Materials).
- Pipette 10 µL of the protoplast solution onto a microscope slide and cover with a coverslip. Visualize using a compound microscope under 40x magnification to ensure only protoplasts have been recovered (Figure 3C).
- Using a hemocytometer, count and calculate the number of protoplasts generated in the above steps. Dilute the protoplast solution into aliquots containing approximately 5 x 106 protoplasts.
NOTE: The protocol can be paused here. Store protoplasts in STC buffer at -80 °C.
5. Protoplast and PEG-assisted transformation and transformant recovery
- To begin the transformation, combine approximately 5 x 106 protoplasts with a single volume of the RNP solution and approximately 6 µg of the dDNA fragment.
NOTE: Protoplasts are very sensitive to mechanical disruption. Be sure to handle them carefully, particularly when pipetting.- Using a pipette, slowly drip 1 mL of a freshly prepared 30% PTC solution onto the protoplast solution and incubate the solution at room temperature for 20 min.
NOTE: This step is a sensitive and very important step. Make sure to use freshly prepared PTC solution and drop the solution over the cells as slowly and evenly as possible, creating a hydrophobic layer across the cell surface. - Add 5 mL of osmotic control medium (OCM) to the protoplast solution and pipette slowly and gently to ensure the solution is thoroughly mixed.
- Incubate the protoplast solution in a shaking incubator at 25 °C with shaking at 80 rpm overnight.
- Using a pipette, slowly drip 1 mL of a freshly prepared 30% PTC solution onto the protoplast solution and incubate the solution at room temperature for 20 min.
- To select the transformed isolates, divide the solution into 5 empty 60 mm culture plates.
- Add 10 mL of OCM agar supplemented with 30 µg/mL hygromycin B to each culture plate and slowly rotate each plate to mix thoroughly.
- Allow the first layer of agar to set before adding 10 mL of osmotic control medium agar supplemented with 40 µg/mL hygromycin B.
- Allow the second layer of agar to set and incubate the cultures at 25 °C until single isolates can be seen growing through both layers of agar.
- To recover successfully transformed isolates, transfer the individual isolates capable of growth through the agar layer supplemented with 40 µg/mL hygromycin B to fresh malt extract agar (MEA) plates supplemented with 50 µg/mL hygromycin B (MEA-50).
- Incubate the fresh cultures at 25 °C for 5 days, checking daily for growth. Cultures capable of sustained growth on this media have successfully been transformed and can be used for further study.
6. Confirmation of the integration and stability of the dDNA
- In order to confirm that the dDNA has been integrated into the genome at the target region, design primers that flank the predicted 5’ and 3’ insert sites (Figure 2).
- Perform two PCRs using these two primer sets and a high-fidelity DNA polymerase. If both PCRs yield amplicons of expected size and sequence, the dDNA was successfully integrated into the target region. Then, assess the mutant stain for stable integration of the dDNA.
- In order to confirm that the dDNA has been stably integrated into the genome and will be maintained during vegetative growth, perform a media transfer test.
- Transfer a block of mycelial-covered agar from an actively growing mutant isolate on MEA-50 medium to unsupplemented MEA medium. Incubate at 25 °C for 3 days.
- Transfer a block of mycelia-covered agar from the isolate growing on MEA medium to MEA-50 medium. Incubate at 25 °C for 3 days.
- Repeat this process, transferring actively growing mycelia from supplemented to unsupplemented medium for at least four rounds.
NOTE: If the isolate is capable of sustained growth on MEA-50 medium after many transfers, the dDNA has been stably integrated into the genome and can be maintained through vegetative growth. The mutant stain can be assessed for the presence of only a single copy of the integrated dDNA.
- In order to confirm that the dDNA has been integrated into the genome at a single location, perform a Southern blot analysis.
- Digest a total of 30 µg of gDNA from each mutant strain using HindIII and EcoRI restriction enzymes in accordance with manufacturer’s protocols.
NOTE: While the choice of restriction enzyme is up to the researcher, ensure that the restriction enzyme’s recognition site is not present in the dDNA sequence. - Separate the digested gDNA on a 0.75% agarose gel and transfer the DNA onto a nylon membrane using standard procedures32.
- Subject the membrane to hybridization using a probe targeting the dDNA sequence.
- Design primers to amplify a short (300 bp) region of the dDNA.
- Using these primers, synthesize the probe using a PCR DIG labeling mix.
- Use the newly synthesized probe for membrane hybridization, treatment and visualization using standard procedures32. If only a single band is seen in each lane, the dDNA is present at only a single location in the genome. The mutant strain can now be used for further phenotypic analysis and functional characterization experiments.
- Digest a total of 30 µg of gDNA from each mutant strain using HindIII and EcoRI restriction enzymes in accordance with manufacturer’s protocols.
7. Phenotypic analysis of the mutant strains
- Conduct mating experiments to determine whether the disruption of the MAT gene had an effect on the sexual capabilities of the fungus being studied.
NOTE: This step is dependent on the particular gene and species being studied. In this case, the gene being targeting is thought to be involved in sexual reproduction and thus mating tests were conducted. If the gene was thought, for example, to be involved in asexual reproduction, then something like conidial production could be measured.- In order to test the heterothallic capabilities of the mutant strain, co-inoculate fresh MEA medium with a mutant strain as well as a strain of opposite mating type. In the case of H. omanensis, keep the lids of the plates closed, but not sealed and incubate at room temperature for 7 days. Visually assess for the production of sexual structures.
- In order to test the homothallic capabilities of the mutant strain, inoculate fresh MEA medium with a mutant strain. In the case of H. omanensis, keep the lids of the plates closed, but not sealed and incubate at room temperature for 7 days. Visually assess for the production of sexual structures.
- Conduct growth rate experiments to determine whether the disruption of the MAT gene had an effect on the growth rate of the fungus being studied.
- Create mycelial-covered agar plugs from the actively growing edge of cultures of the mutant and wildtype strains by inserting the back side of a large, sterile pipette tip into the agar.
- Inoculate fresh MEA medium with these agar plugs. Ensure that at least three replicates per each culture type are made.
- After 3 days of growth at 20 °C, measure the growth on two perpendicular diameters.
- Compare the data from the wildtype and mutant strains.
Results
The protocol described above facilitated the introduction of a premature stop codon into a mating gene from the non-model ascomycete, H. omanensis. This process utilized a version of the CRISPR-Cas9 genome editing system and as such one of the most important steps in this protocol is the design and synthesis of a high quality sgRNA. Figure 1 shows how this molecule was designed in such a way that it A) specifically targets the gene of interest and shows little similarity to other regions in the genome and B) folds correctly in order to bind with the Cas9 protein. The sgRNA must also be capable of effectively cleaving the target region. The ability of the sgRNA to target and allow for the cleavage of the target region was conducted in vitro, yielding two products of the expected size.
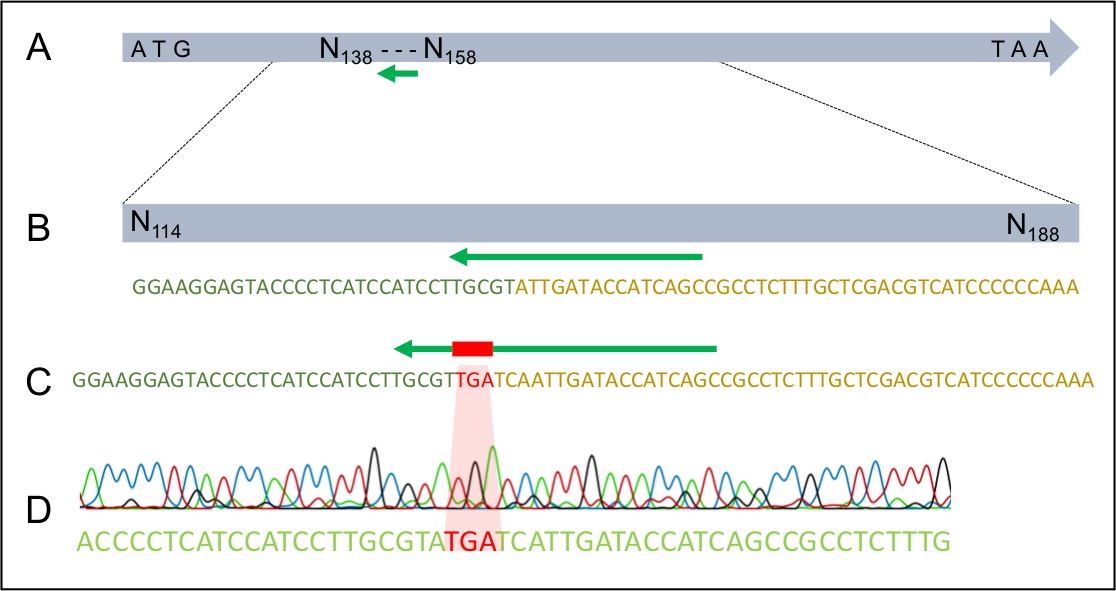
Once successful transformation has taken place, it is important to ensure that the dDNA has integrated into the genome only once and in the expected place. Figure 2 illustrates the design of PCR primers that target the insertion sites, which can be used to screen the potential transformants for the correct integration site. By designing primers that flank the 5’ and 3’ insertion sites, amplification is only possible if the dDNA is inserted at the correct region. Figure 4 illustrates that the premature stop codon was introduced into the MAT1-2-7 gene into the correct reading frame, ensuring that the gene would be truncated in a similar manner to that of H. moniliformis. Furthermore, Southern blot analysis showed that the dDNA construct was only integrated at a single site in the genome.
The success of the protocol was confirmed upon the phenotypic analysis of the mutant strains. In the case of the MAT1-2-7 disruption experiment, two independent mutant strains were developed. In both isolates, the vegetative radial growth rate was significantly reduced, suggesting a pleiotropic effect of the novel mating gene (Figure 5). Furthermore, the mutant isolates were incapable of completing a sexual cycle, producing only immature sexual structures that did not produce sexual spores (Figure 5). This was in contrast to wildtype isolates, which completed the entire sexual cycle within a few days of incubation (Figure 5).

Figure 1: Choosing a suitable sgRNA candidate.
(A) A suitable sgRNA will only have similarity to the target region of the genome (in this case indicated by the MAT locus sequence). (B) A suitable sgRNA will have identical minimal free energy and centroid secondary structures, with the three stem loops and five rings in the primary step loop. Furthermore, the majority of the structure will have high binding probabilities (indicated in dark orange and red) while lower binding probabilities should be seen at the protospacer region (indicated by the black triangles). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Design, amplification and assembly of the dDNA.
The first and second primer pairs (PP1 and PP2) are used to amplify approximately 800 bp upstream (5’) and 800 bp downstream (3’) of the gene of interest. The reverse primer of PP1 and the forward primer of PP2 include regions of homology to the hygromycin resistance cassette. The third primer pair amplifies the entire hygromycin resistance cassette. In a stepwise manner, the various amplicons are assembled until the entire dDNA, comprised of the 5’ region, the hygromycin resistance cassette and 3’ region, is assembled. When transformed into the cell, the dDNA should recombine at the region where the Cas9 enzyme will have been directed to cut, thereby replacing the gene of interest with the hygromycin resistance cassette. PP4 and PP5 can be used to determine if the dDNA has been correctly inserted into the genome at the appropriate location. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: The different cell types important during the protoplast extraction protocol.
(A) Conidia are used as the starting material for the protocol. These conidia are allowed to germinate and grow until they are (B) young germlings. The ideal growth phase of the young germlings are indicated by the two black arrows. Other mycelial strands seen on (B) are too mature for degradation and should not be used. The final step of the protocol is the release of the (C) round protoplasts, indicated by the black, dotted circles. These cells no longer have cell walls and are thus very sensitive to mechanical disruption. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: The successful integration of the TGA stop codon into the MAT1-2-7 gene of H. omanensis.
(A) The full-length H. omanensis MAT1-2-7 gene, with the sgRNA target site indicated by the green arrow. (B) A magnified schematic of the sgRNA target site within the H. omanensis MAT1-2-7 gene. (C) A magnified schematic of a region of the dDNA showing the stop codon flanked by arms homologous to the MAT1-2-7 gene of H. omanensis. (D) Sanger sequence chromatogram indicating the successful integration of the stop codon into the MAT1-2-7 gene. Modified from Wilson et al. 202021. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: The phenotypic differences between (A) wildtype isolates and (B) mutant isolates.
The first three images in each panel show the differences in the sexual capabilities of the two isolate types. While the wildtype isolates form mature ascomata during sexual reproduction, complete with the exudation of spores from the tips of the ascomatal necks, the mutant isolates form only immature sexual structures that do not produce any sexual spores. The fourth image in each panel shows the difference in growth rate and morphology of the two isolate types. While the wildtype isolate grows much faster and with more aerial mycelia, the mutant shows slower and is submerged within the agar. Modified from Wilson et al. 202021. Please click here to view a larger version of this figure.
{kind=link}
| Reaction | Enzyme Concentration | Degradation Time |
| A | 1.250 mg/mL | 180 min |
| B | 1.875 mg/mL | 180 min |
| C | 2.500 mg/mL | 150 min |
| D | 3.750 mg/mL | 150 min |
| E | 4.375 mg/mL | 120 min |
| F | 5.000 mg/mL | 120 min |
Table 1: Degradation of the germling/mycelia solution with lysing enzymes from Trichoderma harzianum. The different enzymes concentrations correspond to different incubation periods, with lower concentrations requiring longer incubations.
Discussion
The protocol for the successful transformation of H. omanensis and editing of the MAT1-2-7 gene was demonstrated by introducing an in-frame premature stop codon along with a gene for resistance to hygromycin B21. This was achieved using a protein-based version of the CRISPR-Cas9 genome editing system. The experiment entailed the in vitro transcription of the sgRNA, PCR-based assembly of the dDNA and the co-transformation of these two nucleic acids with a commercially available Cas9 enzyme into protoplasts extracted from H. omanensis
Unlike other protocols that rely on the availability of many other molecular tools, the protocol described above can be successfully used in species for which the molecular toolbox is still fairly limited21. The protocol relies only on an established transformation system and the availability of NGS data, preferably whole genome sequence. While an effective transformation system may take some optimizing in a species for which this is not available, there are many different protocols available for a variety of species. Furthermore, genome data is becoming increasingly available for even the most obscure of species and is becoming easier to generate de novo if it does not already exist.
Given the length of the protocol, there are many steps at which modifications can be introduced and where troubleshooting may be necessary. This is particularly true of the steps that are considered species specific. For example, there are many incubation steps in this protocol that need to be conducted at specific temperatures and for specific lengths of time in order to generate cell types important for the experiment. These steps would thus require species-specific optimization. Where possible, micrographs of the particular cells or growth phases have been provided to assist in transferring this protocol to a different species (Figure 1). The type and concentration of enzymes used to degrade the cell walls of the fungal cells in order to release the protoplasts will also be specific to the species of fungus being studied. In this protocol, only one source of lysing enzymes from is used, while different enzyme combinations are required for the extraction of protoplasts in species like Fusarium verticillioides33. This step depends entirely on the chemical make of the cell wall and will thus need to be optimized on a species to species basis.
This method is particularly significant to those studying non-model species as there is no reliance on an expression system. A popular method of establishing the CRISPR-Cas9 genome editing system is to express the Cas9 protein, the sgRNA as well as the dDNA from one or two plasmids that are transformed into the cells of choice. In this case, the Cas9 needs to be expressed by a promoter that is capable of high levels of expression in the particular organism being studied. General promoters have been developed for use in filamentous fungi and while they are not compatible in all species, they do allow for low level expression and can successfully be used to express, for example, antibiotic resistance genes. These promoters, however, often do not allow for high levels of expression and thus cannot be used to express the Cas9 protein. Using a protein-based version of the CRISPR-Cas9 genome editing system overcomes this limitation and allows the sgRNA and dDNA to be co-transformed into the cell with an already produced Cas9 enzyme.
The development of this protein-based system for use in H. omanensis came after many unsuccessful attempts at genome editing using both the classical split marker approach as well as the plasmid-based CRISPR-Cas9 system. While efficiencies differ from species to species, the split marker approach has been successfully used with 100% efficiency in species as diverse as Alternaria alternata34,35, and C. nicotianae36. In contrast, the efficiency of this system in H. omanensis was zero, despite more than 80 independent transformation and integration events. Similarly, the plasmid-based CRISPR-Cas9 system has been successfully used with high efficiencies in Trichoderma reesei (>93%)17 and Penicillium chrysogenum (up to 100%)37. This is, again, in contrast to this system’s usefulness in H. omanensis. Sufficient expression of the Cas9 protein was not attainable in H. omanensis despite trying a number of potential promoters, including two species-specific promoters predicted from housekeeping genes. Thus, this system could not be used at all. Using the protein-based version of the CRISPR-Cas9 system, however, yielded many independent transformants, two of which harbored the integrated dDNA in the correct location. Furthermore, this experiment was attempted only once and was successful- further illustrating the ease at which this system can be used.
Future applications of this protocol include its optimization and use in other species of the Ceratocystidaceae. There is already a wealth of NGS data available for these species30,38,39 and studies regarding their host specificity40, growth rate and virulence41 have been conducted. These studies can be strengthened by the functional characterization of the genes that are thought to be involved in these processes, research which will now become possible due to the availability of a transformation and genome editing protocol.
In conclusion, thorough investigation into the genes underlying important biological processes in non-model species is becoming more accessible thanks to the availability of easy-to-use genome editing protocols that do not rely on the existence of extensive biological resources and molecular toolkits. Studying non-model species is becoming easier and will allow for the discovery of novel pathways and interesting deviations from the standard biological processes that have been elucidated in model species.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This project was supported by the University of Pretoria, the Department of Science and Technology (DST)/National Research Foundation (NRF) Centre of Excellence in Tree Health Biotechnology (CTHB). The project was additionally supported by Prof BD Wingfield’s DST/NRF SARChI chair in Fungal Genomics (Grant number: 98353) as well as Dr AM Wilson’s NRF PhD bursary (108548). The grant holders acknowledge that opinions, findings and conclusions or recommendations expressed in this piece of work are that of the researchers and that the funding bodies accept no liability whatsoever in this regard.
Materials
| Name | Company | Catalog Number | Comments |
| EcoRI-HF | New England Biolabs, Ipswich, USA | R3101S | |
| EnGen Spy Cas9 NLS protein | New England Biolabs, Ipswich, USA | M0646T | Used to assemble the RNP |
| Eppendorf 5810 R centrifuge | Eppendorf, Hamberg, Germany | ||
| FastStart Taq DNA Polymerase | Sigma, St Louis, USA | 12032902001 | Standard DNA polyermase |
| GeneJET Gel Extraction Kit | ThermoFisher Scientific, Waltham, USA | K0691 | |
| HindIII-HF | New England Biolabs, Ipswich, USA | R3104S | |
| HiScribeTM T7 Quick High Yield RNA synthesis kit | New England Biolabs, Ipswich, USA | E2050S | |
| Hygromycin B from Streptomyces hygroscopicus | Sigma, St Louis, USA | 10843555001 | |
| Infors HT Ecotron Shaking Incubator | Infors AG, Bottmingen, Switzerland | ||
| LongAmp Taq DNA Polymerase | New England Biolabs, Ipswich, USA | M0323S | Long-range, high-fidelity DNA polymerase |
| Malt extract agar, 2% (MEA) | 20 g ME and 20 g agar in 1 l ddH20 | ||
| Malt extract | Sigma, St Louis, USA | 70167-500G | |
| Agar | Sigma, St Louis, USA | A5306 | |
| Malt Extract broth, 1% (MEB) | Sigma, St Louis, USA | 70167-500G | 2 g ME in 200 ml ddH20 |
| Malt Extract broth, 2% (MEB) | Sigma, St Louis, USA | 70167-500G | 4 g ME in 200 ml ddH20 |
| Miracloth | Merck Millipore, New Jersey, USA | 475855 | |
| Nylon membrane (positively charged) | Sigma, St Louis, USA | 11209299001 | |
| Osmotic control medium (OCM) | 0.3% yeast extract, 20% sucrose, 0.3% casein hydrolysate | ||
| Casein Hydrolysate | Sigma, St Louis, USA | 22090 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Yeast extract | Sigma, St Louis, USA | Y1625 | |
| Osmotic control medium (OCM) agar | Osmotic control medium (OCM) + 1% agar | ||
| Agar | Sigma, St Louis, USA | A5306 | |
| PCR DIG Labeling Mix | Sigma, St Louis, USA | 11585550910 | |
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific, Waltham, USA | F-530XL | High fidelity DNA polymerase |
| Plasmid pcb1004 | N/A | N/A | From: Carroll et al., 1994 |
| Presynthesized sgRNA | Inqaba Biotec, Pretoria, South Africa | Ordered as an synthesized dsDNA with specified sequence | |
| Proteinase K | Sigma, St Louis, USA | P2308 | |
| PTC Solution | 30% polyethylene glycol 8000 in STC buffer from above | ||
| Polyethylene glycol 8000 | Sigma, St Louis, USA | 1546605 | |
| RNase A | ThermoFisher Scientific, Waltham, USA | 12091021 | |
| RNAfold Webserver | Institute for Theoretical Chemistry, University of Vienna | N/A | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| RNAstructure | Mathews Lab | N/A | https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html |
| Sorbitol, 1 M | Sigma, St Louis, USA | 1617000 | 182.17g sorbitol in 1 l ddH20 |
| STC Buffer | 20% sucrose, 50 mM Tris-HCl pH 8.00 and 50 mM CaCl2 | ||
| Calcium chloride | Sigma, St Louis, USA | 429759 | |
| Tris-HCl pH 8.00 | Sigma, St Louis, USA | 10812846001 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Trichoderma harzianum lysing enzymes | Sigma, St Louis, USA | L1412 | |
| Zeiss Axioskop 2 Plus Ergonomic Trinocular Microscope | Zeiss, Oberkochen, Germany |
References
- Ekblom, R., Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 107, 1-15 (2011).
- Russell, J. J., et al. Non-model model organisms. BMC Biology. 15 (55), 1-31 (2017).
- Kück, U., Hoff, B. New tools for the genetic manipulation of filamentous fungi. Applied Microbiology and Biotechnology. 86, 51-62 (2010).
- Li, D., Tang, Y., Lin, J., Cai, W. Methods for genetic transformation of filamentous fungi. Microbial Cell Factories. 16 (168), 1-13 (2017).
- Lorito, M., Hayes, C. K., Di Pietro, A., Harman, G. E. Biolistic transformation of Trichoderma harzianum and Gliocladium virens using plasmid and genomic DNA. Current Biotechnology. 24, 349-356 (1993).
- Taylor, P., et al. Transformation of intact Aspergillus niger by electroporation. Bioscience, Biotechnology, and Biochemistry. 58 (12), 2224-2227 (2014).
- Dhawale, S. S., Paietta, J. V., Marzluf, G. A. A new, rapid and efficient transformation procedure for Neurospora. Current Genetics. 8, 77-79 (1984).
- Sayari, M., Van Der Nest, M. A., Steenkamp, E. T., Adegeye, O. O., Marincowitz, S. Agrobacterium-mediated transformation of Ceratocystis albifundus. Microbiological Research. 226, 55-64 (2019).
- Meyer, V. Genetic engineering of filamentous fungi- Progress, obstacles and future trends. Biotechnology Advances. 26, 177-185 (2008).
- You, B. J., Lee, M. H., Chung, K. R. Gene-specific disruption in the filamentous fungus Cercospora nicotianae using a split-marker approach. Archives of Microbiology. 191, 615-622 (2009).
- Gravelat, F. N., Askew, D. S., Sheppard, D. C. Targeted gene deletion in Aspergillus fumigatus using the hygromycin-resistance split-marker approach. Host-Fungus Interactions. 845, 119-130 (2012).
- Wang, Y., Diguistini, S., Bohlmann, J., Breuil, C. Agrobacterium-meditated gene disruption using split-marker in Grosmannia clavigera, a mountain pine beetle associated pathogen. Current Genetics. 56, 297-307 (2010).
- Wood, A. J., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333, 307 (2011).
- Mahfouz, M. M., Piatek, A., Neal, C. Genome engineering via TALENs and CRISPR/Cas9 systems: Challenges and perspectives. Plant Biotechnology. 12, 1006-1014 (2014).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85 (1), 227-264 (2016).
- Arazoe, T., et al. Tailor-made TALEN system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnology and Bioengineering. 112 (7), 1335-1342 (2015).
- Liu, R., Chen, L., Jiang, Y., Zhou, Z., Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discovery. 1, 1-11 (2015).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-822 (2012).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences. 109 (39), 2579-2586 (2012).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Cell. 8 (11), 2281-2308 (2013).
- Wilson, A. M., Wilken, P. M., Van Der Nest, M. A., Wing, M. J., Wing, B. D. The novel Huntiella omanensis mating gene, MAT1-2-7, is essential for ascomatal maturation. Fungal Genetics and Biology. 137, 103335 (2020).
- Miao, J., et al. Characterization of an N-terminal non-core domain of RAG1 gene disrupted Syrian Hamster model generated by CRISPR Cas9. Viruses. 10 (243), 10050243 (2018).
- Roberts, B., et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Molecular Biology of the Cell. 28 (21), 2854-2874 (2016).
- Schneider, S., Kirchner, M., Kirchner, M., Schneider, S. CRISPR-Cas: From the bacterial adaptive immune system to a versatile tool for genome engineering. Angewandte Chemie International Edition. 54 (46), 13508-13514 (2015).
- Nødvig, C. S., Nielsen, J. B., Kogle, M. E., Mortensen, U. H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 10 (7), 1-18 (2015).
- Wang, Q., Cobine, P. A., Coleman, J. J. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genetics and Biology. 117, 21-29 (2018).
- Nagy, G., et al. Development of a plasmid free CRISPR-Cas9 system for the genetic modification of Mucor circinelloides. Scientific Reports. 7 (16800), 1-10 (2017).
- Al-Subhi, A. M., Al-Adawi, A. O., Van Wyk, M., Deadman, M. L., Wingfield, M. J. Ceratocystis omanensis, a new species from diseased mango trees in Oman. Mycological Research. 110 (2), 237-245 (2006).
- Wilson, A. M., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Pheromone expression reveals putative mechanism of unisexuality in a saprobic ascomycete fungus. PLoS ONE. 13 (3), 0192517 (2018).
- van der Nest, M. A., et al. Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus. 5 (2), 472-485 (2014).
- Wilson, A. M., Godlonton, T., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Unisexual reproduction in Huntiella moniliformis. Fungal Genetics and Biology. 80, 1-9 (2015).
- Sambrook, J., Green, M. . Molecular cloning: A laboratory manual. , (2012).
- Ramamoorthy, V., Govindaraj, L., Dhanasekaran, M., Vetrivel, S., Kumar, K. K., Ebenezar, E. Combination of driselase and lysing enzyme in one molar potassium chloride is effective for the production of protoplasts from germinated conidia of Fusarium verticillioides. Journal of Microbiological Methods. , (2015).
- Lin, C., Yang, S. L., Wang, N., Chung, K. The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata functions independently or cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic processes. Fungal Genetics and Biology. 47 (4), 381-391 (2010).
- Lin, C., Chung, K. Specialized and shared functions of the histidine kinase- and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, a filamentous fungal pathogen of citrus. Fungal Genetics and Biology. 47 (10), 818-827 (2010).
- Choquer, M., et al. The CTB1 gene encoding a fungal polyketide synthase is required for cercosporin biosynthesis and fungal virulence of Cercospora nicotianae. Molecular Plant-Microbe Interactions. 18 (5), 468-476 (2005).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- van der Nest, M. A. M. A., et al. Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans and Ceratocystis moniliformis. IMA Fungus. 5 (1), 135-140 (2014).
- Wingfield, B. D., et al. Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus. 7 (2), 317-323 (2016).
- Fourie, A., Van Der Nest, M. A., De Vos, L., Wingfield, M. J., Wingfield, B. D., Barnes, I. QTL mapping of mycelial growth and aggressiveness to distinct hosts in Ceratocystis pathogens. Fungal Genetics and Biology. 131, 103242 (2019).
- Lee, D. H., Roux, J., Wingfield, B. D., Wingfield, M. J. Variation in growth rates and aggressiveness of naturally occurring self-fertile and self-sterile isolates of the wilt pathogen Ceratocystis albifundus. Plant Pathology. 64 (5), 1103-1109 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved