
Method Article
CrispR-Cas9-Mediato Genome Editing nel Filamentous Ascomycete Huntiella omanensis
In questo articolo
Riepilogo
Il sistema di editing del genoma CRISPR-Cas9 è un editor del genoma facile da usare che è stato utilizzato in specie modello e non modello. Qui presentiamo una versione a base proteica di questo sistema che è stata usata per introdurre un codon di arresto prematuro in un gene di accoppiamento di un fungo ascomycete filamentoso non modello.
Abstract
Il sistema di editing del genoma CRISPR-Cas9 è uno strumento molecolare che può essere utilizzato per introdurre cambiamenti precisi nei genomi di specie modello e non modello. Questa tecnologia può essere utilizzata per una varietà di approcci di editing del genoma, dai knockout geniali e knockin a cambiamenti più specifici come l'introduzione di alcuni nucleotidi in una posizione mirata. L'editing genomico può essere utilizzato per una moltitudine di applicazioni, tra cui la caratterizzazione funzionale parziale dei geni, la produzione di organismi transgenici e lo sviluppo di strumenti diagnostici. Rispetto alle strategie di editing genetico precedentemente disponibili, il sistema CRISPR-Cas9 si è dimostrato facile da stabilire in nuove specie e vanta un'elevata efficienza e specificità. La ragione principale di questo è che lo strumento di editing utilizza una molecola di RNA per indirizzare il gene o la sequenza di interesse, rendendo semplice la progettazione della molecola bersaglio, dato che le regole standard di abbinamento di base possono essere sfruttate. Analogamente ad altri sistemi di editing del genoma, i metodi basati su CRISPR-Cas9 richiedono anche protocolli di trasformazione efficienti ed efficaci, nonché l'accesso a dati di sequenza di buona qualità per la progettazione delle molecole di RNA e DNA di destinazione. Dall'introduzione di questo sistema nel 2013, è stato utilizzato per progettare geneticamente una varietà di specie modello, tra cui Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster e Mus musculus. Successivamente, i ricercatori che lavorano su specie non modello hanno approfittato del sistema e lo hanno utilizzato per lo studio di geni coinvolti in processi diversi come il metabolismo secondario nei funghi, la crescita dei nematodi e la resistenza alle malattie nelle piante, tra molti altri. Questo protocollo descritto di seguito descrive l'uso del protocollo di editing del genoma CRISPR-Cas9 per il troncamento di un gene coinvolto nel ciclo sessuale di Huntiella omanensis, un fungo filamentoso ascomycete appartenente alla famiglia Ceratocystidaceae.
Introduzione
La crescente disponibilità di genomi e trascrimami di alta qualità, completamente assemblati, ha notevolmente migliorato la capacità di studiare un'ampia varietà di processi biologici in una serie di organismi1. Ciò vale sia per le specie modello che per le specie non modello, molte delle quali possono offrire una comprensione più diversificata dei processi biologici. Questi tipi di dati possono essere utilizzati per la scoperta genica, l'identificazione delle reti di trascrizione e confronti sia dell'intero genoma che del trascritoma, ognuno dei quali viene utilizzato con un proprio insieme di applicazioni. Tuttavia, mentre i geni sono previsti, annotati e collegati putativamente a percorsi funzionali diversi a un ritmo mai visto prima, la caratterizzazione funzionale di questi geni rimane indietro, limitata dai toolkit molecolari disponibili per molte specie. Ciò è particolarmente vero per le specie non modello, dove i dati genomici sono relativamente facili da generare, ma dove un'ulteriore caratterizzazione molecolare è stataquasi impossibile 1,2.
La caratterizzazione parziale delle funzioni di geni specifici importanti per la biologia delle specie fungine può essere ottenuta mediante esperimenti di knockout o knockin seguiti da un'analisi fenotipico dei ceppi mutanti3. Questi due sistemi si basano interamente sulla disponibilità di protocolli di ingegneria genetica, tra cui, come minimo, un sistema di trasformazione e un sistema di editing genetico. Ci sono un certo numero di diversi sistemi di trasformazione che sono stati sviluppati in una varietà di funghi filamentosi4. Sistemi fisici come quelli che si basano su biolitica ed elettroporazione sono stati sviluppati rispettivamente in Trichoderma harzianum5 e Aspergillus niger6. Sistemi che utilizzano sostanze chimiche come cloruro di calcio o acetato di litio sono stati sviluppati in Neurospora crassa7. Infine, i sistemi biologici che si basano sull'uso di Agrobacterium tumefaciens per la trasformazione sono stati utilizzati con successo in Ceratocystis albifundus8.
In contrasto con la disponibilità di diversi protocolli di trasformazione, i sistemi di editing del genoma sono meno abbondanti. Molti dei tradizionali esperimenti di caratterizzazione funzionale condotti in funghi filamentosi hanno utilizzato un costrutto di knockout del marcatore diviso sotto forma di un marcatore selezionabile affiancato da regioni di omologia alla regione o gene di destinazione nel genoma3. Il metodo si basa sulla riparazione del DNA orologica diretta (HR), che consente di ricombinare l'omologazione tra il costrutto knockout e la regione di interesse. Questo evento di ricombinazione comporta la sostituzione del gene di interesse con la sequenza del marcatore selezionabile. Purtroppo, mentre questo ha avuto successo in molte specie tra cui Cercospora nicotianae10, Aspergillus fumigatus11 e Grosmannia clamivigera12, i tassi di ricombinazione omologha sono altamente variabili tra diverse specie fungine, rendendo questo un protocollo inefficiente e talvolta inutilizzabile in alcune specie3.
Altri sistemi di editing del genoma, compresi quelli che fanno uso di nucleasi zinco-dita (NF) e nucleasi effettorali di trascrizione-attivatore (TALEN) hanno rappresentato un grande miglioramento sui sistemi più vecchi, in particolare data la loro capacità di apportare modifiche specifiche e mirate13. Sia le FN che i TALEN sono costituiti da una proteina nucleasi e da una proteina in grado di riconoscere specifiche sequenze di nucleotidi13. Dopo il riconoscimento, la nucleasi induce una doppia rottura del DNA incagliato che può facilitare l'introduzione di mutazioni specifiche. Per realizzare cambiamenti del genoma, la regione proteica che riconosce la sequenza dei nucleotidi deve essere progettata specificamente per ogni esperimento. A causa di questa dipendenza dalle interazioni proteina-nucleica per guidare l'editing, la progettazione e la produzione delle molecole di targeting per ogni esperimento di knockout o knockin è difficile elaboriosa 14,15. Illustrativi di queste sfide, pochissimi funghi filamentosi sono stati sottoposti all'editing del genoma utilizzando questi sistemi. Un esempio è il sistema basato su TALENs che è stato sviluppato nel fungo dell'esplosione di riso, Magnaporthe oryzae16.
Probabilmente la più grande rivoluzione nel campo dell'editing del genoma è stata la scoperta e il successivo sviluppo del sistema CRISPR-Cas9, un editor del genoma che consente la scissione mirata di una sequenza di interesse da parte di un endonucleasi guidato da una molecola di RNA. Questo è stato un enorme miglioramento rispetto agli editori del genoma precedentemente sviluppati che si basavano sulle interazioni proteina-nucleico come il principale vantaggio del sistema CRISPR-Cas9 è che si basa su una molecola di RNA per colpire la regione di interesse. Ciò significa che il sistema si basa su un'interazione RNA-DNA e quindi le regole standard di associazione di base possono essere sfruttate durante la progettazione di ogniesperimento 15.
Il sistema CRISPR-Cas9, come descritto qui, è composto da tre componenti principali: un singolo RNA guida (sgRNA), l'enzima Cas9 e un DNA donatore (dDNA)17. Lo sgRNA è composto da una regione di 20 nucleotidi chiamata protospazista e da una regione più lunga chiamata impalcatura18. La regione protospaziare viene utilizzata per guidare il sistema di editing verso la regione di destinazione e viene quindi riprogettata per ogni esperimento. Lo scaffold è la regione dell'RNA che si lega fisicamente all'enzima Cas9 per formare la ribonucleoproteina (RNP) ed è quindi identico indipendentemente dalla regione da prendere di mira. L'enzima Cas9 facilita fisicamente la scissione del DNA bersaglio, utilizzando il protospaziatore come guida per identificare questa regione19. L'ultimo componente, il dDNA, è facoltativo e il suo utilizzo dipende dall'esperimento specifico20. Il dDNA ospita la sequenza che dovrebbe essere specificamente inserita nella regione tagliata dall'enzima Cas9, ed è quindi ideale per esperimenti di knockin genico in cui un gene viene introdotto nel genoma o per esperimenti di knockout genico in cui viene introdotto un gene di resistenza agli antibiotici o un altro marcatore selezionabile per sostituire il gene di interesse. Il dDNA può anche essere progettato in modo tale da introdurre nuove sequenze nel genoma. Ad esempio, come descritto di seguito, è possibile introdurre un codon di arresto in-frame in una particolare regione nel gene di interesse quando è richiesto un troncamento genico21. Altre applicazioni includono la mutazione di regioni specifiche del gene, come un dominiofunzionale 22, o l'introduzione di una sequenza di tagging23.
Uno dei principali vantaggi dell'utilizzo del sistema CRISPR-Cas9 è la sua versatilità24. Un esempio di questa adattabilità è che l'enzima Cas9 può essere introdotto nella cellula ospite in una delle sue tre forme - DNA, RNA o proteina - a seconda del particolare sistema di trasformazione utilizzato. Quando viene introdotto in forma di DNA, il gene cas9 è spesso incluso su un plasmide insieme a un marcatore selezionabile, una cassetta per esprimere lo sgRNA e, se necessario, una cassetta che codifica la sequenza dDNA25. Il vantaggio principale di questo sistema è che solo un singolo costrutto deve essere trasformato nella cellula e la trasformazione di successo assicura che siano presenti tutti i componenti necessari per l'editing del genoma mediato da CRISRP-Cas9. Tuttavia, questo metodo si basa sulla disponibilità di un sistema di espressione per la specie ospite. Affinché Cas9 induca con successo il danno al DNA, deve essere espresso a livelli elevati e, pertanto, è necessario un promotore adatto e potenzialmente specifico. Per le specie non modello in cui tali promotori non sono ancora stati sviluppati, questo può essere un fattore di sminuzione e quindi la capacità di introdurre Cas9 in forma di RNA o proteina può essere un'opzione più attraente. L'introduzione dell'RNA nella cellula porta le sue sfide, in particolare in quanto l'RNA è instabile e potrebbe non sopravvivere al processo di trasformazione. Inoltre, se introdotta in forma di DNA o RNA, la sequenza genica Cas9 potrebbe dover essere ottimizzata per l'uso nel particolare sistema host17. Ad esempio, il gene cas9 di Streptococcus pyogenes potrebbe non funzionare in una cellula ospite di mammiferi e un gene cas9 che è stato ottimizzato per il codon per l'uso in una cellula di mammifero potrebbe non funzionare in una cellula vegetale. Tutte queste sfide possono essere superate utilizzando la forma proteica di Cas9, che, insieme allo sgRNA, può essere assemblata in un RNP e trasformata nella cellula ospite26,27. Questo sistema non si basa su alcun sistema di espressione endogeno o ottimizzazione del codon e dovrebbe quindi funzionare nella maggior parte delle specie non modello. Lo svantaggio del sistema basato sulle proteine è che non è compatibile con i sistemi di trasformazione basati sul DNA come il trasferimento mediato dall'Agrobacterium. Pertanto, affinché il metodo a base di proteine funzioni, deve essere disponibile un protocollo di trasformazione come quelli che si basano su protoplasts o biolistici. Questo sistema a base di RNP è stato utilizzato con successo nei funghi filamentosi, Fusarium oxysporum26 e Mucor circinelloides27.
Huntiella omanensis, un membro della famiglia Ceratocystidaceae, è un fungo cosmopolita spesso trovato su piante legnose appena ferite28. Mentre i dati del genoma e del trascrimama di alta qualità sono disponibili perquesta specie 28,29,30, non sono stati sviluppati protocolli di trasformazione o di editing del genoma. Ad oggi, la ricerca su H. omanensis si è concentrata sui componenti genetici sottostanti del suo ciclosessuale 29,31. Questo fungo presenta un tipico ciclo sessuale eterololico, con riproduzione sessuale che si verifica esclusivamente tra gli isolati del MAT1-1 e MAT1-2 tipi di accoppiamento31. Al contrario, gli isolati MAT1-2 della Huntiella moniliformis strettamente imparentata sono in grado di riproduzione sessuale indipendente e completare un ciclo sessuale in assenza di un partner MAT1-131. Si ritiene che questa differenza nelle capacità sessuali sia, almeno in parte, dovuta a una grande differenza nel gene di accoppiamento, MAT1-2-7, dove H. omanensis ospita una copia completa e intatta, mentre il gene viene gravemente troncato in H. moniliformis29,31. Per caratterizzare ulteriormente il ruolo di questo gene nella riproduzione sessuale, il gene MAT1-2-7 di H. omanensis è stato troncato per imitare il troncamento visto in H. moniliformis21.
Il protocollo seguente descrive in dettaglio la trasformazione di H. omanensis e il troncamento del gene MAT1-2-7 utilizzando una versione a base proteica del sistema di editing del genoma CRISPR-Cas9. Questo protocollo è stato sviluppato dopo che gli approcci della sostituzione genica basata sulla ricombinazione omologa e dell'editing del genoma CRISPR-Cas9 basato sul plasmide non hanno avuto successo.
Protocollo
1. Progettazione e sintesi dello sgRNA
- Per identificare potenziali regioni protospaziali che faranno parte dello sgRNA, cercare manualmente il gene di interesse per 5' NGG 3' triplette, utilizzando la funzione di ricerca in qualsiasi programma viene utilizzato. Annotare queste terzine come sequenze PAM.
NOTA: sono disponibili vari programmi software che cercano e annotano le potenziali sequenze PAM e protospazi.- Selezionare i 20 bp a monte di ciascuna delle sequenze PAM identificate e annotare queste sequenze come potenziali protospazi.
- Per decidere su un singolo protospazista, eseguire i seguenti passaggi di filtraggio per eliminare le sequenze di bassa qualità.
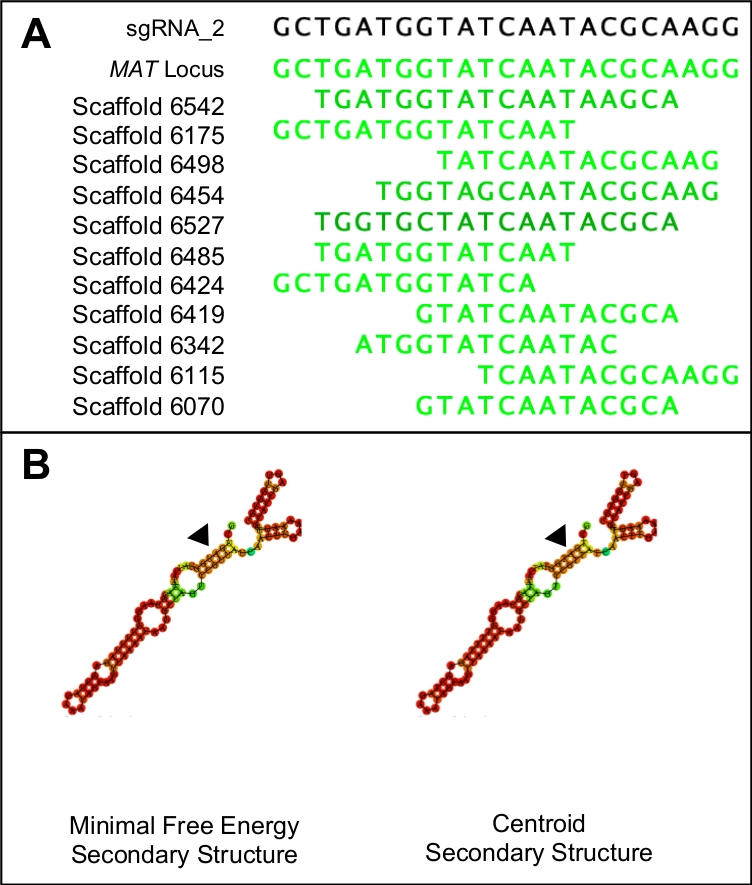
- Per verificare la specificità dei potenziali protospazi, combinare la sequenza PAM e il protospaziatore in un'unica sequenza e utilizzarla come query BLASTn sull'intero genoma. Eliminare tutti i protospazi che mostrano una somiglianza con qualsiasi area del genoma diversa dall'area di destinazione (Figura 1A).
- Per garantire che la molecola di RNA si pieghi nella struttura 3D corretta per il legame con l'enzima Cas9, creare una sequenza combinata che include il protospaziatore e la sequenza di scaffold specifica per l'enzima Cas9.
- Caricare ciascuna delle sequenze combinate protospazial-scaffolding in uno strumento di previsione della struttura secondaria RNA (Tabella dei materiali).
- Analizzare i risultati confrontando l'energia libera minima e le strutture secondarie centroidi.
NOTA: i candidati protospaziali ideali avranno identiche strutture secondarie minime libere e centroidi. Entrambe le strutture secondarie dovrebbero essere costituite da tre anelli di stelo, interrotti da cinque strutture ad anello. Le strutture devono inoltre presentare elevate probabilità di rilegatura (indicate in rosso) in tutta la struttura, ad eccezione della regione che rappresenta il protosppatore (Figura 1B). I valori energetici effettivi non sono rilevanti.
- Scegliere un candidato finale tra quelli che hanno superato i passaggi di filtro precedenti selezionando il candidato più vicino all'area specifica di destinazione.
- Per sintetizzare l'sgRNA come una singola molecola di RNA, utilizzare un kit di sintesi sgRNA(Tabelladei materiali s) compatibile con il particolare enzima Cas9 che verrà utilizzato (ad esempio, Cas9 da Streptococcus pyogenes).
NOTA: I seguenti passaggi possono dipendere dal kit di sintesi sgRNA utilizzato. Nel caso in cui si usi un kit diverso, segui le istruzioni del produttore. In alternativa, lo sgRNA può essere ordinato pre-sintetizzato. Quando si lavora con l'RNA, utilizzare reagenti e usa e getta senza nuclease.- Se il protospaziatore da 20 bp scelto al punto 1.3 precedente non contiene una G all'estremità di 5', aggiungere una G a questa regione.
- Aggiungere la sequenza di promotore T7 alla fine 5' della sequenza di destinazione. Questa sequenza è standard ed è 5' TTCTAATACGACTCACTATAG 3'.
- Aggiungere una sequenza di sovrapposizione di 14 nt alla fine 3' della sequenza di destinazione. Questa sequenza è specifica del kit ed è 5' GTTTTAGAGCTAGA 3' per il kit utilizzato qui.
- Ordinare il frammento risultante 5' TTCTACGACTCACTATAG(N)20 GTTTTAGAGCTAGA, con (N)20 che rappresenta il protospazizzato selezionato presynthesized (Tabella dei materiali).
- Conformemente al protocollo del fabbricante(Tabella dei materiali), combinare i seguenti reagenti a temperatura ambiente: 2 L di acqua, 10 L di buffer di reazione, 5 L della sequenza protospazizzata sintetizzata, 1 L di 0,1 M DTT e 2 0,1 L di trascrizione enzima.
- Incubare questa soluzione a 37 gradi centigradi per 30 minuti e trasferirla nel ghiaccio.
- Aggiungete 30 L di acqua e 2 L di DNase I, mescolate e incubate a 37 gradi centigradi per altri 15 minuti.
- Visualizza l'sgRNA risultante su un gel di agarose del 2%.
2. Testare la capacità di Cleavage in vitro dello sgRNA
NOTA: questo passaggio è facoltativo, ma è consigliato.
- Progetta primer che amplificano un frammento che ospita la sequenza del sito che lo sgRNA scelto si rivolgono e amplificano la regione utilizzando una polimerasi del DNA standard.
NOTA: Se possibile, progettare i primer in modo tale che la scissione nel sito di destinazione produrrà due frammenti di dimensioni molto diverse che possono essere facilmente distinti l'uno dall'altro su un gel di agarose standard. - Assemblare la ribonucleoproteina sgRNA-Cas9 combinata incubando una soluzione composta da 30 nM sgRNA come sintetizzato sopra, 30 nM Cas9 proteina, 10x buffer di reazione e 10 L di acqua a 25 gradi centigradi per 10 min.
- Testare la capacità di scissione dello sgRNA aggiungendo il prodotto PCR della regione bersaglio nella soluzione RNP ad una concentrazione finale di 3 nM.
- Incubare la soluzione a 37 gradi centigradi per 15 minuti.
- Aggiungere 3 g di proteinasi K e 2 g di RNase alla soluzione per fermare la reazione di scissione e incubare a temperatura ambiente per 10 min.
- Visualizza i frammenti di DNA risultanti su un gel di agarose del 2%. Lo sgRNA è adatto per la sperimentazione in vivo se due bande della dimensione prevista sono osservate sul gel.
3. Progettazione e sintesi del dDNA
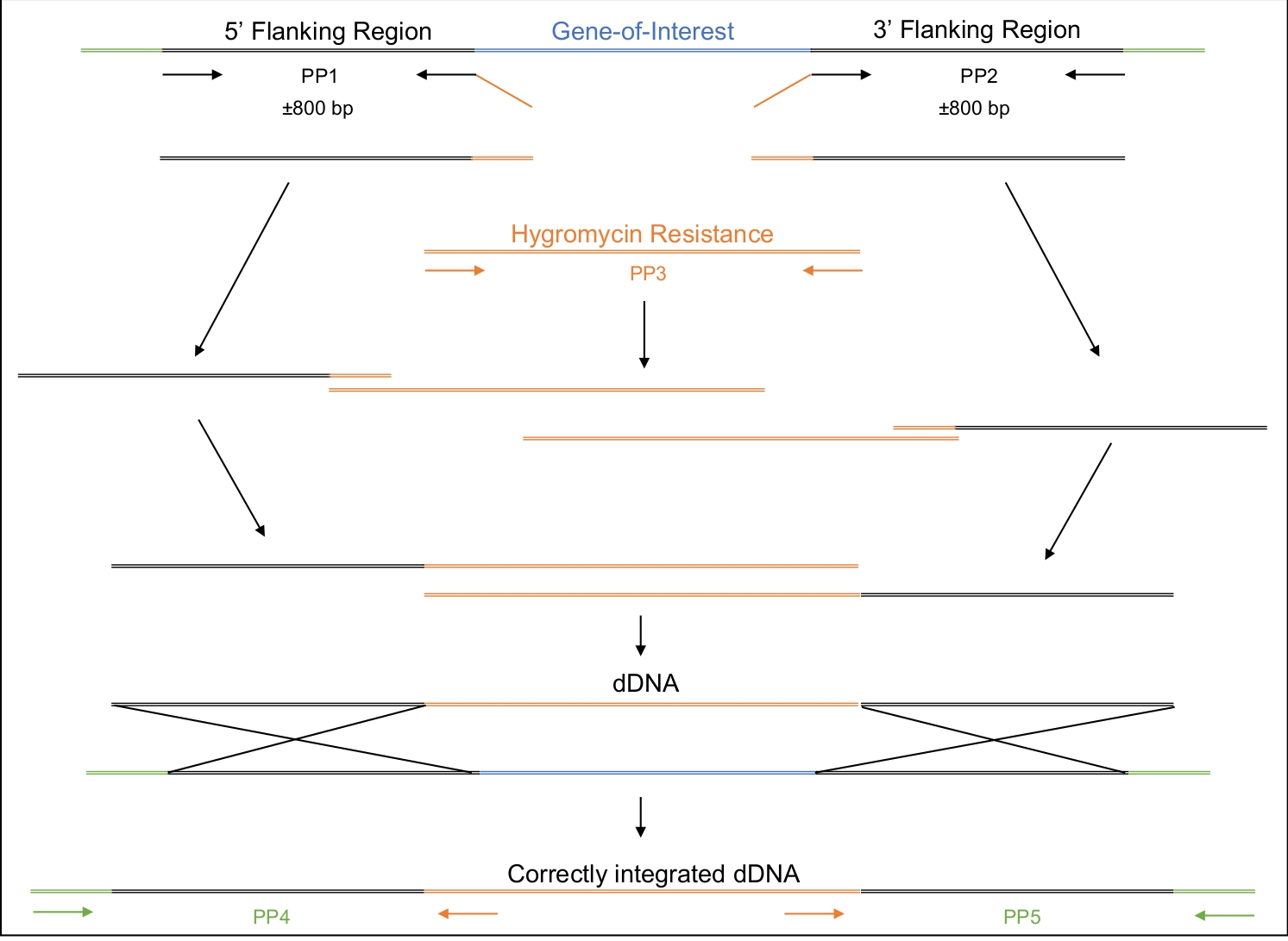
- Progettare il dDNA in modo che sia composto da tre regioni- 5' e 3' regioni con complementarità alla regione genomica di destinazione e una regione centrale che ospita un marcatore selezionabile (Tabella dei materiali, Figura 2).
NOTA: è possibile aggiungere altre sequenze specifiche a questo marcatore selezionabile. Per questo particolare esperimento, è stata aggiunta una sequenza di codon di arresto (5' TGA 3') appena prima del marcatore selezionabile, introducendo così un codon di arresto nel telaio nel gene. Il dDNA può essere ordinato presynthesized. In alternativa, il dDNA può essere amplificato e assemblato utilizzando un approccio PCR graduale e sovrapposto, come descritto di seguito.- Progetta primer per amplificare circa 800 bp delle regioni di fiancheggiamento da 5' e 3'. Aggiungere 20 nt di sequenza che è complementare alla sequenza del marcatore selezionabile al primer inverso della regione 5' e 3' primer in avanti della regione.
- Progetta primer che amplificano il marcatore selezionabile, assicurando che il prodotto amplificato porti il gene della resistenza e un promotore noto per lavorare nella specie di interesse.
- Utilizzando una polimerasi del DNA ad alta fedeltà (Tabella dei Materiali), amplificare le tre regioni dDNA. Amplificare le regioni 5' e 3' dalla gDNA dell'organismo in fase di modifica. Amplificare il marcatore selezionabile da una fonte pertinente.
- In una singola reazione, combinare la regione amplificata di 5' con il marcatore selezionabile e, utilizzando una polimerasi del DNA a lungo raggio e ad alta fedeltà, amplificare l'intera regione.
- In una seconda reazione singola, combinare la regione amplificata di 3' con il marcatore selezionabile e, utilizzando una polimerasi del DNA a lungo raggio e ad alta fedeltà, amplificare l'intera regione.
- Infine, combina i due prodotti PCR precedenti in un'unica reazione e amplifica l'intera sequenza dDNA con una polimerasi del DNA a lungo raggio e ad alta fedeltà.
- Visualizza il frammento di DNA su un gel di agarose dell'1%. Nel caso in cui vengano prodotti due o più frammenti, purificare il frammento di dimensioni corretto dal gel utilizzando un kit di purificazione del gel.
4. Estrazione di protoplasti
- Per produrre conidia, inoculare 200 mL di brodo fresco di estratto di malto (MEB) in una fiaschetta da 500 mL con un blocco di agar ricoperto di micelia di 1 cm x 1 cm.
NOTA: Non tutti i funghi sono in grado di produrre conidia. In tal caso, mycelia può anche essere utilizzato. Questo richiederà in genere concentrazioni più elevate di enzima di lising ulteriormente nel protocollo.- Incubare la coltura liquida in un'incubatrice tremante a 25 gradi centigradi con agitazione a 120 giri/min per 24 - 48 h.
NOTA: Questo tempo di incubazione e la temperatura è stato ottimizzato per H. omanensis. Questo dovrà essere ottimizzato per altre specie. - Per raccogliere la conidia; filtrare la coltura liquida attraverso uno strato di stoffa sterile da laboratorio (ad esempio, Miracloth), trasferire la sospensione coniali in tubi centrifuga da 50 mL e centrifuga a 3.220 x g a 4 gradi centigradi per 10 min. Scarta il supernatant.
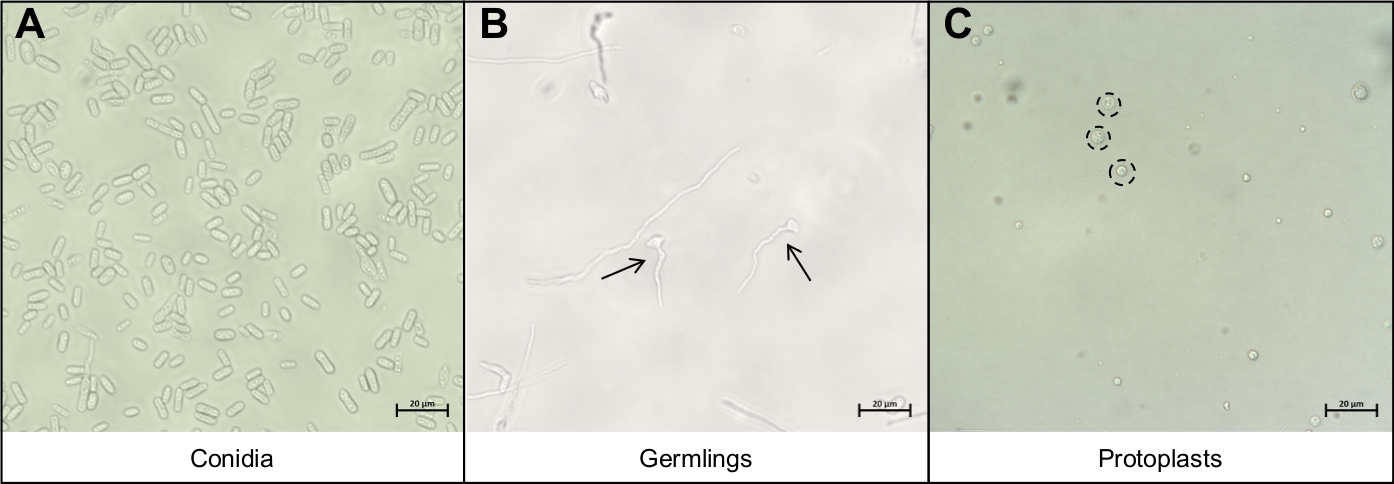
- Rispendere la conidia in 5 mL di acqua e pipette 10 L della soluzione conidia su un faro al microscopio e coprire con un copriposcopio. Visualizzare utilizzando un microscopio composto sotto ingrandimento 40x per garantire che solo conidia sono stati recuperati (Figura 3A).
- In una fiaschetta da 500 mL, inoculare 200 mL di MEB fresco dell'1% con il volume totale di conidia risupesata.
- Incubare la coltura liquida in un'incubatrice di scossa a 25 gradi centigradi con agitazione a 120 giri/min per un massimo di 12 h.
NOTA: Questo tempo di incubazione è stato ottimizzato per H. omanensis. Questo dovrà essere ottimizzato per altre specie. - Per raccogliere i germi, trasferire la coltura liquida in tubi centrifugati da 50 mL e centrifugare a 3.220 x g a 4 gradi centigradi per 10 min. Scarta il supernatant.
- Riespondare i germi in un massimo di 10 mL di 1 M di sorbitolo.
- Pipetta 10 L della soluzione germinale su un condotto al microscopio e coprire con un copriposcopio. Visualizzare utilizzando un microscopio composto sotto ingrandimento 40x per garantire che siano stati recuperati solo i germi (Figura 3B).
NOTA: il protocollo può essere messo in pausa qui. Conservare i germi in 1 M di sorbitolo a -80 gradi centigradi.
- Incubare la coltura liquida in un'incubatrice tremante a 25 gradi centigradi con agitazione a 120 giri/min per 24 - 48 h.
- Per lysire le pareti cellulari dei giovani germi e rilasciare i protoplasti, aggiungere 1 mL della sospensione germinale a 9 mL di enzima liscia a varie concentrazioni in una fiaschetta sterile da 50 mL.
NOTA: Nella tabella 1 vengono utilizzate diverse concentrazioni di enzimi e tempi di incubazione. Gli enzimi e le concentrazioni sono anche suscettibili di variare a seconda del fungo e dovranno essere ottimizzati per ogni specie.- Incubare la soluzione spore-enzima in un'incubatrice di agitazione a 25 gradi centigradi con agitazione a 80 giri/min per 2-3 h.
- Filtrare la soluzione protoplaste attraverso uno strato di stoffa sterile da laboratorio e raccogliere i protoplasti mediante centrifugazione a 1.810 x g a 4 gradi centigradi per 10 min. Scarta il supernatant.
NOTA: I protoplasts sono celle senza pareti cellulari e sono quindi molto sensibili alle interruzioni meccaniche. Assicurarsi di gestirli con attenzione, in particolare durante il pipettamento. - Rispendere con cura il pellet protoplast in 200 L di buffer STC (Tabella dei Materiali).
- Pipette 10 L della soluzione protoplast su un condotto al microscopio e coprire con un coverslip. Visualizzare utilizzando un microscopio composto sotto ingrandimento 40x per garantire che solo i protoplasts sono stati recuperati (Figura 3C).
- Utilizzando un emocitometro, contare e calcolare il numero di protoplasti generati nei passaggi precedenti. Diluire la soluzione protoplast in aliquots contenenti circa 5 x 106 protoplasts.
NOTA: il protocollo può essere messo in pausa qui. Memorizzare i protoplasts nel buffer STC a -80 gradi centigradi.
5. Trasformazione assistita da Protoplast e PEG e recupero trasformatore
- Per iniziare la trasformazione, combinare circa 5 x 106 protoplasts con un singolo volume della soluzione RNP e circa 6 g del frammento dDNA.
NOTA: I protoplasti sono molto sensibili alle interruzioni meccaniche. Assicurarsi di gestirli con attenzione, in particolare durante il pipettamento.- Utilizzando una pipetta, gocciolare lentamente 1 mL di una soluzione PTC appena preparata al 30% sulla soluzione protoplast e incubare la soluzione a temperatura ambiente per 20 min.
NOTA: Questo passaggio è un passo delicato e molto importante. Assicurarsi di utilizzare la soluzione PTC appena preparata e rilasciare la soluzione sulle celle il più lentamente e uniformemente possibile, creando uno strato idrofobico sulla superficie della cella. - Aggiungere 5 mL di supporto di controllo osmotico (OCM) alla soluzione protoplast e pipetta lentamente e delicatamente per garantire che la soluzione sia accuratamente miscelata.
- Incubare la soluzione protoplast in un'incubatrice di agitazione a 25 gradi centigradi con agitazione a 80 giri/min durante la notte.
- Utilizzando una pipetta, gocciolare lentamente 1 mL di una soluzione PTC appena preparata al 30% sulla soluzione protoplast e incubare la soluzione a temperatura ambiente per 20 min.
- Per selezionare gli isolati trasformati, dividere la soluzione in 5 piastre di coltura vuote da 60 mm.
- Aggiungere 10 mL di agar OCM integrato con 30 g/mL hygromycin B ad ogni piastra di coltura e ruotare lentamente ogni piastra per mescolare accuratamente.
- Lasciare prescinoso al primo strato di agar prima di aggiungere 10 mL di agar medio di controllo osmotico integrato con 40 g/mL di igromicina B.
- Lasciare che il secondo strato di agar possa impostare e incubare le colture a 25 gradi centigradi fino a quando non si possono vedere singoli isolati crescere attraverso entrambi gli strati di agar.
- Per recuperare gli isolati trasformati con successo, trasferire i singoli isolati in grado di crescere attraverso lo strato di agar integrato con 40 g/mL hygromycin B a piastre fresche di estratto di malto (MEA) integrate con 50 g/mL di igromicina B (MEA-50).
- Incubare le colture fresche a 25 gradi centigradi per 5 giorni, controllando la crescita giornaliera. Le culture in grado di crescere a lungo su questi mezzi di comunicazione sono state trasformate con successo e possono essere utilizzate per ulteriori studi.
6. Conferma dell'integrazione e della stabilità del dDNA
- Per confermare che il dDNA è stato integrato nel genoma nella regione di destinazione, progettare primer che fiancheggiano i siti di inserto previsti 5' e 3' (Figura 2).
- Eseguire due PCR utilizzando questi due set primer e una polimerasi del DNA ad alta fedeltà. Se entrambi i PCR producono ampliconi di dimensioni e sequenza previste, il dDNA è stato integrato con successo nell'area di destinazione. Quindi, valutare la macchia mutata per l'integrazione stabile del dDNA.
- Al fine di confermare che il dDNA è stato stabilmente integrato nel genoma e sarà mantenuto durante la crescita vegetativa, eseguire un test di trasferimento dei media.
- Trasferire un blocco di agar ricoperto di miceli da un isolato mutante in crescita attiva su un mezzo MEA medio-50 meA medio-morbido. Incubare a 25 gradi centigradi per 3 giorni.
- Trasferire un blocco di agar ricoperto di micelia dall'isolato che cresce su media MEA a media MEA-50. Incubare a 25 gradi centigradi per 3 giorni.
- Ripetere questo processo, trasferendo micelia in crescita attiva da mezzo integrato a non elastico per almeno quattro turni.
NOTA: Se l'isolato è in grado di una crescita sostenuta sul mezzo MEA-50 dopo molti trasferimenti, il dDNA è stato stabilmente integrato nel genoma e può essere mantenuto attraverso la crescita vegetativa. La macchia mutata può essere valutata per la presenza di una sola copia del dDNA integrato.
- Per confermare che il dDNA è stato integrato nel genoma in un'unica posizione, eseguire un'analisi delle macchie del sud.
- Digerire un totale di 30 g di gDNA da ogni ceppo mutante utilizzando gli enzimidi restrizione Hin dIII ed EcoRI in conformità con i protocolli del produttore.
NOTA: Mentre la scelta dell'enzima di restrizione è all'interno del ricercatore, assicurarsi che il sito di riconoscimento dell'enzima di restrizione non sia presente nella sequenza dDNA. - Separare il gDNA digerito su un gel di agarose dello 0,75% e trasferire il DNA su una membrana di nylon utilizzando le procedure standard32.
- Sottopone la membrana all'ibridazione utilizzando una sonda mirata alla sequenza dDNA.
- Progetta primer per amplificare una regione corta (300 bp) del dDNA.
- Utilizzando questi primer, sintetizzare la sonda utilizzando un mix di etichettatura PCR DIG.
- Utilizzare la sonda appena sintetizzata per l'ibridazione, il trattamento e la visualizzazione della membrana utilizzando le procedure standard32. Se in ogni corsia viene vista una sola banda, la dDNA è presente in una sola posizione nel genoma. Il ceppo mutante può ora essere utilizzato per ulteriori esperimenti di analisi fenotipico ed esperimenti di caratterizzazione funzionale.
- Digerire un totale di 30 g di gDNA da ogni ceppo mutante utilizzando gli enzimidi restrizione Hin dIII ed EcoRI in conformità con i protocolli del produttore.
7. Analisi fenotippica dei ceppi mutanti
- Condurre esperimenti di accoppiamento per determinare se l'interruzione del gene MAT ha avuto un effetto sulle capacità sessuali del fungo in fase di studio.
NOTA: Questo passaggio dipende dal particolare gene e dalle specie in fase di studio. In questo caso, si pensa che il gene di destinazione sia coinvolto nella riproduzione sessuale e quindi sono stati condotti test di accoppiamento. Se si pensasse, ad esempio, che il gene sia coinvolto nella riproduzione asessuata, si potrebbe misurare qualcosa come la produzione coniettiale.- Al fine di testare le capacità eterothalliche del ceppo mutante, co-inoculare il mezzo MEA fresco con un ceppo mutante e un ceppo di tipo accoppiamento opposto. Nel caso di H. omanensis, tenere i coperchi delle piastre chiusi, ma non sigillati e incubati a temperatura ambiente per 7 giorni. Valutare visivamente per la produzione di strutture sessuali.
- Al fine di testare le capacità omotaliche del ceppo mutante, inoculare il mezzo MEA fresco con un ceppo mutante. Nel caso di H. omanensis, tenere i coperchi delle piastre chiusi, ma non sigillati e incubati a temperatura ambiente per 7 giorni. Valutare visivamente per la produzione di strutture sessuali.
- Condurre esperimenti sul tasso di crescita per determinare se l'interruzione del gene MAT ha avuto un effetto sul tasso di crescita del fungo in fase di studio.
- Creare tappi di agar ricoperti di miceli dal bordo attivamente crescente delle colture dei ceppi mutanti e wildtype inserendo il lato posteriore di una grande punta di pipetta sterile nell'agar.
- Inoculare il mezzo MEA fresco con queste spine di agar. Assicurarsi che vengano effettuate almeno tre repliche per ogni tipo di impostazioni cultura.
- Dopo 3 giorni di crescita a 20 gradi centigradi, misurare la crescita su due diametri perpendicolari.
- Confrontare i dati del tipo selvatico e dei ceppi mutanti.
Risultati
Il protocollo sopra descritto ha facilitato l'introduzione di un codon di arresto prematuro in un gene di accoppiamento dall'ascomycete non modello, H. omanensis. Questo processo ha utilizzato una versione del sistema di editing del genoma CRISPR-Cas9 e come tale uno dei passaggi più importanti di questo protocollo è la progettazione e la sintesi di un sgRNA di alta qualità. La figura 1 mostra come questa molecola sia stata progettata in modo tale che si rivolge specificamente al gene di interesse e mostra poca somiglianza con altre regioni del genoma e B) si piega correttamente per legarsi con la proteina Cas9. L'sgRNA deve anche essere in grado di fendere efficacemente la regione bersaglio. La capacità dell'sgRNA di colpire e consentire la scissione della regione bersaglio è stata condotta in vitro,producendo due prodotti della dimensione prevista.
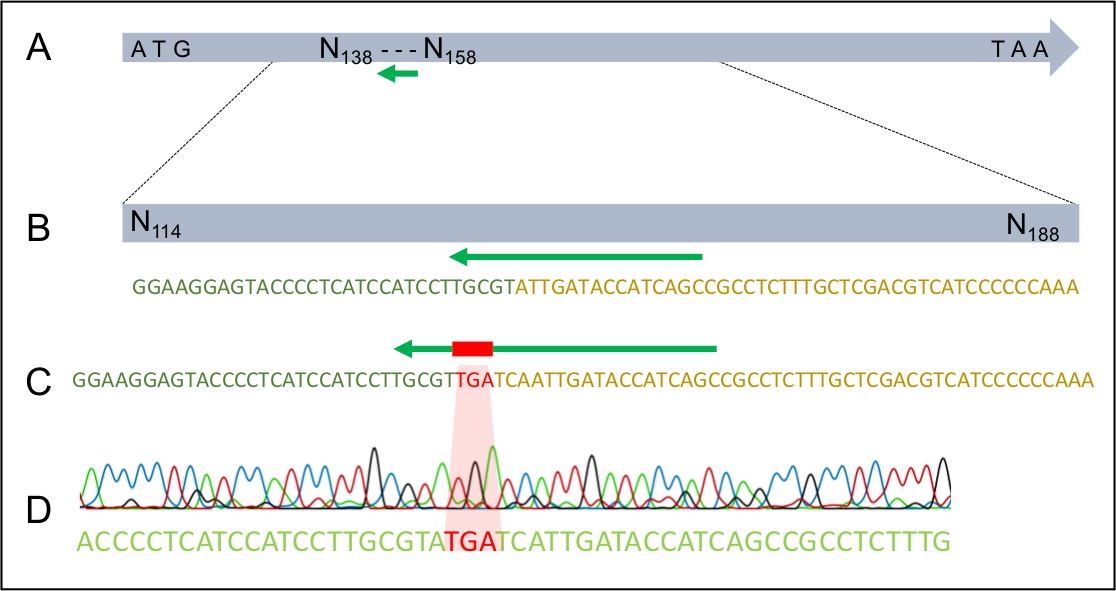
Una volta che la trasformazione ha avuto luogo con successo, è importante assicurarsi che il dDNA si sia integrato nel genoma solo una volta e nel luogo previsto. Figura 2 illustra la progettazione di primer PCR che si rivolgono ai siti di inserimento, che possono essere utilizzati per lo screening dei potenziali trasformatori per il sito di integrazione corretto. Progettando primer che fiancheggiano i siti di inserimento da 5' e 3', l'amplificazione è possibile solo se il dDNA viene inserito nella regione corretta. La figura 4 illustra che il codon arresto prematuro è stato introdotto nel gene MAT1-2-7 nel quadro di lettura corretto, assicurando che il gene sarebbe stato troncato in modo simile a quello di H. moniliformis. Inoltre, l'analisi delle macchie del sud ha mostrato che il costrutto dDNA era integrato solo in un singolo sito nel genoma.
Il successo del protocollo è stato confermato dopo l'analisi fenotipico dei ceppi mutanti. Nel caso dell'esperimento di interruzione MAT1-2-7, sono stati sviluppati due ceppi mutanti indipendenti. In entrambi gli isolati, il tasso di crescita radiale vegetativa è stato significativamente ridotto, suggerendo un effetto pleiotropico del nuovo gene di accoppiamento (Figura 5). Inoltre, gli isolati mutanti non erano in grado di completare un ciclo sessuale, producendo solo strutture sessuali immature che non producevano spore sessuali (Figura 5). Ciò è stato in contrasto con gli isolati wildtype, che hanno completato l'intero ciclo sessuale entro pochi giorni dall'incubazione (Figura 5).

Figura 1: Scelta di un candidato sgRNA adatto.
(A) Un sgRNA adatto avrà solo somiglianza con la regione bersaglio del genoma (in questo caso indicato dalla sequenza di locus MAT). (B) Un sgRNA adatto avrà identica energia libera minima e strutture secondarie centroidi, con i tre anelli steli e cinque anelli nel ciclo di passo primario. Inoltre, la maggior parte della struttura avrà alte probabilità di legame (indicate in arancione scuro e rosso) mentre le probabilità di legame più basse dovrebbero essere viste nella regione protospaziare (indicata dai triangoli neri). Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Progettazione, amplificazione e assemblaggio del dDNA.
La prima e la seconda coppia di primer (PP1 e PP2) vengono utilizzate per amplificare circa 800 bp a monte (5') e 800 bp a valle (3') del gene di interesse. Il primer inverso di PP1 e il primer in avanti di PP2 includono regioni di omologia alla cassetta di resistenza alla igromicina. La terza coppia primer amplifica l'intera cassetta di resistenza alla igromicina. In modo graduale, i vari ampliconi sono assemblati fino a quando non viene assemblato l'intero dDNA, composto dalla regione 5 ', la cassetta di resistenza alla igromicina e la regione 3'. Quando trasformato nella cellula, il dDNA dovrebbe ricombinarsi nella regione in cui l'enzima Cas9 sarà stato diretto a tagliare, sostituendo così il gene di interesse con la cassetta di resistenza alla igromicina. PP4 e PP5 possono essere utilizzati per determinare se il dDNA è stato inserito correttamente nel genoma nella posizione appropriata. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: I diversi tipi di cellule importanti durante il protocollo di estrazione protoplast.
(A) Conidia sono utilizzati come materiale di partenza per il protocollo. Queste conidia sono autorizzate a germinare e crescere fino a quando non sono (B) giovani germi. La fase di crescita ideale dei giovani germi sono indicate dalle due frecce nere. Altri fili miceli visti su ( B )sonotroppo maturi per il degrado e non devono essere utilizzati. La fase finale del protocollo è il rilascio dei protoplastsrotondi( C ), indicati dai cerchi neri punteggiati. Queste cellule non hanno più pareti cellulari e sono quindi molto sensibili alle interruzioni meccaniche. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: La riuscita integrazione del codon arresto TGA nel gene MAT1-2-7 di H. omanensis.
(A) Il gene H. omanensis MAT1-2-7 a lunghezza intera, con il sito bersaglio sgRNA indicato dalla freccia verde. (B) Uno schema ingrandito del sito bersaglio sgRNA all'interno del gene H. omanensis MAT1-2-7. (C) Uno schema ingrandito di una regione del dDNA che mostra il codon di arresto affiancato da braccia omologhe al gene MAT1-2-7 di H. omanensis. (D) Cromatogramma della sequenza Sanger che indica la riuscita integrazione del codon di arresto nel gene MAT1-2-7. Modificato da Wilson et al. 202021. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Le differenze fenotipi tra (A) isola il tipo selvatico e gli isolati mutanti (B).
Le prime tre immagini in ogni pannello mostrano le differenze nelle capacità sessuali dei due tipi isolati. Mentre gli isolati wildtype formano maturo comecomata durante la riproduzione sessuale, completo di esudamento di spore dalle punte dei colli ascomatal, gli isolati mutanti formano solo strutture sessuali immature che non producono spore sessuali. La quarta immagine in ogni pannello mostra la differenza nel tasso di crescita e nella morfologia dei due tipi di isolamento. Mentre l'isolato di tipo selvatico cresce molto più velocemente e con più micelia aerea, il mutante mostra più lento e viene immerso all'interno dell'agar. Modificato da Wilson et al. 202021. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Reazione | Concentrazione degli enzimi | Tempo di degradazione |
| Un | 1,250 mg/mL | 180 min |
| B | 1,875 mg/mL | 180 min |
| C | 2.500 mg/mL | 150 min |
| D | 3,750 mg/mL | 150 min |
| E | 4,375 mg/mL | 120 min |
| F | 5.000 mg/mL | 120 min |
Tabella 1: Degradazione della soluzione germinale/micelia con enzimi di lissidazione di Trichoderma harzianum. Le diverse concentrazioni di enzimi corrispondono a diversi periodi di incubazione, con concentrazioni più basse che richiedono incubazioni più lunghe.
Discussione
Il protocollo per la trasformazione di successo di H. omanensis e l'editing del gene MAT1-2-7 è stato dimostrato introducendo un codon di arresto prematuro in-frame insieme a un gene per la resistenza alla megromicina B21. Ciò è stato ottenuto utilizzando una versione basata sulle proteine del sistema di editing del genoma CRISPR-Cas9. L'esperimento ha comportato la trascrizione in vitro dello sgRNA, l'assemblaggio basato su PCR del dDNA e la co-trasformazione di questi due acidi nucleici con un enzima Cas9 disponibile in modo commerciale in protoplasti estratti da H. omanensis
A differenza di altri protocolli che si basano sulla disponibilità di molti altri strumenti molecolari, il protocollo sopra descritto può essere utilizzato con successo in specie per le quali la cassetta degli attrezzi molecolare èancora abbastanza limitata 21. Il protocollo si basa solo su un sistema di trasformazione consolidato e sulla disponibilità di dati NGS, preferibilmente sulla sequenza dell'intero genoma. Mentre un sistema di trasformazione efficace può richiedere alcune ottimizzazione in una specie per la quale questo non è disponibile, ci sono molti protocolli diversi disponibili per una varietà di specie. Inoltre, i dati del genoma stanno diventando sempre più disponibili anche per le specie più oscure e stanno diventando più facili da generare de novo se non esistono già.
Data la lunghezza del protocollo, ci sono molti passaggi in cui è possibile introdurre modifiche e dove potrebbe essere necessaria la risoluzione dei problemi. Ciò è particolarmente vero per i passi che sono considerati specifici delle specie. Ad esempio, ci sono molti passaggi di incubazione in questo protocollo che devono essere condotti a temperature specifiche e per specifiche lunghezze di tempo al fine di generare tipi di cellule importanti per l'esperimento. Questi passaggi richiederebbero quindi un'ottimizzazione specifica delle specie. Ove possibile, sono state fornite micrografie delle cellule o delle fasi di crescita specifiche per contribuire al trasferimento di questo protocollo a una specie diversa (Figure 1). Il tipo e la concentrazione di enzimi utilizzati per degradare le pareti cellulari delle cellule fungine al fine di rilasciare i protoplasts saranno specifici anche per le specie di funghi in fase di studio. In questo protocollo, viene utilizzata solo una fonte di enzimi di llysing da, mentre diverse combinazioni di enzimi sono necessari per l'estrazione di protoplasti in specie come Fusa verticillioides33. Questo passo dipende interamente dalla produzione chimica della parete cellulare e dovrà quindi essere ottimizzato su base da specie a specie.
Questo metodo è particolarmente significativo per coloro che studiano specie non modello in quanto non vi è alcuna dipendenza da un sistema di espressione. Un metodo popolare per stabilire il sistema di editing del genoma CRISPR-Cas9 è quello di esprimere la proteina Cas9, l'sgRNA e il dDNA da uno o due plamidi che si trasformano nelle cellule di scelta. In questo caso, il Cas9 deve essere espresso da un promotore che è in grado di alti livelli di espressione nel particolare organismo in fase di studio. I promotori generali sono stati sviluppati per l'uso in funghi filamentosi e, sebbene non siano compatibili in tutte le specie, consentono un'espressione di basso livello e possono essere utilizzati con successo per esprimere, ad esempio, i geni della resistenza agli antibiotici. Questi promotori, tuttavia, spesso non consentono alti livelli di espressione e quindi non possono essere utilizzati per esprimere la proteina Cas9. L'utilizzo di una versione a base proteica del sistema di editing del genoma CRISPR-Cas9 supera questa limitazione e consente allo sgRNA e al dDNA di essere co-trasformati nella cellula con un enzima Cas9 già prodotto.
Lo sviluppo di questo sistema a base proteica per l'uso in H. omanensis è arrivato dopo molti tentativi falliti di editing del genoma utilizzando sia il classico approccio split marker che il sistema CRISPR-Cas9 basato sul plasmide. Mentre l'efficienza varia da specie a specie, l'approccio split marker è stato utilizzato con successo con il 100% di efficienza in specie diverse come Alternaria alternata34,35e C. nicotianae36. Al contrario, l'efficienza di questo sistema in H. omanensis era pari a zero, nonostante più di 80 eventi indipendenti di trasformazione e integrazione. Allo stesso modo, il sistema CRISPR-Cas9 basato su plasmide è stato utilizzato con successo con elevate efficienze in Trichoderma reesei (>93%)17 e Penicillium chrysogenum (fino al 100%)37. Questo è, ancora una volta, in contrasto con l'utilità di questo sistema in H. omanensis. Una sufficiente espressione della proteina Cas9 non è stata raggiunta in H. omanensis nonostante abbia provato un certo numero di potenziali promotori, tra cui due promotori specifici delle specie previsti dai geni delle pulizie. Pertanto, questo sistema non poteva essere utilizzato affatto. Utilizzando la versione basata sulle proteine del sistema CRISPR-Cas9, tuttavia, ha prodotto molti trasformatori indipendenti, due dei quali ospitavano il dDNA integrato nella posizione corretta. Inoltre, questo esperimento è stato tentato una sola volta e ha avuto successo, illustrando ulteriormente la facilità con cui questo sistema può essere utilizzato.
Le future applicazioni di questo protocollo includono la sua ottimizzazione e l'uso in altre specie del Ceratocystidaceae. C'è già una ricchezza di dati NGS disponibili per queste specie30,38,39 e studi riguardanti la loro specificità ospite40, tasso di crescita e virulenza41 sono stati condotti. Questi studi possono essere rafforzati dalla caratterizzazione funzionale dei geni che si pensa siano coinvolti in questi processi, ricerca che diventerà ora possibile grazie alla disponibilità di un protocollo di trasformazione e di modifica del genoma.
In conclusione, un'analisi approfondita dei geni alla base di importanti processi biologici nelle specie non modello sta diventando più accessibile grazie alla disponibilità di protocolli di editing del genoma facili da usare che non si basano sull'esistenza di ampie risorse biologiche e toolkit molecolari. Lo studio delle specie non modello sta diventando più facile e permetterà la scoperta di nuovi percorsi e interessanti deviazioni dai processi biologici standard che sono stati chiariti nelle specie modello.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo progetto è stato sostenuto dall'Università di Pretoria, dal Dipartimento di Scienza e Tecnologia (DST)/National Research Foundation (NRF) Centre of Excellence in Tree Health Biotechnology (CTHB). Il progetto è stato inoltre sostenuto dalla cattedra DST/NRF SARChI del professor BD Wingfield in Fungal Genomics (numero Grant: 98353) e dalla borsa di studio NRF PhD del dottor AM Wilson (108548). I titolari della sovvenzione riconoscono che i pareri, i risultati e le conclusioni o le raccomandazioni espresse in questo lavoro sono i ricercatori e che gli organismi di finanziamento non si accettano alcuna responsabilità al riguardo.
Materiali
| Name | Company | Catalog Number | Comments |
| EcoRI-HF | New England Biolabs, Ipswich, USA | R3101S | |
| EnGen Spy Cas9 NLS protein | New England Biolabs, Ipswich, USA | M0646T | Used to assemble the RNP |
| Eppendorf 5810 R centrifuge | Eppendorf, Hamberg, Germany | ||
| FastStart Taq DNA Polymerase | Sigma, St Louis, USA | 12032902001 | Standard DNA polyermase |
| GeneJET Gel Extraction Kit | ThermoFisher Scientific, Waltham, USA | K0691 | |
| HindIII-HF | New England Biolabs, Ipswich, USA | R3104S | |
| HiScribeTM T7 Quick High Yield RNA synthesis kit | New England Biolabs, Ipswich, USA | E2050S | |
| Hygromycin B from Streptomyces hygroscopicus | Sigma, St Louis, USA | 10843555001 | |
| Infors HT Ecotron Shaking Incubator | Infors AG, Bottmingen, Switzerland | ||
| LongAmp Taq DNA Polymerase | New England Biolabs, Ipswich, USA | M0323S | Long-range, high-fidelity DNA polymerase |
| Malt extract agar, 2% (MEA) | 20 g ME and 20 g agar in 1 l ddH20 | ||
| Malt extract | Sigma, St Louis, USA | 70167-500G | |
| Agar | Sigma, St Louis, USA | A5306 | |
| Malt Extract broth, 1% (MEB) | Sigma, St Louis, USA | 70167-500G | 2 g ME in 200 ml ddH20 |
| Malt Extract broth, 2% (MEB) | Sigma, St Louis, USA | 70167-500G | 4 g ME in 200 ml ddH20 |
| Miracloth | Merck Millipore, New Jersey, USA | 475855 | |
| Nylon membrane (positively charged) | Sigma, St Louis, USA | 11209299001 | |
| Osmotic control medium (OCM) | 0.3% yeast extract, 20% sucrose, 0.3% casein hydrolysate | ||
| Casein Hydrolysate | Sigma, St Louis, USA | 22090 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Yeast extract | Sigma, St Louis, USA | Y1625 | |
| Osmotic control medium (OCM) agar | Osmotic control medium (OCM) + 1% agar | ||
| Agar | Sigma, St Louis, USA | A5306 | |
| PCR DIG Labeling Mix | Sigma, St Louis, USA | 11585550910 | |
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific, Waltham, USA | F-530XL | High fidelity DNA polymerase |
| Plasmid pcb1004 | N/A | N/A | From: Carroll et al., 1994 |
| Presynthesized sgRNA | Inqaba Biotec, Pretoria, South Africa | Ordered as an synthesized dsDNA with specified sequence | |
| Proteinase K | Sigma, St Louis, USA | P2308 | |
| PTC Solution | 30% polyethylene glycol 8000 in STC buffer from above | ||
| Polyethylene glycol 8000 | Sigma, St Louis, USA | 1546605 | |
| RNase A | ThermoFisher Scientific, Waltham, USA | 12091021 | |
| RNAfold Webserver | Institute for Theoretical Chemistry, University of Vienna | N/A | http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi |
| RNAstructure | Mathews Lab | N/A | https://rna.urmc.rochester.edu/RNAstructureWeb/Servers/Predict1/Predict1.html |
| Sorbitol, 1 M | Sigma, St Louis, USA | 1617000 | 182.17g sorbitol in 1 l ddH20 |
| STC Buffer | 20% sucrose, 50 mM Tris-HCl pH 8.00 and 50 mM CaCl2 | ||
| Calcium chloride | Sigma, St Louis, USA | 429759 | |
| Tris-HCl pH 8.00 | Sigma, St Louis, USA | 10812846001 | |
| Sucrose | Sigma, St Louis, USA | 84097 | |
| Trichoderma harzianum lysing enzymes | Sigma, St Louis, USA | L1412 | |
| Zeiss Axioskop 2 Plus Ergonomic Trinocular Microscope | Zeiss, Oberkochen, Germany |
Riferimenti
- Ekblom, R., Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 107, 1-15 (2011).
- Russell, J. J., et al. Non-model model organisms. BMC Biology. 15 (55), 1-31 (2017).
- Kück, U., Hoff, B. New tools for the genetic manipulation of filamentous fungi. Applied Microbiology and Biotechnology. 86, 51-62 (2010).
- Li, D., Tang, Y., Lin, J., Cai, W. Methods for genetic transformation of filamentous fungi. Microbial Cell Factories. 16 (168), 1-13 (2017).
- Lorito, M., Hayes, C. K., Di Pietro, A., Harman, G. E. Biolistic transformation of Trichoderma harzianum and Gliocladium virens using plasmid and genomic DNA. Current Biotechnology. 24, 349-356 (1993).
- Taylor, P., et al. Transformation of intact Aspergillus niger by electroporation. Bioscience, Biotechnology, and Biochemistry. 58 (12), 2224-2227 (2014).
- Dhawale, S. S., Paietta, J. V., Marzluf, G. A. A new, rapid and efficient transformation procedure for Neurospora. Current Genetics. 8, 77-79 (1984).
- Sayari, M., Van Der Nest, M. A., Steenkamp, E. T., Adegeye, O. O., Marincowitz, S. Agrobacterium-mediated transformation of Ceratocystis albifundus. Microbiological Research. 226, 55-64 (2019).
- Meyer, V. Genetic engineering of filamentous fungi- Progress, obstacles and future trends. Biotechnology Advances. 26, 177-185 (2008).
- You, B. J., Lee, M. H., Chung, K. R. Gene-specific disruption in the filamentous fungus Cercospora nicotianae using a split-marker approach. Archives of Microbiology. 191, 615-622 (2009).
- Gravelat, F. N., Askew, D. S., Sheppard, D. C. Targeted gene deletion in Aspergillus fumigatus using the hygromycin-resistance split-marker approach. Host-Fungus Interactions. 845, 119-130 (2012).
- Wang, Y., Diguistini, S., Bohlmann, J., Breuil, C. Agrobacterium-meditated gene disruption using split-marker in Grosmannia clavigera, a mountain pine beetle associated pathogen. Current Genetics. 56, 297-307 (2010).
- Wood, A. J., et al. Targeted genome editing across species using ZFNs and TALENs. Science. 333, 307 (2011).
- Mahfouz, M. M., Piatek, A., Neal, C. Genome engineering via TALENs and CRISPR/Cas9 systems: Challenges and perspectives. Plant Biotechnology. 12, 1006-1014 (2014).
- Wang, H., La Russa, M., Qi, L. S. CRISPR/Cas9 in genome editing and beyond. Annual Review of Biochemistry. 85 (1), 227-264 (2016).
- Arazoe, T., et al. Tailor-made TALEN system for highly efficient targeted gene replacement in the rice blast fungus. Biotechnology and Bioengineering. 112 (7), 1335-1342 (2015).
- Liu, R., Chen, L., Jiang, Y., Zhou, Z., Zou, G. Efficient genome editing in filamentous fungus Trichoderma reesei using the CRISPR/Cas9 system. Cell Discovery. 1, 1-11 (2015).
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337, 816-822 (2012).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences. 109 (39), 2579-2586 (2012).
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. Genome engineering using the CRISPR-Cas9 system. Cell. 8 (11), 2281-2308 (2013).
- Wilson, A. M., Wilken, P. M., Van Der Nest, M. A., Wing, M. J., Wing, B. D. The novel Huntiella omanensis mating gene, MAT1-2-7, is essential for ascomatal maturation. Fungal Genetics and Biology. 137, 103335 (2020).
- Miao, J., et al. Characterization of an N-terminal non-core domain of RAG1 gene disrupted Syrian Hamster model generated by CRISPR Cas9. Viruses. 10 (243), 10050243 (2018).
- Roberts, B., et al. Systematic gene tagging using CRISPR/Cas9 in human stem cells to illuminate cell organization. Molecular Biology of the Cell. 28 (21), 2854-2874 (2016).
- Schneider, S., Kirchner, M., Kirchner, M., Schneider, S. CRISPR-Cas: From the bacterial adaptive immune system to a versatile tool for genome engineering. Angewandte Chemie International Edition. 54 (46), 13508-13514 (2015).
- Nødvig, C. S., Nielsen, J. B., Kogle, M. E., Mortensen, U. H. A CRISPR-Cas9 system for genetic engineering of filamentous fungi. PLoS ONE. 10 (7), 1-18 (2015).
- Wang, Q., Cobine, P. A., Coleman, J. J. Efficient genome editing in Fusarium oxysporum based on CRISPR/Cas9 ribonucleoprotein complexes. Fungal Genetics and Biology. 117, 21-29 (2018).
- Nagy, G., et al. Development of a plasmid free CRISPR-Cas9 system for the genetic modification of Mucor circinelloides. Scientific Reports. 7 (16800), 1-10 (2017).
- Al-Subhi, A. M., Al-Adawi, A. O., Van Wyk, M., Deadman, M. L., Wingfield, M. J. Ceratocystis omanensis, a new species from diseased mango trees in Oman. Mycological Research. 110 (2), 237-245 (2006).
- Wilson, A. M., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Pheromone expression reveals putative mechanism of unisexuality in a saprobic ascomycete fungus. PLoS ONE. 13 (3), 0192517 (2018).
- van der Nest, M. A., et al. Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus. 5 (2), 472-485 (2014).
- Wilson, A. M., Godlonton, T., van der Nest, M. A., Wilken, P. M., Wingfield, M. J., Wingfield, B. D. Unisexual reproduction in Huntiella moniliformis. Fungal Genetics and Biology. 80, 1-9 (2015).
- Sambrook, J., Green, M. . Molecular cloning: A laboratory manual. , (2012).
- Ramamoorthy, V., Govindaraj, L., Dhanasekaran, M., Vetrivel, S., Kumar, K. K., Ebenezar, E. Combination of driselase and lysing enzyme in one molar potassium chloride is effective for the production of protoplasts from germinated conidia of Fusarium verticillioides. Journal of Microbiological Methods. , (2015).
- Lin, C., Yang, S. L., Wang, N., Chung, K. The FUS3 MAPK signaling pathway of the citrus pathogen Alternaria alternata functions independently or cooperatively with the fungal redox-responsive AP1 regulator for diverse developmental, physiological and pathogenic processes. Fungal Genetics and Biology. 47 (4), 381-391 (2010).
- Lin, C., Chung, K. Specialized and shared functions of the histidine kinase- and HOG1 MAP kinase-mediated signaling pathways in Alternaria alternata, a filamentous fungal pathogen of citrus. Fungal Genetics and Biology. 47 (10), 818-827 (2010).
- Choquer, M., et al. The CTB1 gene encoding a fungal polyketide synthase is required for cercosporin biosynthesis and fungal virulence of Cercospora nicotianae. Molecular Plant-Microbe Interactions. 18 (5), 468-476 (2005).
- Pohl, C., Kiel, J. A. K. W., Driessen, A. J. M., Bovenberg, R. A. L., Nygård, Y. CRISPR/Cas9 based genome editing of Penicillium chrysogenum. ACS Synthetic Biology. 5 (7), 754-764 (2016).
- van der Nest, M. A. M. A., et al. Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans and Ceratocystis moniliformis. IMA Fungus. 5 (1), 135-140 (2014).
- Wingfield, B. D., et al. Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus. 7 (2), 317-323 (2016).
- Fourie, A., Van Der Nest, M. A., De Vos, L., Wingfield, M. J., Wingfield, B. D., Barnes, I. QTL mapping of mycelial growth and aggressiveness to distinct hosts in Ceratocystis pathogens. Fungal Genetics and Biology. 131, 103242 (2019).
- Lee, D. H., Roux, J., Wingfield, B. D., Wingfield, M. J. Variation in growth rates and aggressiveness of naturally occurring self-fertile and self-sterile isolates of the wilt pathogen Ceratocystis albifundus. Plant Pathology. 64 (5), 1103-1109 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati