È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Fluorescenza base tecnica Primer Extension determinare trascrizionali punti di partenza e siti di taglio di RNasi
In questo articolo
Riepilogo
We here describe a fluorescence based primer extension method to determine transcriptional starting points from bacterial transcripts and RNA processing in vivo using an automated gel sequencer.
Abstract
Fluorescenza estensione primer a base (FPE) è un metodo molecolare per determinare i punti di partenza di trascrizione o siti di trasformazione di molecole di RNA. Ciò è ottenuto mediante trascrizione inversa di RNA di interesse utilizzando specifici primer fluorescente e successiva analisi dei frammenti di cDNA risultante dalla elettroforesi su gel denaturante di poliacrilammide. Contemporaneamente, un tradizionale reazione di sequenziamento Sanger viene eseguito sul gel per mappare le estremità dei frammenti di cDNA alle loro basi corrispondenti esatti. In contrasto con 5'-RACE (Rapid Amplification di cDNA Ends), in cui il prodotto deve essere clonato e sequenziato più candidati, la maggior parte dei frammenti di cDNA generato dall'estensione del primer può essere simultaneamente rilevata in una corsa di gel. Inoltre, l'intera procedura (da trascrizione inversa di analisi finale dei risultati) può essere completato in un giorno lavorativo. Usando primer fluorescente, l'uso di isotopi radioattivi pericolosi reagenti etichettatopossono essere evitati e tempi di lavorazione sono ridotti come i prodotti possono essere rilevati durante la procedura di elettroforesi.
Nella seguente protocollo, si descrive un metodo di estensione in vivo di primer fluorescente per rilevare in modo affidabile e rapido l'estremità 5 'di RNA dedurre trascrizionale punti di partenza e siti di lavorazione RNA (ad esempio, dai componenti di sistema tossina-antitossina) a S. aureus, E. coli e altri batteri.
Introduzione
Estensione Primer 1 è un metodo molecolare per determinare l'estremità 5 'di molecole di RNA specifici fino ad una risoluzione di una base. Il vantaggio di altri metodi come 5'-RACE (amplificazione rapida delle estremità del cDNA) è il tempo di ritorno veloce e la capacità di analizzare facilmente una miscela di diverse lunghezze di molecole di RNA.
Questo metodo funziona sottoponendo molecole di RNA per invertire le reazioni di trascrizione utilizzando primer fluorescenti specifiche, generando frammenti di cDNA di alcune lunghezze. Queste molecole di cDNA vengono eseguiti insieme reazioni di sequenziamento tradizionali Sanger 2 su gel di poliacrilammide denaturante e possono essere rilevate da loro fluorescenza a causa dell'uso di primer fluorescente. Le lunghezze dei frammenti di cDNA vengono quindi valutati rispetto alla scala sequenziamento, consentendo la mappatura delle estremità 5 'RNA.
Tradizionalmente, le reazioni di estensione dei primer sono utilizzati in combinazionecon isotopi radioattivi per rilevare molecole di cDNA su film X-ray. A causa di rischi per la salute, problemi di smaltimento e maneggevolezza, protocolli più recenti utilizzano fluorescenza per la rilevazione del primer extension con sequenziatori automatici, benché la loro sensibilità è leggermente inferiore. Utilizzando primer fluorescente, la procedura di radio-marcatura ricorrenti può essere omesso, come primer fluorescenti sono stabili per un lungo periodo di tempo (più di un anno nelle nostre mani).
Il metodo che descriviamo qui utilizza un sequenziatore automatizzato gel, ma con lievi modifiche, sequenziatori capillari può essere utilizzato anche per la separazione cDNA e rilevamento 3. Il parallelismo tra analisi del gel permette di rilevare anche una piccola quantità di RNA scissione o trasformazione. Un altro vantaggio è l'alta risoluzione di questo metodo, come scissione o la trasformazione anche di una sola base del terminale può essere rilevato.
Per quanto riguarda la rilevazione di RNA scissione o trasformazione, typically due diversi tipi di estensioni di primer si distinguono. In un caso, il trattamento enzimatico è fatto in vitro utilizzando RNA purificato e l'enzima purificato, mentre nell'altro caso, la lavorazione avviene in vivo e l'RNA risultante viene purificato. In entrambi i casi l'RNA viene sottoposto ad un primer extension effettuata in vitro, tuttavia, a seconda della fonte di RNA, il metodo o è chiamato un primer extension vivo o in vitro. Nel protocollo presentiamo, ci concentriamo esclusivamente sulla vivo primer extension in, a causa della facilità d'uso (non proteine purificate necessario) e la possibilità di determinare punti di partenza trascrizionali e trasformazione in una sola volta. Tuttavia, in vitro estensioni di primer sono in linea di principio configurare allo stesso modo e questo protocollo può servire come punto di partenza.
Il metodo qui illustrato può essere applicato a molte specie batteriche purché siano suscettibili di altapurezza e alto rendimento preparazione di acidi nucleici.
La ricerca nel nostro laboratorio si concentra sulla portata normativa di tossina-antitossina (TA) Sistemi di 4,5, un campo in cui è ampiamente utilizzato il metodo primer extension. TA-sistemi sono piccoli elementi genetici presenti nei genomi procariotici che consistono di una proteina tossica stabile e endogenamente attiva e una proteina o RNA antitossina lo più instabile che contrasta la tossicità 6,7. Attività di tossina è a volte esercitata da inibizione della replicazione, sintesi della parete cellulare o altri meccanismi, ma il più delle volte per attività RNasi 8,9. Tipicamente, RNasi specificità è determinata effettuando diverse prove, una delle quali è il metodo di primer extension. Reazioni primer extension sono adatti per questa applicazione, come miscela di frammenti di lunghezza clivati e completi possono essere analizzati simultaneamente per determinare la loro 5 'estremità. Utilizzando un mix di in vitro e in vivo estensioni di primer, laspecifica RNasi clivaggio tossina, ad esempio, specificità di sequenza può essere determinata 10-13.

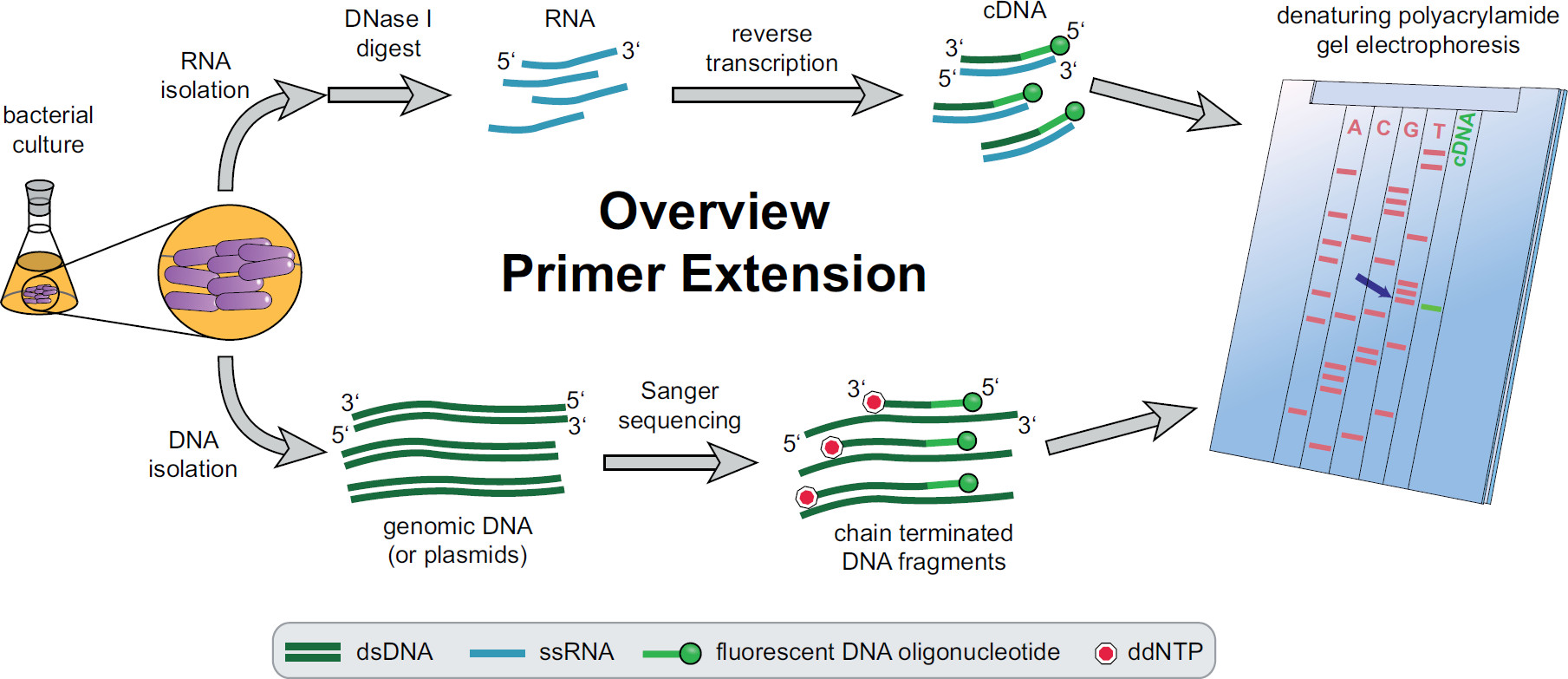
Figura 1. Panoramica della procedura di primer extension. Le colture batteriche sono incubate e trattati in base alle esigenze sperimentali. L'RNA totale viene estratto dalle cellule, trattate con DNasi I per rimuovere tracce di DNA e sottoposti ad una reazione di trascrizione inversa utilizzando bersaglio specifici primer di DNA fluorescenti rendimento cDNA. Il DNA genomico o plasmidi vengono estratti e successivamente utilizzati per fluorescenti reazioni di sequenziamento Sanger per il confronto dimensioni con i frammenti di cDNA. Prodotti primer extension vengono eseguiti insieme a prodotti di sequenziamento Sanger su un gel denaturante di poliacrilammide di urea e analizzati con un laser automatico e microscopio. La base sequenziamento che si allinea con la band cDNA è il lav base 5 'del cDNA (freccia blu). Maggiori informazioni in Fekete, et al. 3 Fate clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Una panoramica di tutta la procedura di primer extension può essere trovato in Figura 1. In breve, le cellule batteriche vengono coltivate, raccolte, il pellet cellulare lisato e l'RNA estratto. RNA purificato viene quindi trattato con DNasi I per rimuovere tracce di molecole di DNA che potrebbero agire come modelli per la trascrittasi inversa. Primer fluorescenti specifici sono aggiunti al RNA, ibridato alla regione di interesse e successivamente trascrizione inversa, con conseguente DNA a singolo filamento complementare (cDNA). Una scaletta sequenziamento è stato creato da sequenziamento Sanger tradizionale impiegando primer fluorescenti e separati su un gel di poliacrilammide denaturante a fianco del primer frammenti di estensione di cDNA. La risultantegel viene analizzato confrontando le bande fluorescenti, che permette l'identificazione delle estremità 5 'di interesse. Punti di partenza di trascrizione e siti di lavorazione sono poi valutato individualmente in sequenza comparazioni.
Protocollo
1. High Yield RNA Preparazione
- RNA Isolation
NOTA: Elevate concentrazioni di RNA totale sono necessari per la reazione di primer extension. Kit colonna Spin di solito non danno la quantità di RNA necessario (~ 5 - 16 mg in 5 ml di volume). Pertanto si raccomanda di purificazione con il metodo di estrazione tiocianato-fenolo-cloroformio guanidina acido, riportate qui di seguito.
NOTA: Il fenolo è cancerogeno, tossico e corrosivo. Si prega di leggere le schede di sicurezza e utilizzare sotto una cappa di aspirazione con protezione adeguata!- Crescere o trattare le cellule batteriche (S. aureus o di E. coli in questo esempio), se lo desideri e la raccolta di 10 min centrifugazione a 4.600 × g e 4 ° C. Nota: In genere abbiamo raccolto un totale di 600 OD di 20 - 70. I pellet cellulari possono essere conservati per diverse settimane a -20 ° C.
- Risospendere il pellet di cellule in 1 ml di soluzione di acido guanidina tiocianato-fenolo-cloroformio e versare in un2 ml a vite tazza contenente 0,5 ml di 0,1 mm perle di vetro di zirconio / silice.
- Lyse le cellule tre volte in un battitore di preparazione veloce / tallone a 6,5 m / sec per 30 sec per tre turni, raffreddamento i campioni in ghiaccio per 5 minuti dopo ogni esecuzione. Nota: campione omogeneizzato può essere conservato a -80 ° C per diverse settimane.
- Incubare lisato per 5 min a temperatura ambiente e quindi aggiungere 200 ml di cloroformio.
- Agitare vigorosamente o vortice del campione per 30 secondi per estrarre l'RNA.
- Incubare a temperatura ambiente per 3 minuti quindi si centrifuga per 15 minuti a 13.000 - 15.000 × g e 4 ° C. Nota: I solventi sono separati in una fase organica inferiore (rosa, contiene proteine), l'interfase (bianco, contiene DNA) e una fase acquosa superiore (trasparente, contiene RNA).
NOTA: Da questo punto in uso solo RNase reagenti liberi e plastica ware! - Preparare fresco RNasi-free da 1,5 ml provette, etichette in modo appropriato e aggiungere circa 500 ml di 100% RNasi isopropanolo gratuita ogni (utilizzare circa la stessa volume come fase acquosa nel tubo precedente).
- Tenere il tubo ad angolo e trasferire la fase acquosa (circa 500 ml) per provette preparate utilizzando RNasi punte libere. Non disturbare l'interfase.
- Precipitare l'RNA capovolgendo più volte e incubando per 10 minuti a RT.
- Centrifugare i campioni per 15 minuti a 13.000 - 15.000 × g e 4 ° C e rimuovere il surnatante con una pipetta o di aspirazione (pompa a getto d'acqua e il biberon). Non disturbare il pellet di RNA bianco trasparente sul fondo.
- Aggiungere 1 ml di etanolo al 70-80% RNasi libero (non vortex) per lavare. Nota: RNA in etanolo può essere conservato per diverse settimane a -20 ° C.
- Centrifugare per 5 minuti a 7500 × g e 4 ° C e scartare il surnatante con una pipetta o, preferibilmente, l'aspirazione.
- Aria secca pellet di RNA per 15 - 30 min sotto la cappa. Non asciugare eccessivamente, altrimenti pellet potrebbero essere difficili da ridisciogliere.
- Risospendere il pellet in 50 ml di RNasi libero DDH 2 O o buffer di stoccaggio RNA.
- Misurare la concentrazione di RNA con uno spettrofotometro microvolumi UV-Vis o una cuvetta di quarzo (e fotometro convenzionali) e procedere alla DNasi I digestione.
- DNasi I digestione del RNA per rimuovere tracce di DNA
NOTA: Poiché DNA può agire come modello spuri nella reazione di trascrizione inversa (primer extension), dovrebbe essere rimosso dal campione. Vari metodi per rimuovere il DNA dal RNA sono disponibili soluzioni che di solito si basano su DNasi digestione. Un metodo efficiente semplice ma efficace ed economico per la rimozione del DNA è indicato di seguito.- Bagno d'acqua Preriscaldare a 37 ° C.
- Mescolare i composti elencati nella Tabella 1 in una provetta da 1,5 ml di reazione.
- Incubare la miscela per 1 ora a 37 ° C in un bagno d'acqua, e poi procedere direttamente alla estrazione con fenolo / cloroformio. Nota: inattivazione di calore della DNasi I non è raccomandato, in quanto questo potrebbe degradare l'RNA.
- Estrazione con fenolo / cloroformio di RNA dopo DNasi I digestione
NOTA: L'RNA deve essere purificato per rimuovere nucleotidi liberi, frammenti di DNA e componenti tampone dalla digestione DNasi I. Estrazione con fenolo / cloroformio permette elevato recupero e la concentrazione del campione di RNA ed è quindi indicato di seguito. Altri metodi per la purificazione dell'RNA possono anche essere utilizzati, se soddisfano questi requisiti.- Dividere la DNasi 500 microlitri mi digestione mescolare in due campioni di 250 microlitri in 2 ml provette di reazione.
- Aggiungere 1 volume (250 ml) di acido P / C soluzione / I (in acqua satura di fenolo, cloroformio e isopentanol, rapporto di 25: 24: 1, pH 4,5-5).
NOTA: P soluzione / C / I è cancerogeno, tossico e corrosivo. Si prega di leggere le schede di sicurezza e utilizzare sotto una cappa di aspirazione con protezione adeguata! - Vigorosamente vortice o posto in una piattaforma vortex per 1 - 3 min.
- Centrifugare per 30 minuti a 13.000 - 15.000 xg e 4 ° C.
- Raccogliere la fase superiore (acquosa) e trasferimento in nuova provetta (250 microlitri).
- Aggiungere 1/9 del volume (28 ml) di 3 M di sodio acetato pH 5.2.
- Aggiungi 2,5-3 volumi di etanolo puro (700 ml).
- Mescolare nel vortex brevemente e posto a -80 ° C per 30 minuti oa -20 ° C per 2 - 3 ore. Se necessario, memorizzare il RNA O / N a -20 ° C.
- Centrifuga per il 30 - 60 min a 13.000 - 15.000 × g e 4 ° C.
- Rimuovere il surnatante pipettando o aspirazione.
- Lavare pellet aggiungendo 1 ml di etanolo al 70% sul pellet. Non fare del campione vortice.
- Campione Centrifugare per 5 minuti a 13.000 - 15.000 × g e 4 ° C.
- Rimuovere il surnatante pipettando o aspirazione.
- Aria pellet asciutto sotto il cofano fumi. Conservare il pellet a -20 ° CO / N se necessario.
- Sciogliere il precipitato in 30 ml DEPC trattati H 2 O con il vortex per 2 minuti e utilizzare questa soluzione per sciogliere il pellet del corrispondente secondo tubo per campione (30 microlitri soluzione per una coppia di estrazione).
- Misurare la concentrazione di RNA, e assicurarsi che sia superiore a 1 mg / mL per l'uso in primer extension di mRNA mediamente espressi.
- Se necessario, conservare l'RNA a -20 ° C per diverse settimane fino a pochi mesi.
Reazione 2. Primer Extension
- Design Primer
NOTA: Quando si progetta primer per un esperimento di primer extension, obbedire linee guida generali della progettazione di primer PCR (vedi manuale che accompagna il sequenziatore automatico gel per ulteriori informazioni e la discussione sezione di questo documento).- In particolare, assicurano che i primer (i) non contengono sequenze di basi, (ii) possedere una G o C all'estremità 3 ', (iii) hanno un GC equilibrata: AT rapporto, (iv) avere una temperatura di ricottura di circa 55-60 ° C e (v) si legano almeno 50 bp, meglio 100 bp a valle della regione di interesse per ricevere immagini chiare.
- Reazione di primer extension
NOTA: La reazione di primer extension (sintesi del DNA) richiede elevate quantità di stampo di RNA. Se la quantità di RNA utilizzati sono scelti in basso, il segnale potrebbe essere troppo bassa per rilevare! Si consiglia pertanto di purificazione dell'RNA come descritto sopra.
NOTA: ATTENZIONE: Utilizzare RNase reagenti liberi e articoli in plastica !!!- Riscaldare il termo-cycler ad una temperatura di 95 ° C ed effettuare tutte le ulteriori fasi di incubazione in un termo-cycler per facilità di uso e riproducibilità.
- Mescolare i composti dalla tabella 2 in un tubo di PCR per ciascun campione di RNA.
- Denaturare i campioni per 1 min a 95 ° C.
- Posizionare le provette su ghiaccio e freddo per 5 minuti per ibridare RNA e primer.
- Impostare la macchina PCR a 47 ° C.
- Nel frattempo preparare la miscela di trascrizione master inversa come descritto nella Tabella 3.
- Aggiungere 4 ml di trascrizione inversa master mix per ogni RNA ibridadel campione.
- Incubare le provette per 1 ora a 47 ° C. Nota: La temperatura ottimale per AMV RT è 42 ° C, temperature più elevate ma aiutano a superare le strutture secondarie delle molecole di RNA.
- Arrestare la reazione riscaldando i campioni a 95 ° C per 2 min.
NOTA: La formammide è corrosivo, tossico e può essere dannoso per il feto. Si prega di leggere le schede di sicurezza, maneggiare con cura e indossare una protezione adeguata! - Aggiungere 6 ml di formammide colorante di caricamento (95% (v / v) formammide deionizzata, EDTA 10 mM, 0,05% (w / v) di blu di bromofenolo) e conservare per O / N a due settimane a -20 ° C al buio.
3. Preparazione del Sequencing Ladder
NOTA: La reazione di sequenziamento scala richiede o moderate quantità di plasmidi o elevate quantità di DNA genomico. Quando possibile, l'uso di plasmidi nella reazione di sequenza è raccomandato a causa della facilità di isolamento e alta sigintensità finale. In altri casi, si usa normalmente un metodo adottato da Marmur 5,14 per preparare il DNA genomico di E. coli e S. cellule aureus senza la necessità di utilizzare fenolo. In linea di principio può essere utilizzato qualsiasi metodo che permetta di ottenere elevate quantità e la purezza del DNA genomico.
- Isolamento del DNA genomico
- Crescere 10 ml di E. coli o S. cellule aureus O / N in LB, BM 5 o medio TSB.
- Celle di raccolta per centrifugazione per 10 minuti a 4600 xg in una provetta Falcon da 15 ml.
- Pellet risospeso in 2 ml di tampone P1 come si trova in alcuni mini kit di preparazione (50 mM Tris-HCl pH 8,0, EDTA 10 mM, 100 ug / ml RNasi A).
- Cellule Lisare per 45 - 60 min con 20 - 40 microlitri lysostaphin (0,5 mg / ml, conservazione a -20 ° C). Nota: per E. coli il enzimatico pre-trattamento può essere omesso o lisozima utilizzato.
- Aggiungere 100 ml di soluzione satura SDS-(nel 45% etanolo) alla sospensione e incubate per 5 minuti a 37 ° C.
- Aggiungere 650 ml 5 M NaClO 4 e le cellule brevemente vortice.
NOTA: cloroformio è un potenziale cancerogeno. Si prega di leggere le schede di sicurezza e utilizzare sotto una cappa di aspirazione con protezione adeguata !!! - Aggiungere 3 ml di cloroformio / isopentanol (24: 1 ratio) alla miscela e agitare per almeno 60 sec. Nota: Il liquido deve trasformarsi in una emulsione bianca omogenea.
- Centrifugare campione per 10 minuti a 4.600 xg e RT per separare le fasi.
- Trasferire con cautela la fase chiara superiore (acquosa) in una nuova provetta. Se la soluzione è torbida, ripetere l'estrazione con cloroformio / isopentanol. Misurare il volume della soluzione di DNA e preparare una nuova provetta con 2 volumi di etanolo (100%).
- Lentamente decantare o pipetta la soluzione di DNA in etanolo contenente tubo. Nota: DNA dovrebbe precipitare come trasparenti, dense spire sul fondo o quando completamente disidratato come cluster bianco galleggiante.
- Recuperare il DNA utilizzando ganci realizzati pipette Pasteur in vetro (Figura 2) e lavare ogni campione due volte mediante immersione in un singolo tubo di 1 ml di etanolo al 70%.
- Posizionare i ganci in posizione verticale in un rack e asciugare il pellet per 60 min. Se necessario, memorizzare il DNA essiccati per diversi giorni a temperatura ambiente.
- Sciogliere DNA rompendo i DNA coperti ganci di vetro e messa in una provetta 2,0 ml di reazione contenente 100 - 500 ml DDH 2 O. Regolare il volume ad una concentrazione finale di DNA di 1.000 - 1.500 ng / ml. Per una reazione di sequenziamento, utilizzare 10 - 18 mg di DNA genomico.

Figura 2. istruzioni su come creare una canna da pesca DNA. Tenere la punta di una pipetta Pasteur di vetro nella fiamma di un becco Bunsen. Ciò causa il vetro per avviare la fusione dopo alcuni secondi, creando un piccolo gancio a tha fine. Rimuovere rapidamente dal fuoco e lasciate raffreddare per 1 min.
- Plasmide Isolamento
- Preparare plasmidi utilizzando kit mini preparazione standard e sciogliere in tampone di eluizione (10 mM Tris-Cl, pH 8,5). A seconda delle dimensioni plasmide, utilizzare 100 - 500 ng di plasmide per una scaletta sequenziamento.
- Reazione di sequenziamento Sanger
NOTA: disponi un semplice protocollo che utilizza un kit di sequenziamento di primer fluorescente con 7-deaza-dGTP che funziona bene ai fini delle estensioni di primer. Fare riferimento al manuale del kit di sequenziamento per informazioni dettagliate. Si noti che la reazione di sequenziamento deve utilizzare lo stesso innesco come la reazione di primer extension per creare prodotti della stessa lunghezza.- Mescolare 12 ml di DNA genomico (~ 10 - 15 mg) con 1 ml di DMSO e 1 ml di primer fluorescente (2 pmol / ml).
- Per ogni 1 ml di quattro miscele di reazione di sequenziamento (A, C, G o T), aggiungere 3 ml di DNA / DMSO / miscela di primer.
- Mettere i campioni in una macchina PCR, ed eseguire il seguente programma PCR: 95 ° C per 2 min; 35 cicli di 95 ° C per 20 sec, 54 ° C per 20 sec, 70 ° C per 30 sec; mantenere a 4 ° C per sempre.
- Dopo la corsa, togliere i campioni dalla macchina, aggiungere 6 ml di colorante di caricamento e conservare in ghiaccio (a breve termine) oppure a -20 ° C per diversi giorni o settimane.
4. Gel Setup e Apparato Run
NOTA: Informazioni dettagliate su come l'apparato gel di sequenziamento è assemblato, il gel viene preparato e come il gel viene eseguito può essere trovato nel protocollo del produttore.
- Preparativi
- Preparare 10x TBE, come indicato nella tabella 4.
- Il giorno della corsa gel di preparare 1 L di tampone TBE 1x con ultrapura DDH 2 O.
- Preparare 10% (w / v) APS. Nota: Può essere conservato in 200 microlitri aliquote a -20 ° C per diversi mesi, ma l'attività potrebbe ridursi nel tempo <./ Li>
- Montaggio di gel camera di colata
- Evitare la polvere e altri residui tra le lastre di vetro. Superfici di lavoro Quindi pulire accuratamente utilizzando salviettine umidificate.
- Pulire una coppia di piastre in vetro da 25 cm usando salviette di carta usa e getta e acqua distillata su entrambi i lati e poi isopropanolo per il lato interno delle lastre di vetro.
- Inserire 0,25 millimetri distanziali sul vetro posteriore e abbassare la lastra di vetro dentata sopra (Figura 3).
- Fissare le guide di gel per entrambi i lati delle lastre di vetro con l'estremità del modulo e dei piloti di ingresso ferroviario rivolte verso l'alto e stringere leggermente le manopole.

Figura 3. vista delle lastre di vetro elettroforesi di gel esplosa. Lastre di vetro devono essere utilizzati direzionalmente. Fare attenzione ad affrontare il lato interno delle lastre di vetro verso l'interno e l'esternolato verso l'esterno.

Figura 4. Vista di un apparato gel assemblato. Dopo l'iniezione della soluzione di gel, il distanziatore tasca è posizionata nella soluzione tra le lastre di vetro. La piastra di fusione viene poi scivolò tra la lastra di vetro frontale e le rotaie gel e fissato stringendo le manopole ferroviari.
- Casting il gel
NOTA: acrilamide non polimerizzato è neurotossico! Si prega di leggere le schede di sicurezza e utilizzare una protezione adeguata !!!- Aggiungere i composti elencati nella Tabella 5 in un becher e miscelare con una barra agitatrice e un agitatore magnetico.
- Immediatamente dopo l'aggiunta di APS e TEMED, raccogliere la soluzione di gel in una siringa da 50 ml e posizionare un filtro da 0,45 nm sulla punta.
- O tenere il bordo superiore della lastra di vetro con una mano o collocare il sandwich in una colata di gel basamento per creare un pendio inclinato 10 - 20 °.
- Lentamente erogare la soluzione di gel tra le lastre di vetro mentre continuo movimento della siringa da un lato all'altro e fermare una volta che la soluzione di gel incontra l'estremità inferiore.
- Spostare di lato o rimuovere eventuali bolle formate utilizzando un gancio bolla.
- Inserire il distanziale tasca gel (0,25 mm) tra le lastre di vetro alla estremità dentellata, immergere nella soluzione di gel e fissare collegando la piastra di colata.
- Viti ferroviari superiore Fissare leggermente (vedi figura 4 per apparecchi completamente assemblato).
- Lasciate set gel per 1 - 2 ore.
- Togliere la lastra e tasca colata distanziale e pulire la tasca da residui di sale e gel.
- Risciacquare con DDH 2 O e asciugare la soluzione in eccesso con carte veline.
- L'esecuzione e la visualizzazione del gel
NOTA: I gel di sequenziamento sono direttamente sottoposti ad elettroforesi in gel di imager, mentrela fluorescenza viene simultaneamente rilevata da un microscopio laser. In contrasto con elettroforesi su gel convenzionale, in cui il gel viene eseguito prima e poi macchiato e visualizzato, l'unità di rilevazione è fisso e analizza le bande in tempo reale mentre passano il laser. Qui di seguito una procedura software di raccolta dati ImagIR in OS / 2 è delineato, che possono essere adottate per versioni più recenti. Per ulteriori informazioni, consultare il manuale dell'utente.- Far scorrere il supporto serbatoio di accumulo nelle guide di gel sulle lastre di vetro anteriore e serrare le manopole.
- Posizionare il gel nel serbatoio di gel inferiore della termocamera gel automatizzato contro la piastra di riscaldamento e fissare facendo scorrere l'entrata pilota ferroviario nelle staffe apparecchi.
- Riempire tampone TBE 1x nelle camere di tampone di gel inferiore e superiore, chiudere la camera di buffer inferiore e collegare la camera tampone superiore alla potenza utilizzando il cavo di alimentazione.
- Se presente, pulire la tasca gel da saline residui ripetutamente pipettando tampone nella tasca.
- Chiudere la camera di serbatoio di accumulo in alto con il coperchio superiore del buffer.
- Chiudere lo sportello della macchina e accendere la termocamera e il computer e avviare il software di raccolta dei dati di base ImagIR.
- Creare un nuovo file di progetto (File-> Nuovo ...), immettere un nome file di progetto, selezionare gli intervalli di laser (700 o 800 nm) e confermare con OK.
- Selezionare Opzioni-> Auto gain ... dal menu immagine in alto, fare clic su Auto per avviare la misurazione automatica di guadagno e di accettare le impostazioni facendo clic su OK.
- Mettere a fuoco il laser selezionando Opzioni-> Fuoco ... dal menu di controllo dello scanner, fare clic sul pulsante Auto e accettando le impostazioni facendo clic su OK.
- Ripetere la procedura di guadagno automatico per regolare l'area che viene messo a fuoco.
- Imposta il controllo dello scanner in base a queste impostazioni: 2.000 V, 35 mA, 45 W, 45 ° C, filtro di scansione: 3, Velocità di scansione: 3.
- Anticipo del gel vuoto per 20 minuti (selezione tensione ON e premere ).
- Nel frattempo, riscaldare la scala sequenziamento ed i prodotti di estensione di primer in una macchina PCR per 2 minuti a 90 ° C, poi raffreddare su ghiaccio.
- Arrestare il elettroforesi, aprire il sequencer gel automatico e rimuovere la parte superiore del coperchio del serbatoio di accumulo.
- Inserire il dente di squalo-pettine tra le lastre di vetro e leggermente perforare il gel con i denti di squalo (vedi Figura 5).

Figura 5. Vista del primo piano di gel con dente di squalo pettine. Esempio (viola) è applicato tra i denti di squalo.
- Pipetta o 1 - 2 ml di primer prodotti di estensione o reazioni di sequenziamento scaletta in ciascuna tasca gel (formata dai denti di squalo).
- Se sono necessari non tutte le tasche, riempire le tasche vuote con loading dye per evitare incomportamento in esecuzione costante.
- Chiudere il serbatoio tampone e porta del sequencer gel.
- Inizia elettroforesi e accendere il laser (Select tensione ON e Laser ON e premere ).
- Arrestare elettroforesi una regione di interesse è passato il laser.
Risultati
Come illustrato nella figura 6, una reazione di primer extension può essere utilizzato per determinare i punti di partenza trascrizionali di trascritti di interesse e può aiutare a dedurre regioni promotrici (tipicamente identificati da -10 e -35 elementi). Il più alto (più lunga) frammento di cDNA rappresenta il 'estremità del mRNA 5 e quindi può essere facilmente mappato rispetto alla scala sequenziamento.
Discussione
Primer extension fluorescente è un metodo semplice e rapido per determinare l'estremità 5 'di RNA, sia per TSP- o l'identificazione di trasformazione secondaria dell'RNA. Grazie all'uso di primer fluorescenti, le reazioni possono essere impostati e gestiti senza misure di sicurezza supplementari (a differenza in caso di primer marcati radioattivamente). Poiché i campioni vengono rilevati mediante fluorescenza, possono essere esposte mentre l'elettroforesi è in corso che consente l'analisi...
Divulgazioni
The authors have nothing to disclose that would present a conflict of interest.
Riconoscimenti
We thank Anne Wochele for her assistance in the laboratory and Vera Augsburger for help with the automated gel sequencer. We thank the Deutsche Forschungsgemeinschaft for funding by grants BE4038/2 and BE4038/5 within the “priority programmes” SPP1316 and SPP1617.
Materiali
| Name | Company | Catalog Number | Comments |
| Name | Supplier | Catalog Number(s) | Comment |
| AMV Reverse Transcriptase (20-25 U/µl) | NEB / Roche | NEB: M0277-T / Roche: 10109118001 | |
| DNase I (RNase free) | Ambion (life technologies) | AM2222 | |
| FastPrep-24 Instrument | MPBio | 116004500 | |
| Fluorescently labeled primers | Biomers | n/a | 5’ DY-681 modification of “ordinary” DNA oligonucleotides. Compatible dyes such as the LICOR IRDye 700/800 are also available from other suppliers such as IDTdna. |
| Li-Cor 4200 Sequencer incl. ImagIR Data collection software | Li-Cor | Product discontinued | |
| NanoDrop 2000 | Thermo Scientific | ||
| Nuclease free water | Ambion (life technologies) | AM9915G | |
| Plasmid mini preparation kit | QIAGEN | 12125 | |
| RapidGel-XL-40% Concentrate | USB | US75863 | |
| RNA STORAGE BUFFER | Ambion (life technologies) | AM7000 | |
| Roti-Aqua-P/C/I | Carl Roth | X985.3 | Alternative: “Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1)” from Ambion (AM9720) |
| SUPERase•In RNase Inhibitor | Ambion (life technologies) | AM2696 | |
| Thermo Sequenase fluorescently labelled primer cycle sequencing kit with 7-deaza-dGTP | GE Healthcare | RPN2538 | |
| TRIzol reagent | life technologies | 15596-026 | |
| Zirconia/Silica Beads 0.1 mm | BioSpec | 11079101z |
Riferimenti
- Simpson, C. G., Brown, J. W. Primer extension assay. Methods Mol. Biol. 49, 249-256 (1995).
- Sanger, F., Nicklen, S., Coulson, A. R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74, 5463-5467 (1977).
- Fekete, R. A., Miller, M. J., Chattoraj, D. K. Fluorescently labeled oligonucleotide extension: a rapid and quantitative protocol for primer extension. Biotechniques. 35, 90-94 (2003).
- Schuster, C. F., et al. Characterization of a mazEF Toxin-Antitoxin Homologue from Staphylococcus equorum. J. Bacteriol. 195, 115-125 (2013).
- Nolle, N., Schuster, C. F., Bertram, R. Two paralogous yefM-yoeB loci from Staphylococcus equorum encode functional toxin-antitoxin systems. Microbiology. 159, 1575-1585 (2013).
- Yamaguchi, Y., Park, J. H., Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 45, 61-79 (2011).
- Schuster, C. F., Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 340, 73-85 (2013).
- Goeders, N., Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 6, 304-324 (2014).
- Yamaguchi, Y., Inouye, M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Progress in molecular biology and translational science. 85, 467-500 (2009).
- Park, J. H., Yamaguchi, Y., Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 585, 2526-2532 (2011).
- Zhu, L., et al. et al.Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J. Bacteriol. 191, 3248-3255 (2009).
- Zhu, L., et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 69, 559-569 (2008).
- Fu, Z., Donegan, N. P., Memmi, G., Cheung, A. L. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871-8879 (2007).
- Marmur, J. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3, 208-218 (1961).
- Emory, S. A., Belasco, J. G. The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J. Bacteriol. 172, 4472-4481 (1990).
- Cole, S. T., Bremer, E., Hindennach, I., Henning, U. Characterisation of the promoters for the ompA gene which encodes a major outer membrane protein of Escherichia coli. Mol. Gen. Genet. 188, 472-479 (1982).
- Schleifer, K. H., Kilpper-Bälz, R., Devriese, L. Staphylococcus arlettae sp. nov., S. equorum sp. nov. and S. kloosii sp. nov.: Three New Coagulase-Negative, Novobiocin-Resistant Species from Animals. Syst. Appl. Microbiol. 5, 501-509 .
- Yu, H., Goodman, M. F. Comparison of HIV-1 and avian myeloblastosis virus reverse transcriptase fidelity on RNA and DNA templates. J. Biol. Chem. 267, 10888-10896 (1992).
- Ying, B. W., Fourmy, D., Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA. 13, 2042-2050 (2007).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati