
Method Article
Digital Polymerase Chain Reaction Assay for the Genetic Variation in a Sporadic Familial Adenomatous Polyposis Patient Using the Chip-in-a-tube Format
In This Article
Summary
Digital polymerase chain reaction (PCR) is a useful tool for the high-sensitivity detection of single nucleotide variants and DNA copy number variants. Here, we demonstrate key considerations for measuring rare variants in the human genome using digital PCR with the chip-in-a-tube format.
Abstract
The quantitative analysis of human genetic variation is crucial for understanding the molecular characteristics of serious medical conditions, such as tumors. Because digital polymerase chain reactions (PCR) enable the precise quantification of DNA copy number variants, they are becoming an essential tool for detecting rare genetic variations, such as drug-resistant mutations. It is expected that molecular diagnoses using digital PCR (dPCR) will be available in clinical practice in the near future; thus, how to efficiently conduct dPCR with human genetic material is a hot topic. Here, we introduce a method to detect Adenomatous polyposis coli (APC) somatic mosaicism using dPCR with the chip-in-a-tube format, which allows eight dPCR reactions to be simultaneously conducted. Care should be taken when filling and sealing the reaction mixture on the chips. This article demonstrates how to avoid the over- and underestimation of positive partitions. Furthermore, we present a simple procedure for collecting the dPCR product from the partitions on the chips, which can then be used to confirm the specific amplification. We hope that this methods report will help promote the dPCR with the chip-in-a-tube method in genetic research.
Introduction
Quantitative PCR (qPCR) is frequently used to quantify human genetic variation, including single nucleotide variants (SNVs) and DNA copy number variations (CNVs). In qPCR, a polymerase reaction is performed in each tube in the same way as in the conventional end-point PCR, and an amplification signal is acquired from the tube after every thermal cycle. By contrast, in dPCR, meaning end-point dPCR in this report, the PCR mixture is loaded into many microscopic chambers, termed partitions, where DNA templates are present or absent at a limiting dilution, and every partition containing PCR mixture is assessed as negative or positive after the complete PCR. While it is easy to estimate that there are no DNA templates in the negative partitions, it is unclear how many copies of DNA templates are present in the positive partitions. Therefore, the number of DNA templates in the positive partitions is estimated based on the Poisson distribution, using the count from the negative partitions1. dPCR is more expensive and has a smaller dynamic range compared to qPCR, but this method enables an absolute quantification and offers a higher sensitivity and greater precision2.
One of the main applications of dPCR is the validation of the variants identified by next-generation sequencing (NGS). Especially in the case of rare variants, meaning low variant fractions, validation is crucial due to sequencing errors that may occur in NGS3. While Sanger sequencing and qPCR are useful tools for the validation of SNVs and CNVs, their sensitivity is low compared to dPCR. Therefore, dPCR technology is in demand for genetic studies dealing with rare variants. Recently, the detection by liquid biopsy of rare variants related to cancer characteristics, such as drug-resistance, has become a hot topic in molecular diagnosis and therapy4. The dPCR technology appears suitable for liquid biopsy studies and is expected to have important clinical applications in the near future, although improvements related to the dynamic range and cost are still required to be implemented.
Commercially-available dPCR technology can be roughly categorized into droplet-based and chip-based platforms; the difference is how the DNA templates are partitioned5,6,7. In the dPCR with chip-in-a-tube format, the PCR mixture is distributed by the capillary action into the partitions on a chip. The chips are built in eight-tube strips, which many lab staff will already be familiar with, and, thus, eight samples can be treated at one time8. In the reading step, it takes < 1 h to treat 96 samples by detecting the whole image of the chip and not each partition. Since dPCR with the chip-in-a-tube format has a high throughput compared with other dPCR systems, usability and productivity are the key advantages for its users.
In this report, somatic mosaicism of the APC gene in a patient with sporadic familial adenomatous polyposis is used as a representative case and the results of dPCR and NGS are compared. The main purpose of this report is to clearly quantify a single nucleotide variant using dPCR with the chip-in-a-tube format. We hope that this report is helpful for researchers interested in adopting the dPCR platform for their own work.
Protocol
The study design was approved by the Institutional Review Boards of the Hamamatsu University School of Medicine (G-260-4). Written informed consent was obtained from the patient and his parents.
1. Quality Control of Genomic DNA
NOTE: Genomic DNA (gDNA) was extracted from peripheral blood using the well-established silica-membrane-based DNA purification method. In advance of the below procedures, the concentration of gDNA was determined using a spectrophotometer.
- Prepare the gDNA sample at a concentration of 10–100 ng/µL.
- Add 10 µL of DNA sample buffer to an 8-tube strip.
- Add 1 µL of DNA ladder or gDNA to the tubes. Vortex the mixture for 1 min using the microplate attachment.
- Load the tube, the gel device and pipette tips into the electrophoresis instrument (Table of Materials) and start the run by pressing the 'Start' button.
NOTE: The electrophoresis system will automatically operate with one click on the Start button. - Confirm that the lower marker contained in the DNA sample buffer is correctly assigned on the electropherograms. If it is incorrectly assigned, manually assign the marker in the 'Electroherogram mode' of the software.
NOTE: The DNA integrity number (DIN) will be automatically calculated. The DIN value comes from the DNA integrity. High and low DIN values indicate highly intact and strongly degraded gDNA, respectively. - Specify the size region of the gDNA (> 200 bp) in the 'Region mode' of the software to automatically calculate the concentration of gDNA (> 200 bp).
2. Primer and Probe Design
- Calculate the melting temperature (Tm) values using any open access tool under the following conditions: 10–25 bases, 50 mM Na+/K+, 0.80 mM dNTPs, and 3 mM Mg2+ (Table of Materials). Design the forward and reverse primers to amplify the genomic region containing target alleles with the Tm value around 60 °C and the amplicon length at 100–300 bp.
NOTE: Refer to Table 1 for the concentration of oligonucleotides. - Design the locked nucleic acid (LNA) probes for the reference and variant alleles based on the following conditions: (i) 1–6 LNAs are present in each probe; (ii) a fluorescent dye and a quencher are present at the 5'- and 3'-terminuses of each probe, respectively; (iii) the difference between the Tm values of the matched and mismatched probes is > 10 °C; (iv) Tm values of the matched and mismatched probes are higher and lower than those of the primers, respectively [e.g., forward primer: 60.8 °C, reverse primer: 59.8 °C, reference allele probe (matched/mismatched): 62.6/45.3 °C, variant allele probe (matched/mismatched): 61.7/50.5 °C]. The Tm values were calculated using the open access tool also used for step 2.1.
- Ensure that the designed primers and probes do not encompass the single nucleotide polymorphisms with a frequency of > 0.1% as determined from databases [e.g., the Single Nucleotide Polymorphism database (dbSNP)].
3. Digital PCR
- Prepare the primers, probes, and gDNA stock solutions in the concentrations described in Table 1 with a TE buffer and store them at -20–4 °C.
- Mix the reagents at room temperature in a total volume of 15 µL to a final concentration as described in Table 1.
- Add 3 µL of distilled water instead of gDNA in a no-template control (NTC).
- Prepare 3.5x the amount of the PCR mixture in triplicate to account for the pipetting error.
- Pipette the PCR mixture up and down to mix it.
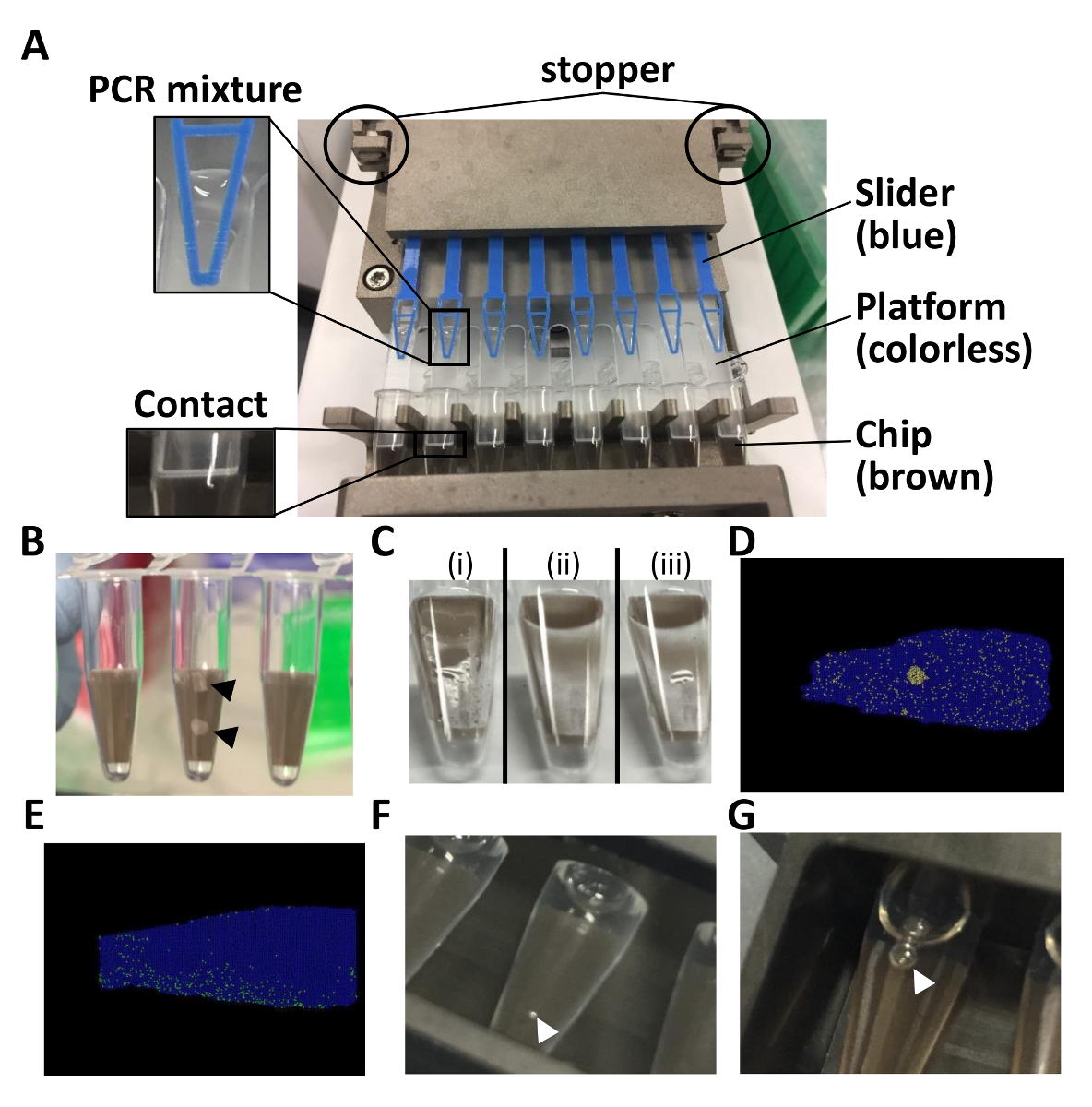
- Set the loading platform onto the chips built in the 8-tube strip and set them on the autoloader. Ensure that there is a contact between the chip and the loading platform.
- Fit a loading slider on the platform and hold the slider with the stopper off of the loader.
- Pipette 15 µL of the PCR mixture on the platform near the tip of the slider (Figure 1A).
- Run the loader by pressing the button of the loader (for approximately 1 min).
NOTE: It is not a problem if a small amount of PCR mixture remains on the platform. It is possible to sequentially rerun the loader. - Set the chip-in-a-tube filled with PCR mixture in the side slot of the sealing enhancer. Push the slide lid and the edge of the top lid to not break the chip (Figure 1B).
- Run the sealing enhancer (approximately 2 min). Sequentially rerun the sealing enhancer for 1 min if a puddle of liquid is still visible because an incomplete sealing causes a cross-contamination of the positive signals (Figures 1C and 1D).
- Add 230 µL of sealing fluid, an oil-based reagent, to the tubes.
NOTE: The chips should now be immersed in the fluid. - Set the tubes in the thermal cycler. Run the thermal cycler as described in Table 2 (for approximately 2 h). Tune the temperature or the duration of the PCR if there is an uneven distribution of positive partitions (Figure 1E).
NOTE: It is recommended to set the ramp rate to around 1 °C/s. - Leave the tubes in the thermal cycler for at least 15 min to reduce the baseline noise.
NOTE: The tubes can be left overnight as well. Fluorescence signals were detected even the following day. - Set the tubes on the detection jig and pour 6 mL of distilled water into the jig. If air bubbles are visible inside and outside of the tubes, clear them (Figures 1F and 1G).
- Load the jig into the detector and run it with a click on the 'Run' button of the software after the selection of Fluorescene, Experiment and Sample/NTC tabs (Table of Materials).
NOTE: The dual color detection of eight samples with a default intensity takes approximately 4 - 5 min. - Confirm the position plot, histogram and 2D scatter plot.
NOTE: iIf the fluorescence intensity of the negative partitions is high, adjust the intensity with a click on the Settings button of the software so that it falls within the range of 30–70. - Calculate the variant allele fraction (VAF) using the following formula:

Here, Cv and Cr are the copy numbers of the variant and the reference allele, respectively.
NOTE: They are automatically calculated in the software program based on Poisson statistics.
4. Collection of PCR Product
- Remove the sealing fluid from the tube.
- Add 100 µL of TE buffer to the tube. Vortex vigorously for 30 s and briefly centrifuge the tube.
- Transfer the TE solution to another tube and add 30 µL of 3 M sodium acetate and 200 µL of ethanol.
- Cool the solution at -20 °C overnight.
- Centrifuge the solution for 30 min at 16,000 x g and remove the supernatant.
- Add 300 µL of 80% ethanol to the tube and vortex.
- Centrifuge the solution for 15 min at 16,000 x g and remove the supernatant. Allow the precipitate to air-dry for 5 min.
- Dissolve the precipitate in 1–5 µL of TE buffer after air-drying.
- If necessary, assess the product size using an electrophoresis instrument with reference to step 1. Load 1 µL of the solution to the gel device for a high sensitivity (Table of Materials).
Results
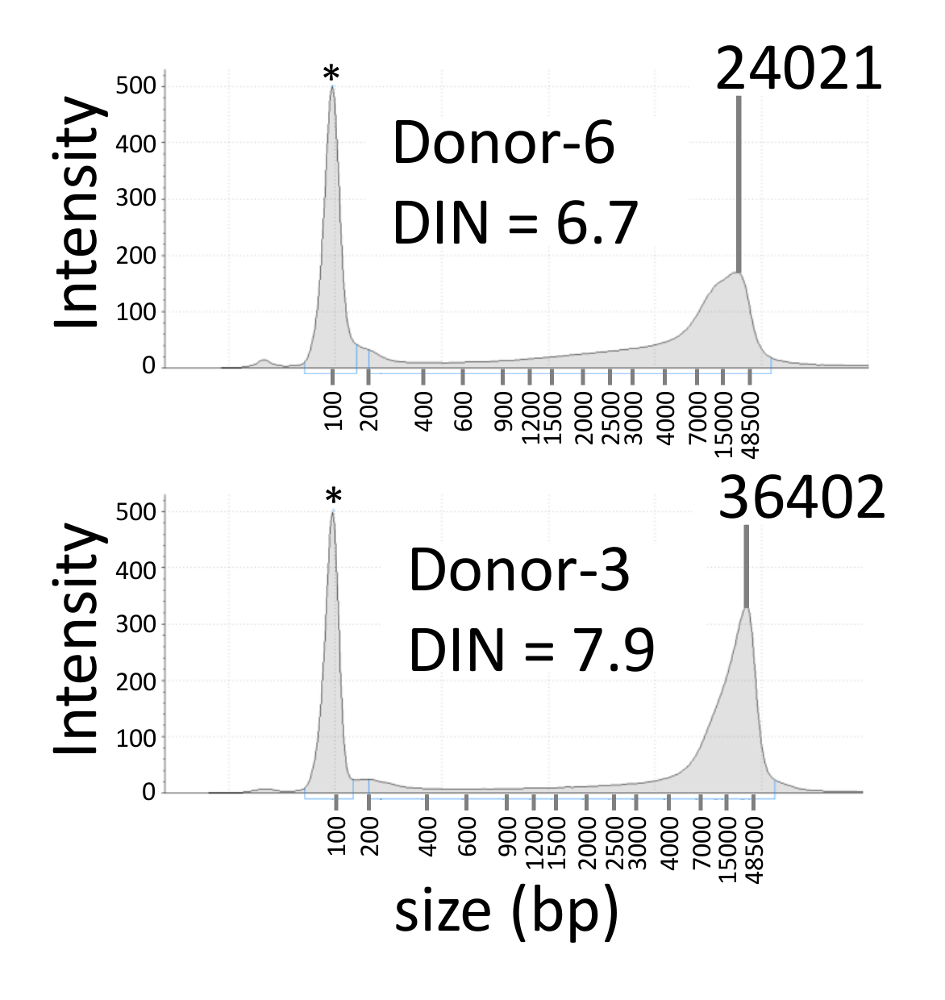
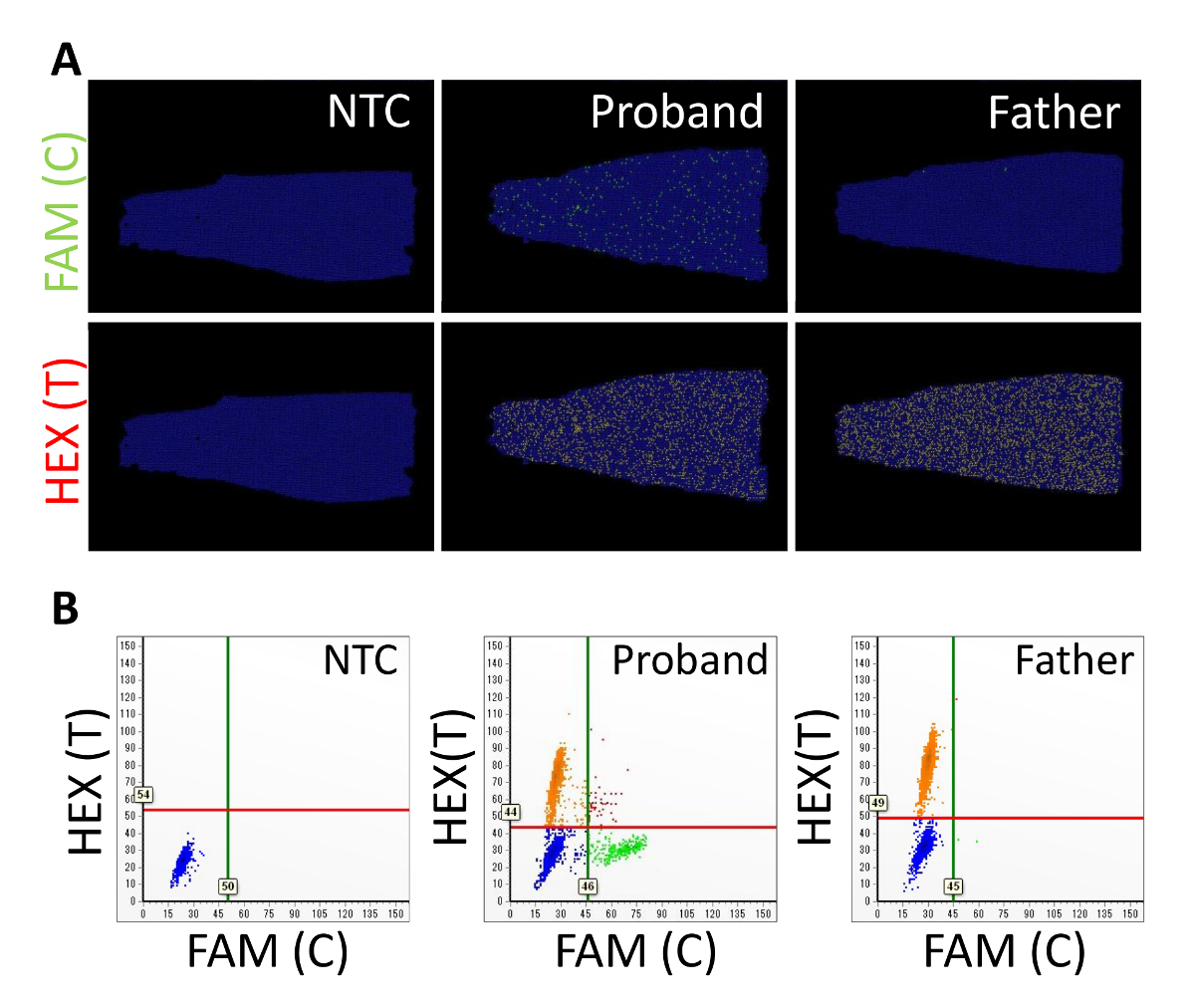
We reviewed somatic mosaicism of the APC gene in a patient with sporadic familial adenomatous polyposis according to the procedures described above. In a previous study, the APC c.834 + 2T > C mutation was found in the patient, but not in their parents, using Sanger sequencing and NGS9. The designs of the primers and probes used in the representative results are shown in Table 3. Genomic DNA was extracted from the blood of the proband, the parents, and six healthy donors. The DIN values of the nine gDNA samples ranged to 6.7–7.9 (Figure 2 and Table 4). FAM and HEX were used as the fluorescence dyes for the C and T alleles, respectively. The green and yellow spots on the position plots represent FAM-positive and HEX-positive partitions, respectively (Figure 3A). The blue spots represent negative partitions for both FAM and HEX. The black background corresponds to partitions where the reaction mixture was not added. Many positive partitions of the C and T alleles were detected in the proband, while few positive partitions of the C allele were detected in the proband's father or the NTC and, in the latter, positive partitions of the T allele were not detected. The signal separation between the C and T alleles was clear on the two-dimensional (2-D) scatterplot, and the most positive signals of HEX and FAM were exclusive of each other (Figure 3B). The VAF of the proband was 13.2%, which is similar to that determined using NGS (12.7%) (Table 4). The VAFs of the proband's parents and healthy donors were < 0.1%. The detection limit for the APC c.834 + 2T > C mutation was estimated to be 0.3% using 3x the standard deviation for the repeated measurements with 10–11 ng of the total gDNA10.
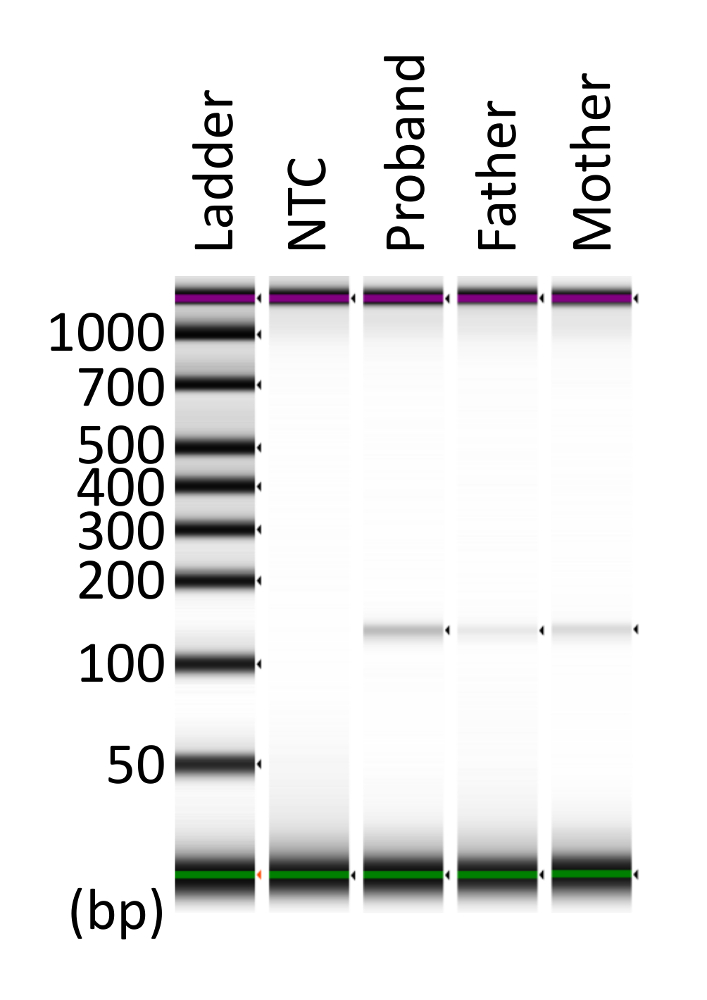
The amplicon size in the dPCR assay for APC c.834 + 2T > C was predicted to be 123 bp. To confirm that the dPCR product was amplified as a single band, the dPCR product was collected from the assayed chip. Using electrophoresis, a single band was clearly detected, and the product size was consistent with the prediction (Figure 4).

Figure 1: Points to note for the dPCR assay with chip-in-a-tube format. (A) This panel shows how to set up the 8-tube strip in the autoloader. (B) This panel shows broken chips. (C) This panel shows how the chip surfaces just after (i) loading and (ii) sealing. (iii) A puddle of liquid is visible in the case of an incomplete sealing. (D) This panel shows the position plot in the case of a cross-contamination. (E) This panel shows the position plot in the case of an uneven distribution. (F) This panel shows how air bubbles on the tube surface immersed in water. (G) This panel shows air bubbles inside of the tube. Throughout the entire figure, black and white arrowheads indicate broken chip pieces and air bubbles, respectively. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative electropherograms of blood gDNA. Electropherograms of blood gDNA with DIN values of 6.7 and 7.9 are shown in the upper and bottom panel, respectively. The asterisk indicates a lower marker. DIN: DNA integrity number. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative results of APC somatic mosaicism using dPCR with the chip-in-a-tube format. (A) The position plots and (B) scatterplots of the no-template control (NTC), the proband, and the father are shown here. HEX and FAM correspond to the reference and variant alleles, respectively. Vertical and horizontal lines indicate the thresholds of HEX and FAM intensities, respectively. This figure has been modified from Kahyo et al.10. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Collection of dPCR products from the assayed chip. The collected and concentrated dPCR products were subjected to electrophoresis. The predicted target size was 123 bp. This figure has been modified from Kahyo et al.10. NTC: no-template control. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Concentration of stock solution | Volume (μL) | Final concentration | Recommended final concentration |
| DNase/RNase-Free Distilled Water | - | 1.75 | - | - |
| 2x PCR Master mix | - | 7.5 | - | - |
| LNA probe for major allele | 2 μM | 0.5 | 66.7 nM | 3.33–100 nM |
| LNA probe for minor allele | 2 μM | 0.5 | 66.7 nM | 3.33–100 nM |
| Forward primer | 1 μM | 0.5 | 33.3 nM | 3.33–50.0 nM |
| Reverse primer | 1 μM | 0.5 | 33.3 nM | 3.33–50.0 nM |
| 20x dPCR solution | - | 0.75 | - | - |
| gDNA | 3.33 ng/μL | 3 | 666 pg/μL | 20 pg/μL–2,000 pg/μL1 |
Table 1: Reagents used for the dPCR assay. 1Depending on expected precision and sensitivity.
| Step | Temperature | Time | Cycles |
| Initial denature | 95 °C | 5 m | 1 |
| Denature Annealing and extension | 95 °C 58 °C | 50 s 90 s | 42 |
| Final extension | 70 °C | 5 m | 1 |
Table 2: Thermal cycler conditions.
| Name | Sequence1 | Tm value2 |
| Forward primer | 5'-GGTCAAGGAGTGGGAGAAATC-3′ | 60.8 °C |
| Reverse primer | 5'-TCTTAGAACCATCTTGCTTCATACT-3′ | 59.8 °C |
| LNA probe for T allele | 5'-HEX-ATTT[A]CCTGACCA-IBFQ-3' | Matched: 62.6 °C Mismatched: 45.3 °C |
| LNA probe for C allele | 5'-FAM-TTT[G]CCTGACC-IBFQ-3' | Matched: 61.7 °C Mismatched: 50.5 °C |
Table 3: Design of the primers and probes. 1Underlined and bracketed letters are the LNA and targeted variant, respectively. 2Calculated according to step 2 of the protocol.
| NGS3 | dPCR4 | |||||||
| Sample1 | Tissue | Input DNA (ng) | DIN2 | VAF (%) | Variant (copies) | Reference (copies) | VAF (%) | |
| Mean | RSD | |||||||
| NTC | - | - | - | - | 1.23 | 0 | N/A | N/A |
| Proband | Blood | 11.3 | 7.6 | 12.7 | 379 | 2.48 x 103 | 13.2 | 0.0353 |
| Father | Blood | 10.3 | 7.7 | 0.0167 | 1.08 | 3.42 x 103 | 0.0341 | 0.439 |
| Mother | Blood | 10.8 | 7.6 | 0.0136 | 0.956 | 3.04 x 103 | 0.0313 | 0.637 |
| Donor-1 | Blood | 11.7 | 7.3 | - | 1.64 | 3.01x 103 | 0.0543 | 0.414 |
| Donor-2 | Blood | 10.5 | 7.6 | - | 1.06 | 2.90 x 103 | 0.0406 | 0.539 |
| Donor-3 | Blood | 17.6 | 7.9 | - | 4.24 | 5.65 x 103 | 0.0713 | 0.783 |
| Donor-4 | Blood | 12.3 | 7.6 | - | 2.38 | 4.16 x 103 | 0.0666 | 0.557 |
| Donor-5 | Blood | 19.2 | 7 | - | 1.48 | 6.79 x 103 | 0.0213 | 0.571 |
| Donor-6 | Blood | 14.4 | 6.7 | - | 3.44 | 4.67 x 103 | 0.0728 | 0.729 |
Table 4: Results of the dPCR assay for APC somatic mosaicism. 1NTC: no-template control; Donor: healthy donor. 2DIN: DNA integrity number. 3Iwaizumi et al.9, Hum Genome Var. 2 15057 (2015). 4The experiments were run in triplicate and independently repeated 3x; the data are expressed as the mean value; RSD: relative standard deviation; N/A: not applicable. The table has been modified from Kahyo et al.10.
Discussion
The DIN value is often used to assess damaged DNA (e.g., gDNA from formalin-fixed paraffin-embedded tissue) before quantification or sequencing, because an advanced degradation of gDNA can result in low-quality data. DIN assessment is, thus, becoming an important step in the quality control of human gDNA11. Since the gDNA used in the representative experiment had been stored at 4 °C for 4–11 years after its extraction from blood, its DIN values were determined for a quality control before the dPCR assay. This step is especially recommended if the materials are suspected to be damaged. The concentration of gDNA was also determined by electrophoresis. Because the dynamic range of dPCR is not as wide as for qPCR due to the limitations of the partitions, a measurement of the gDNA concentration is an important step for a successful dPCR assay. As the amount of input DNA was correlated with the total copy number of the variant and reference alleles in the dPCR assay (R-squared = 0.935; Table 4), electrophoresis is useful for measuring the gDNA concentration before the dPCR assay.
The loading and sealing processes are important steps for successful results. It is crucial to confirm the appropriate mounting of the PCR mixture on the platform and ensure contact between the chip and the platform (Figure 1A). Protrusion of PCR mixture from the slider can cause invalid distribution on the chip. Confirming the distribution of the positive and negative partitions on a position plot is also necessary for a precise analysis, because it is assumed in Poisson statistics that DNA templates are randomly partitioned into chambers1. In the representative results, positive partitions were distributed throughout the chip (Figure 3). This outcome meets the requirement for Poisson statistics and reflects that the loading and sealing procedures were effective. When setting the tubes in the sealing enhancer, the chips could be broken if the central portion of the top lid is pushed too strongly (Figure 1B). To avoid this, gently push the edge of the top lid. If there is an artificial cluster of positive partitions (Figure 1D), the copy number may be overestimated due to a cross-contamination between partitions. The sealing procedure should be improved if a puddle of liquid is visible on the surface (Figure 1C) (e.g., by sequentially rerunning the sealing enhancer for 1 min). If there is an uneven distribution of positive partitions (Figure 1E), the copy number may be underestimated due to insufficient amplification or contamination of the sealing fluid and water. The PCR conditions should be adjusted in this case [e.g., by tuning the temperature or the duration of the PCR (step 3.11 of the protocol), or by ensuring that the sealing fluid and water poured into the jig are clean]. Fluorescence signals are detected from the 8-tube strip immersed in distilled water. If air bubbles adhere to the tube surface, there is the possibility that they may interfere with the signal detection (Figure 1F). Therefore, bubbles should be cleared using a tool, such as a fine pipette tip. Additionally, air bubbles may form inside the tubes when they are set in the jig (Figure 1G). If the bubbles inside the tubes are large enough to cover the chip, they should also be cleared. The bubbles may clear if they are left for several minutes at room temperature.
Some positive partitions might be detected in the NTC. To avoid a contamination of the DNA templates, keep the bench clean and, if possible, make a clean space with a fan filter unit. If a contamination of the DNA templates is suspected, thoroughly clean the space and/or discard the used reagents. By contrast, if the positive partitions of the reference allele are not detected in the presence of gDNA, reconfirm the concentrations of the gDNA, primers, and probes. Notably, the primers were used at a lower concentration in the representative experiment, compared to conventional PCR (Table 1)12. It is also crucial to confirm the ramp rate, which is rarely altered in conventional PCR.
What makes dPCR with the chip-in-a-tube format unique is the partitioning of the PCR mixture inside the universal 8-tube strip8,13. A transfer of the partitioning chambers into a PCR tube is not required in this system, thus reducing the contamination risk. There is also an advantage in the dPCR system with chip-in-a-tube format; it takes < 4 h to finish 96 assays in that dPCR system, while it takes at least 5 h to finish the same number of assays in droplet-based dPCR14. Furthermore, the universal 8-tube strip enables the use of a conventional thermal cycler, unlike another chip-based dPCR15, which requires a thermal cycler with a flat block. Also, the accumulated knowledge and reagents for conventional PCR can be applied to the dPCR system. This will be helpful for expanding the applications of dPCR.
It should be noted that dPCR has several limitations. In dPCR with the chip-in-a-tube format, the partition number of the chip is relatively low compared with those in other dPCR platforms. Further improvement to the chip device will be required to increase the dynamic range, which depends on the number of partitions. Because the internal space of the universal tube is limited, a breakthrough design for the chip device is required to increase the number of partitions. The automation of the entire dPCR procedure in the future would greatly reduce human error, resulting in even more reliable results. Because of its high-throughput and high-sensitivity nature, the dPCR with chip-in-a-tube format is expected to be applied to treat a number of samples for liquid biopsies and environmental DNA.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) (No. 15K08397) to Tomoaki Kahyo, and by grants from the Japan Agency for Medical Research and Development (AMED) (No. 927960719), Grant-in-Aid for Exploratory Research (No. 16K15256), Expenditure for Associated Projects of an Incentive Special Budget for the Promotion of a National University Reform in Management Expenses Grants (No. 1019253), and the Smoking Research Foundation to Haruhiko Sugimura. The authors thank Dr. Iwaizumi and Dr. Kurachi for their clinical support. The funders had no role in the preparation of the manuscript. Figure 3, Figure 4, and Table 4 are adapted and reprinted from an article by Kahyo et al.10, with permission from Elsevier.
Materials
| Name | Company | Catalog Number | Comments |
| QIAamp DNA Blood Maxi Kit | Qiagen | 51194 | Before Protocol 1: Extraction of genomic DNA |
| NanoDrop 1000 | ThermoFisher SCIENTIFIC | ND-8000 | Before Protocol 1: Spectrophotometer |
| 8-strip tube | Agilent Technologies | 401428 | Protocol 1 |
| Genomic DNA sample buffer | Agilent Technologies | 5067-5366 | Protocol 1: A component of Genomic DNA Screen Tape Assay |
| DNA ladder | Agilent Technologies | 5067-5366 | Protocol 1: A component of Genomic DNA Screen Tape Assay |
| MS3 Basic Small Shaker | IKA | 3617000 | Protocol 1: Vortex mixer |
| Genomic DNA ScreenTape | Agilent Technologies | 5067-5365 | Protocol 1: Gel device |
| 2200 TapeStation system | Agilent Technologies | G2965AA | Protocol 1: Electrophoresis instrument |
| TapeStation Analysis Software | Agilent Technologies | Bundled with G2965AA | Protocol 1: Analysis software |
| DNA oligo primers | IDT | Custom order | Protocol 2 |
| LNA probes | IDT | Custom order | Protocol 2 |
| Software tool | IDT | Web site: http://biophysics.idtdna.com/ | Protocol 2 |
| dbSNP database | NCBI | Web site: https://www.ncbi.nlm.nih.gov/projects/SNP/ | Protocol 2 |
| TE buffer | ThermoFisher SCIENTIFIC | 12090015 | Protocol 3 |
| DNase/RNase-Free Distilled Water | ThermoFisher SCIENTIFIC | 10977015 | Protocol 3 (Table 1) |
| Clarity Digital PCR Probe Mastermix | JN Medsys | 12013 | Protocol 3 (Table 1): 2xPCR Master Mix A component of #10011 |
| Clarity Sealing Fluid | JN Medsys | 12005 | Protocol 3: Sealing fluid A component of #10011 |
| Clarity JN Solution | JN Medsys | 12006 | Protocol 3 (Table 1): 20xdPCR solution A component of #10011 |
| Clarity Tube-strip | JN Medsys | 12007 | Protocol 3: Chip-in-a-tube A component of #10011 |
| Clarity Sample Loading Kit | JN Medsys | 12008 | Protocol 3: Loading platform and slider A component of #10011 |
| Clarity Auto Loader | JN Medsys | 11002 | Protocol 3: Auto loader A component of #10001 |
| Clarity Sealing Enhancer | JN Medsys | 11003 | Protocol 3: Sealing enhancer A component of #10001 |
| Clarity Reader | JN Medsys | 11004 | Protocol 3: Reader A component of #10001 |
| Life Eco Thermal Cycler | Bioer Technology | TC-96GHbC | Protocol 3: Thermal cycler |
| Clarity Software | JN Medsys | Bundled with #10001 | Protocol 3: Analysis software |
| High Sensitivity D1000 screen tape | Agilent Technologies | 5067-5584 | Protocol 4: Gel device for high sensitivity |
References
- Majumdar, N., Wessel, T., Marks, J. Digital PCR modeling for maximal sensitivity, dynamic range and measurement precision. PLoS One. 10 (3), 0118833(2015).
- Huggett, J. F., et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clinical Chemistry. 59 (6), 892-902 (2013).
- Chen, L., Liu, P., Evans, T. C., Ettwiller, L. M. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 355 (6326), 752-756 (2017).
- Alix-Panabières, C., Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discovery. 6 (5), 479-491 (2016).
- Kiss, M. M., et al. High-throughput quantitative polymerase chain reaction in picoliter droplets. Analytical Chemistry. 80 (23), 8975-8981 (2008).
- Hindson, B. J., et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical Chemistry. 83 (22), 8604-8610 (2011).
- Ottesen, E. A., Hong, J. W., Quake, S. R., Leadbetter, J. R. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science. 314 (5804), 1464-1467 (2006).
- Low, H., Chan, S. J., Soo, G. H., Ling, B., Tan, E. L. Clarity™ digital PCR system: a novel platform for absolute quantification of nucleic acids. Analytical and Bioanalytical Chemistry. 409 (7), 1869-1875 (2017).
- Iwaizumi, M., et al. A novel APC mosaicism in a patient with familial adenomatous polyposis. Human Genome Variation. 2, 15057(2015).
- Kahyo, T., et al. Application of digital PCR with chip-in-a-tube format to analyze Adenomatous polyposis coli (APC) somatic mosaicism. Clinica Chimica Acta. 475, 91-96 (2017).
- Kanai, Y., et al. The Japanese Society of Pathology Guidelines on the handling of pathological tissue samples for genomic research: Standard operating procedures based on empirical analyses. Pathology International. 68 (2), 63-90 (2018).
- Lorenz, T. C. Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. Journal of Visualized Experiments. (63), e3998(2012).
- Cao, L., et al. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosensors and Bioelectronics. 90, 459-474 (2017).
- Bio-Rad Laboratories. , Available from: https://www.bio-rad.com/webroot/web/pdf/Isr/literature/Bulletin_6311.pdf (2018).
- Thermo Fisher Scientific. , Available from: https://www.garvan.org.au/research/capabilities/molecular-genetics/documents/qs-3d-user-guide.pdf (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved