
Method Article
Visualizing the Node and Notochordal Plate In Gastrulating Mouse Embryos Using Scanning Electron Microscopy and Whole Mount Immunofluorescence
In This Article
Summary
The node and notochordal plate are transient signaling organizers in developing mouse embryos that can be visualized using several techniques. Here, we describe in detail how to perform two of the techniques to study their structure and morphogenesis: 1) scanning electron microscopy (SEM); and 2) whole mount immunofluorescence (WMIF).
Abstract
The post-implantation mouse embryo undergoes major shape changes after the initiation of gastrulation and morphogenesis. A hallmark of morphogenesis is the formation of the transient organizers, the node and notochordal plate, from cells that have passed through the primitive streak. The proper formation of these signaling centers is essential for the development of the body plan and techniques to visualize them are of high interest to mouse developmental biologists. The node and notochordal plate lie on the ventral surface of gastrulating mouse embryos around embryonic day (E) 7.5 of development. The node is a cup-shaped structure whose cells possess a single slender cilium each. The proper subcellular localization and rotation of the cilia in the node pit determines left-right asymmetry. The notochordal plate cells also possess single cilia albeit shorter than those of the node cells. The notochordal plate forms the notochord which acts as an important signaling organizer for somitogenesis and neural patterning. Because the cells of the node and notochordal plate are transiently present on the surface and possess cilia, they can be visualized using scanning electron microscopy (SEM). Among other techniques used to visualize these structures at the cellular level is whole mount immunofluorescence (WMIF) using the antibodies against the proteins that are highly expressed in the node and notochordal plate. In this report, we describe our optimized protocols to perform SEM and WMIF of the node and notochordal plate in developing mouse embryos to help in the assessment of tissue shape and cellular organization in wild-type and gastrulation mutant embryos.
Introduction
Gastrulation and the accompanying morphogenetic movements are crucial for shaping the mouse embryo1. The changes in cellular shape and organization during morphogenesis dictate positional information to regulate cell fate and also allow the ensuing signaling pathways to precisely perform their functions to diversify the newly formed germ layers1. The formation of transient organizing structures and signaling centers such as the node and notochord is essential for the execution of the developmental program2. Developmental biologists have used a variety of techniques to study the morphogenesis of these structures, most notable of which is the use of cellular reporters and live ex vivo imaging to follow the dynamics in cellular and subcellular behavior2,3,4. In this report, we focus on describing the details of our optimized protocols for two of these techniques: scanning electron microscopy (SEM) and whole mount immunofluorescence (WMIF), which were and are still instrumental in studying the morphogenesis of the node and the notochordal plate, the precursor of the notochord.
The mouse embryonic node is a teardrop-shaped cup of cells that is located on the ventral surface of the mouse embryo around the early to late head fold stages during gastrulation and morphogenesis (embryonic day, E7.5-E8)2,5,6,7. The notochordal plate morphologically emanates anteriorly from the node3. Each cell in the node and notochordal plate is characterized by a single cilium that protrudes to the outside, which is longer in node cells but whose length varies with the developmental stage2. The rotation of cilia in the node pit has been shown to be important for signaling that determines left-right asymmetry4. The notochordal plate is the precursor of the notochord, the signaling center that is important for the patterning of the adjacent somites and the overlying neural tube3.
Because of the attributes of location (surface), shape (cup) and possessing distinct outer cellular structures (cilia), SEM has traditionally been used to visualize the node and notochordal plate and study their formation and structure2,7. SEM is also used to study the changes in the structure of the node itself or the cilia on its cells in mutations that affect gastrulation, morphogenesis, as well as cilia formation8,9,10. SEM is a technique that utilizes a focused beam of electrons to interrogate the topological ultrastructure of the outer surface of materials such as biological specimens11. The sample is usually fixed, dried and then sputter-coated with metals for observation under a scanning electron microscope as we describe in Step 1.
WMIF is a staining technique to visualize gene products, such as proteins, in three-dimensions (3D). WMIF of tissues, organs or even whole organisms provides spatial information about the distribution of the signal and the shape of the resulting structure in 3D. The technique is based on fixing the sample then staining it with fluorescent conjugates. Mouse embryos ~ E7.5 are small and transparent and therefore ideal for WMIF protocols to visualize the node and notochordal plate. For example, the transcription factor Barchyury (T) is expressed in the nuclei of the node and notochordal plate, and to a lesser extent in the primitive streak, around E7.5-E8 of embryonic development and good working antibodies against T by WMIF are commercially available and make the staining procedure possible. The cells of the node and notochordal plate are also characterized by constricted apical surfaces, which face the outside and thus can be stained with fluorescence-conjugated Phalloidin to mark F-Actin at the apical constrictions. Using these reagents as examples, the combination of T and F-Actin staining by WMIF provides a representation of the node and notochordal plate in 3D in gastrulating mouse embryos as we demonstrate in Step 2 8. However, markers of cilia, such as ARL13B or acetylated tubulin, as well as other markers of the node and notochordal plate, such as FOXA2, can also be used to perform WMIF on developing mouse embryos3,4.
We have shown that striatin-interacting protein 1 (STRIP1) is essential for normal gastrulation and morphogenesis in the mouse embryo8. STRIP1 is a core component of the striatin-interacting phosphatases and kinases complexes (STRIPAK), which we and others have implicated in the actin cytoskeleton organization8,12. A major defect in Strip1 mutant embryos is in the formation of the axial mesoderm (node and notochordal plate) and extension of the antero-posterior body axis. We have used SEM and WMIF to analyze the node and notochordal plate in wild-type (WT) and Strip1 mutant embryos as we show in the Representative Results and corresponding figures.
Protocol
All experiments involving animal experiments were approved by the responsible authorities in North Rhein Westphalia (LANUV-NRW).
1. Scanning Electron Microscopy of the Mouse Embryonic Node
- Sacrifice the pregnant female mouse at ~ E7.5 (2-4 somite stage) by cervical dislocation. A detailed explanation with diagrams of Steps 1.1 - 1.7 is available in mouse embryo laboratory manuals13.
- Open the abdomen through the skin and mesenteries and remove the uterus using scissors and fine forceps.
- Rinse the uterus briefly in distilled water and place it in a small clean Petri dish (6 cm) containing 1x phosphate-buffered saline (PBS).
- Under a dissecting microscope and using fine forceps, remove the uterine muscles to free the individual deciduae or implantation sites.
- Hold each decidua with one pair of forceps and use the other pair to make a longitudinal full-thickness incision between the red part (future placenta) and white part (where the embryo is located). Make superficial perforations vertically along the white part of the decidua contiguous with the incision. Pull the decidua apart horizontally into two halves and carefully scoop out the embryo in the white part of the decidua.
- Transfer the embryo to a new Petri dish (35 mm) with fresh sterile-filtered PBS. Repeat for all deciduae/embryos.
- Remove Reichert's membrane, a relatively opaque membrane engulfing the embryo, from each embryo by teasing it away like a sock starting at the ectoplacental cone (reddish implantation site). For genotyping, take a little piece (~ 0.1 mm2) of the yolk sac at this stage.
- Under a chemical hood and wearing appropriate protection (gloves), transfer the embryos to EM grade fixative composed of 2.5% glutaraldehyde in sterile-filtered PBS in a microcentrifuge (1.5 mL) tube at room temperature. Fix the embryos overnight at 4 °C.
- Carefully remove the glutaraldehyde fixative from the tube without touching the embryos and discard in a proper waste container. Wash the embryos three times in sterile-filtered PBS, for 15 min each at room temperature.
- Dehydrate the embryos in an ethanol series for 5 min each: 50%, 70%, 85%, and three times in 100% or absolute ethanol. Store the embryos at −20 °C in ethanol or proceed directly to the next step.
- Transfer the embryos in ethanol to baskets for critical point drying (CPD) in a critical point dryer machine. Fill the chamber with ethanol to cover the baskets completely.
- Exchange the ethanol by carefully flushing with liquid CO2 for ten times at 10 °C. Drain off liquid CO2 after the last step until the chamber is half full. Heat up to 40 °C until the pressure reaches 80 bars (the critical point) and the liquid CO2 changes to gas. Wait for 10 min then slowly blow off the gas over approximately 45 min.
- As an easier alternative to CPD for drying, add hexamethyldisilazane (HMDS) at a ratio of 1:1 to the embryos in ethanol for 30 min. Then transfer the embryos to pure HMDS for 30 min. Remove the embryos out of the liquid using a pipette and leave them to dry for 30 min.
NOTE: Both drying methods worked equally well in our hands. - Use a fine brush to mount the dried embryos with the ventral side (node) up on an SEM stub with double sided tape.
- Insert the stubs with the embryos into a sputter coating machine for gold particle coating, which is preferred to charge the long thin cilia. Apply a layer of 120-150 Å; the time is dependent on the current, which will vary with each sample.
- Place the coated stubs with embryos into an SEM microscope, apply vacuum and observe the embryonic node and notochordal plate cells with cilia at magnifications ranging from 1000X to 15,000X.
2. Whole Mount Immunofluorescence of the Mouse Node and Notochordal Plate
- Using ice-cold PBS with 0.05% Tween 20 (PBSTw), follow Steps 1.1-1.7 above to remove the embryos at E7.75 and place them in PBSTw in a 35 mm Petri dish on ice.
- Under a chemical hood and wearing appropriate protection (gloves), transfer the embryos to a fixative solution of 4% paraformaldehyde in PBS in a microcentrifuge tube. Fix the embryos overnight at 4 °C.
- Carefully remove the paraformaldehyde fixative from the tube without touching the embryos and discard in a proper waste container. Wash the embryos three times in PBS containing 0.2% Triton X-100 (PBSTr) for 5 min each at room temperature. Perform all the wash and next incubation steps on a nutating shaker.
- Remove the last wash and add blocking solution containing PBSTr with 10% heat-inactivated serum (from the host species of the secondary antibody). Block from 2 h to overnight (or longer) on a nutator at 4 °C.
- Remove the blocking and add ~ 1 mL of the primary antibody diluted in blocking solution, for example an anti-T antibody at 1:500 dilution. Incubate overnight (or longer) on a nutator at 4 °C.
- Remove the primary antibody and save it for later use by adding sodium azide to a final concentration of 0.02% (1 µL of 20% stock to 1 mL of antibody solution). The antibody can be reused ~ 10 times. Rinse the embryos twice with PBSTr and then wash them three times for 30 min each on a nutator at 4 °C.
- Replace the wash with a fluorescence-conjugated secondary antibody, against the primary antibody host species, diluted at ~ 1:1000 overnight (or longer) on a nutator at 4 °C.
- Remove the secondary antibody and rinse twice with PBSTr, then wash three times for 30 min with PBSTr.
- Replace the last wash with PBSTr containing 1:500 fluorescence-conjugated phalloidin, to stain F-Actin, and 1:1000 DAPI, to stain nuclei, for 1 h at room temperature.
- Rinse twice in PBSTr and wash once with PBSTr for 30 min at room temperature.
- Replace PBSTr with PBS and leave the embryos on ice. Prepare clean positively-charged slides (60 x 24 mm) and coverslips (24 x 24 mm) and aqueous glycerol-based mounting media, for example, 90% glycerol in 1xPBS and an antifade reagent, to mount the embryos.
- Put two pieces of clear tape at a distance of ~ 15 mm from each other on the clear part of the slide. This will create enough 3D space (in the Z dimension) that would allow flattening the embryos but not completely squishing them.
- Under a dissecting microscope, carefully move the embryos using a cut P200 pipette (to allow enough space for the embryo to be transferred and not damaged) to the slide.
- Using fine forceps, make two full cuts on the lateral sides of the yolk sac to unfold the embryo. Place the embryo with the ventral side (node and notochordal plate) up (dorsal of neural tube down on the slide).
- Add 50 µL of mounting media on the embryo. Place 4-5 embryos per slide. Add a dab of mounting media to the side of the coverslip that will first touch the slide (top or bottom side), then place it straddling the two pieces of tape and lower it slowly onto the embryos using fine forceps or a bent fine needle while avoiding creating air bubbles.
- Clean the excess mounting media by using an absorbent wipe. Be careful not to move the coverslip in the process.
- Using a generous amount of nail polish, seal the sides of the coverslip without moving it.
- Observe under a scanning confocal microscope.
Results
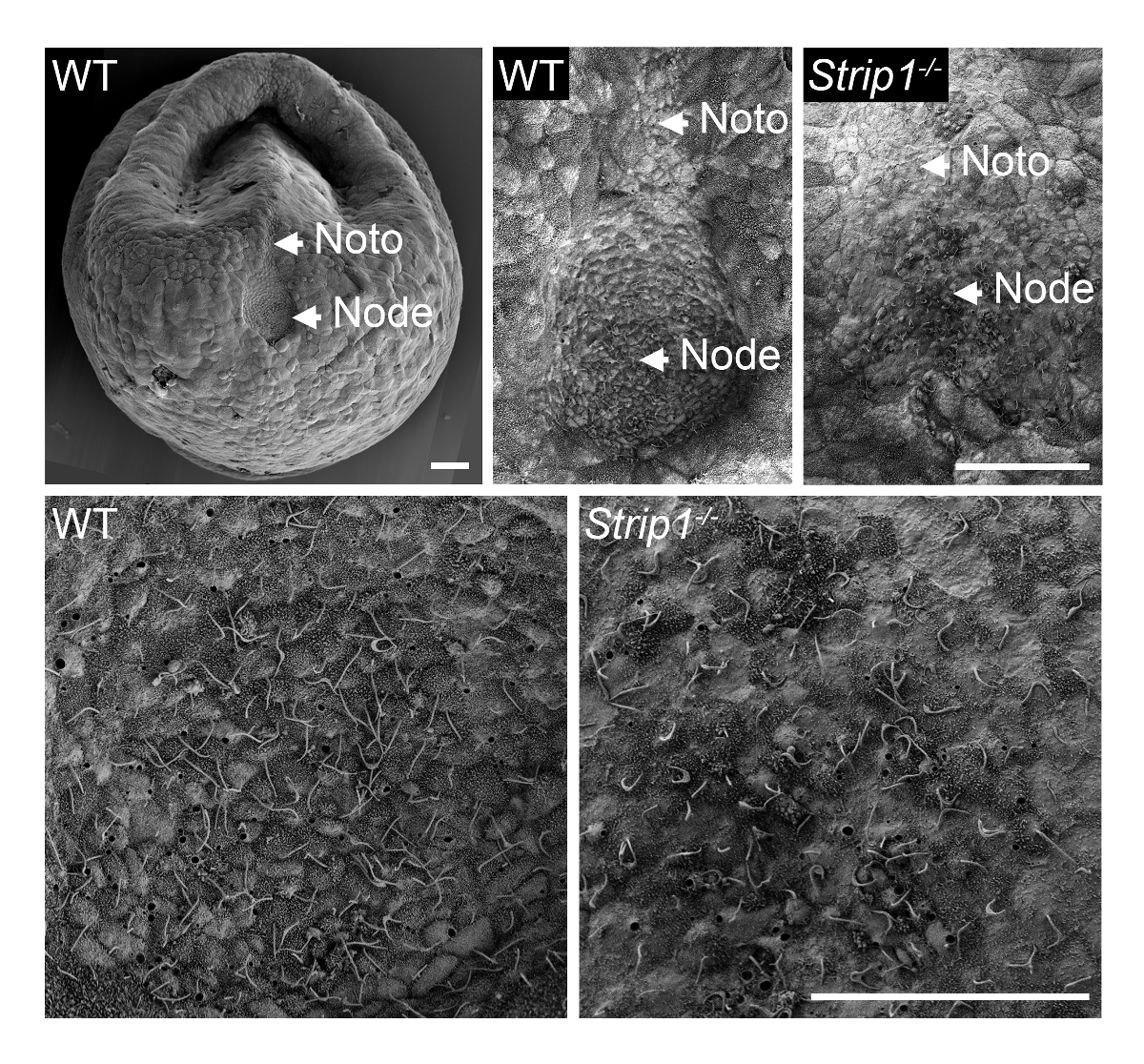
In order to examine the formation of the node in WT and Strip1 mutant embryos at ~ E7.5, we used SEM as described in Step 1 and shown in Figure 18. The ultrastructural details of the outside topology using SEM were quite informative and it was immediately clear that unlike the pit-shaped node in WT embryos, the mutant embryos had a flattened and irregular node. Higher magnification of the embryos showed the characteristic cilia on node cells that identified them unambiguously. The apparent lower density of cilia in the mutant might be attributable to the loss of node pit structure and curvature or a lower number of node cells. The notochordal plate which appears emanating from the node was also irregular in the mutant embryos. They were identifiable with their shorter cilia. Therefore, SEM was important to reveal the node morphogenesis defects in Strip1 mutants8. We have also used SEM in previous studies to show the absence of cilia in the embryonic node of mutants that lacked centrioles, which provide the template for cilia9.
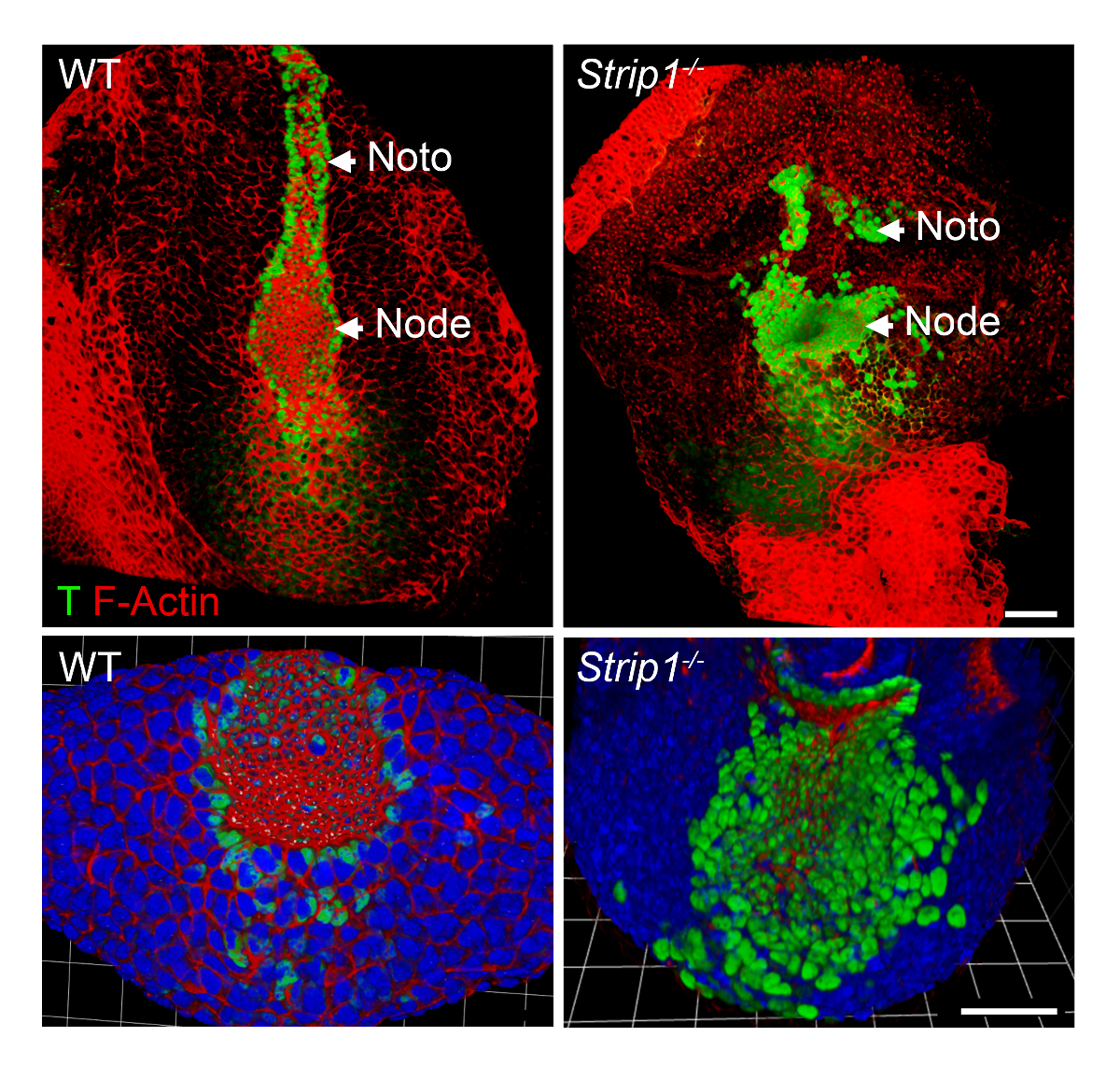
To study the axial mesoderm formation defects in Strip1 mutant embryos at the cellular level, we used WMIF as described in Step 2 and shown in Figure 2. Using this technique, the node and notochordal plate were easily identified by F-Actin and T staining. WT node and notochordal plate cells have constricted apical domains where F-Actin was enriched, and nuclear T staining was evident. The notochordal plate extended rostrally in the WT but was short and irregular in the mutant. The data showed that F-Actin organization is abnormal in the different germ layers of the mutant embryos including the axial mesoderm8. Thus, WMIF was instrumental to study the defects in node and notochordal plate formation in Strip1 mutant embryos.

Figure 1. Scanning electron microscopy reveals the defects in node morphogenesis in Strip1 mutant mouse embryos. (Top) SEM analyses of WT and Strip1 mutant ventral embryonic nodes and notochordal plates (Noto)8. An example of a low magnification image of a WT embryo is shown on the left. (Bottom) Higher magnifications of the center of the nodes shown on top revealing the long monocilia projecting from the node cells. Anterior is up in all panels. Scale bars: 30 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Whole mount immunofluorescence shows the abnormal node and notochordal plate at the cellular level in Strip1 mutant embryos. (Top) Ventral 3D rendering (Volocity software) of WMIF on WT and Strip1 mutant embryos using a combination of fluorescence-conjugated phalloidin (F-Actin, red) and T antibody (green) staining. (Bottom) More examples of the staining shown above focusing on the node with higher zoom and including DAPI. Anterior is up in all panels. Scale bars: 30 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
In this work, we demonstrate how to perform SEM and WMIF to visualize the mouse embryonic node and notochordal plate. The small size of gastrulating mouse embryos ~ E7.5 and the presence of these structures on the surface make them ideal to study using the techniques described2,7,8. The availability of good antibodies, such as T and cilia markers, gives excellent 3D information using WMIF on the structure, organization and formation of these essential embryonic organizers8.
Because mouse embryonic development proceeds at a very rapid pace and the node and notochordal plate are only transiently present on the surface of the embryo, timing is essential for the success of these experiments2,3. For example, 2-4 somite embryos are good for SEM analysis of a mature node pit with long cilia. In much earlier or later embryos (for example, 12 h before or after), the node might not be present on the surface. WMIF is a little more flexible in this regard but the structures themselves are also transient during development and the timing in this case depends on the researchers' interests.
The purity of the reagents is also essential for the success of these techniques, especially in probing the ultrastructure by SEM. Tiny impurities that stick to the embryos usually result in huge artifacts.
We have tested two different methods of embryo fixation for SEM one using half Karnovsky's fixative (2.5% glutaraldehyde, 2% paraformaldehyde and 0.1 M cacodylate buffer) and a simpler 2.5% glutaraldehyde in 1x PBS. We prefer to use the glutaraldehyde and PBS fixative as described in Step 1, however, we and others have also used the half Karnovsky's fixative successfully for SEM.
We have also compared two methods of drying the embryos for SEM and found no difference in the quality of the sample by using either a critical point dryer or HMDS as described in Step 1 and reported elsewhere14.
For Step 2, we tested embedding the embryos after the final washing steps in 1% low melting agarose mounted on a 35 mm glass-bottom dish and then topping it with ~ 10 µL of mounting medium. This embedding method works and preserves the original 3D structure of the embryo and associated structures; however, a multiphoton microscope is required to image the specimen because a regular confocal microscope cannot reach as deep into the intact embryos (~ 1 mm).
We believe that using these two techniques gives complementary information on the structure of the node and the notochordal plate during normal development and in mutants which show defects in the formation of these structures.
Disclosures
The authors have nothing to disclose.
Acknowledgements
H.B. is supported by startup funding from the Medical Faculty and SFB829 of the University of Cologne. C.X. is supported by DFG grant BA 5810/1-1. We would like to thank the Imaging Facilities at the CECAD research center and Memorial Sloan Kettering Cancer Center (New York, USA). We thank Joaquín Grego-Bessa (Spanish National Center for Cardiovascular Research, Madrid, Spain) for his insight on mounting the embryos for WMIF.
Materials
| Name | Company | Catalog Number | Comments |
| 1,1,1,3,3,3 Hexamethyldisilazane (HMDS) | Carl Roth | 3840 | |
| Anti-T antibody | R&D Systems | AF2058 | |
| Critical Point Dryer | Blazers Union | CPD 020 | |
| DAPI | AppliChem | A4099,0005 | |
| Glutardialdehyde solution 25% | Merck | 1042390250 | |
| Triton X-100 | Sigma Aldrich | X100-100ML | |
| Tween 20 | AppliChem | A4974,0500 | |
| SEM coating unit PS3 | Agar Aids for Electron Microscopy | PS3 | |
| SEM microscope Quantum FEG 250 | ThermoFisher Scientific (FEI) | Quantum FEG 250 |
References
- Rivera-Pérez, J. A., Hadjantonakis, A. K. The dynamics of morphogenesis in the early mouse embryo. Cold Spring Harbor Perspectives in Biology. , (2015).
- Lee, J. D., Anderson, K. V. Morphogenesis of the node and notochord: The cellular basis for the establishment and maintenance of left-right asymmetry in the mouse. Developmental Dynamics. 237 (12), 3464-3476 (2008).
- Balmer, S., Nowotschin, S., Hadjantonakis, A. K. Notochord morphogenesis in mice: Current understanding and open questions. Developmental Dynamics. 245 (5), 547-557 (2016).
- Yoshiba, S., et al. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science. , (2012).
- Jurand, A. Some aspects of the development of the notochord in mouse embryos. Journal of Embryology and Experimental Morpholog. 32 (1), 1-33 (1974).
- Poelmann, R. E. The head-process and the formation of the definitive endoderm in the mouse embryo. Anatomy and Embryology (Berl). , 41-49 (1981).
- Sulik, K., et al. Morphogenesis of the murine node and notochordal plate. Developmental Dynamics. 201 (3), 260-278 (1994).
- Bazzi, H., Soroka, E., Alcorn, H. L., Anderson, K. V. STRIP1, a core component of STRIPAK complexes, is essential for normal mesoderm migration in the mouse embryo. Proceedings of the National Academy of Sciences of the United States of America. 114 (51), 10928-10936 (2017).
- Bazzi, H., Anderson, K. V. Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proceedings of the National Academy of Sciences of the United States of America. 111 (15), 1491-1500 (2014).
- Huangfu, D., Liu, A., Rakeman, A. S., Murcia, N. S., Niswander, L., Anderson, K. V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 426 (6962), 83-87 (2003).
- McMullan, D. Scanning electron microscopy 1928-1965. Scanning. 17 (3), 175-185 (2006).
- Bai, S. W., et al. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biology. 9, (2011).
- Behringer, R., Gertsenstein, M., Vintersen Nagy, K., Nagy, A. Manipulating the Mouse Embryo: A Laboratory Manual, Fourth Edition. Cold Harb Lab Press. , (2014).
- Braet, F., De Zanger, R., Wisse, E. Drying cells for SEM, AFM and TEM by hexamethyldisilazane: a study on hepatic endothelial cells. Journal of Microscopy. 186, Pt 1 84-87 (1997).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved