
Method Article
Measuring Dengue Virus RNA in the Culture Supernatant of Infected Cells by Real-time Quantitative Polymerase Chain Reaction
In This Article
Summary
Real-time quantitative polymerase chain reaction analysis combined with reverse transcription (RT-qPCR) has been widely used to measure the level of RNA virus infections. Here we present a direct RT-qPCR assay, which does not require an RNA purification step, developed for the quantification of several RNA viruses, including dengue virus.
Abstract
At present, real-time polymerase chain reaction (PCR) technology is an indispensable tool for the detection and quantification of viral genomes in research laboratories, as well as for molecular diagnosis, because of its sensitivity, specificity, and convenience. However, in most cases, the quantitative PCR (qPCR) assay generally used to detect virus infection has relied on the purification of viral nucleic acid prior to the PCR step. In this study, the fluorescence-based reverse transcription qPCR (RT-qPCR) assay is developed through the combination of a processing buffer and a one-step RT-PCR reagent so that the whole process, from the harvest of the culture supernatant of virus-infected cells until real-time detection, can be performed without viral RNA purification. The established protocol enables the quantification of a wide range of RNA concentrations of dengue virus (DENV) within 90 min. In addition, the adaptability of the direct RT-qPCR assay to the evaluation of an antiviral agent is demonstrated by an in vitro experiment using a previously reported DENV inhibitor, mycophenolic acid (MPA). Moreover, other RNA viruses, including yellow fever virus (YFV), Chikungunya virus (CHIKV), and measles virus (MeV), can be quantified by direct RT-qPCR with the same protocol. Therefore, the direct RT-qPCR assay described in this report is useful for monitoring RNA virus replication in a simple and rapid manner, which will be further developed into a promising platform for a high-throughput screening study and clinical diagnosis.
Introduction
The genus Flavivirus of the Flaviviridae family comprises more than 70 enveloped, positive-stranded RNA viruses that are transmitted by mosquitoes and ticks. Importantly, flavivirus infection in humans often leads to severe clinical illness, such as hemorrhagic fever and encephalitis1. Indeed, a recent outbreak of the Zika virus (ZIKV), a flavivirus, has spread explosively throughout the Americas and has been shown to be associated with neurological complications, including microcephaly2,3. Flaviviruses, therefore, have significant clinical and economic impact on modern society.
An important flavivirus is dengue virus (DENV), a mosquito-borne virus widely distributed in the tropical and subtropical areas of the world. DENV is transmitted by Aedes mosquitos, including Aedes albopictus and Aedes aegypti, resulting in the global spread of dengue outbreaks4. Currently, the World Health Organization (WHO) classifies dengue disease as three categories: dengue without warning signs, dengue with warning signs, and severe dengue4. Although primary infection with one of the four serotypes of DENV (DENV-1 to -4) is often asymptomatic or self-limiting, epidemiologic studies have shown that the secondary infection of different serotypes increases the risk of more serious forms of dengue. It is worth noting that antibody-dependent enhancement of DENV infection caused by the non- or subneutralizing antibodies produced during the primary infection has been proposed as a potential mechanism of severe dengue which occurred as the secondary infection. However, no specific antiviral drug is available for DENV infection5.
For the development of antiviral agents against DENV, routine and robust assays, which are amenable to a high-throughput setting, are essential for detecting virus replication quantitatively. Conventionally, biological assays (e.g., plaque assay) for quantifying infectious viruses and immunological assays (e.g., enzyme-linked immunosorbent assay [ELISA]) for detecting viral antigens have been used to monitor DENV replication in vitro and in vivo6,7. However, these assays are often laborious and require several days to accomplish, which impedes the handling of large numbers of samples. In this regard, RT-qPCR is a reliable assay for detecting DENV infection: it is specific, sensitive, and relatively speedy7. In addition, the RT-qPCR technique has more advantages in a high-throughput format. Nevertheless, the typical procedure of RT-PCR for RNA viruses demands the recovery of viral nucleic acids from collected samples. Although various extraction methods can be employed for RT-qPCR, multiple steps are still needed to obtain purified viral RNA. Also, RT-qPCR assays usually require expensive spin columns or hazardous organic solvents, thereby making the preparation process more cumbersome. Since avoiding mishandling and/or cross-contamination between samples is a critical factor for the successful quantification of viral genomes, a less laborious and time-consuming method is preferred, particularly if the RT-qPCR is to be applied to high-throughput format.
Here, we report a simple RT-qPCR procedure for measuring DENV RNA in a culture supernatant of infected cells that does not involve viral RNA purification. This optimized RT-qPCR protocol is composed of two steps: i) the incubation of a supernatant of DENV-infected cell culture with a processing buffer, and ii) one-step RT-qPCR on a real-time PCR instrument. Accordingly, DENV RNA in a culture supernatant can be detected within 90 min (Figure 1). We also describe the application of the direct RT-qPCR to other RNA virus infections.
Protocol
1. Preparation of the DNA Template of RNA Standard
- Prepare a PCR mix (total volume of 50 μL, Table 1) using a plasmid DNA vector containing DENV-2 New Guinea C (NGC, accession number: AF038403.1) 3’ untranslated region (3’UTR)8 and primers (T7-DENV 3’UTR Fwd and DENV 3’UTR Rvs) in a 0.2 mL tube, as described in Table 2.
NOTE: This step is to be conducted under a clean hood (or in a clean room) to avoid the contamination of any amplicon-related products. - PCR-amplify the cDNA of T7 sequence-fused DENV 3’UTR in a thermocycler, using the following conditions: 30 cycles (of 98 °C for 10 s, 55 °C for 5 s, and 72 °C for 5 s).
- Mix the PCR reaction with 10 μL of 6x gel-loading dye and load it onto a 1% agarose gel with a DNA molecular ladder ranging from 0.1 to 10 kilobase pairs (kbp).
- Visualize the PCR product (479 base pairs [bp]) under long-wavelength UV light using a DNA visualization method and a gel imaging system. Cut out the DNA band using a clean scalpel.
- Extract DNA using a DNA purification kit (Table of Materials).
NOTE: This purification step can be performed outside the clean hood, but it is essential to keep a clean environment during the process to avoid RNase contamination, which is a major problem in the following DENV RNA standard synthesis steps.- Weigh the gel slice and add 100 μL of capture buffer per 100 mg of gel slice in a 1.5 mL microcentrifuge tube.
- Dissolve the gel by incubation at 60 °C for 5–15 min.
- Assemble a DNA purification column with a collection tube and transfer 600 μL of the dissolved sample to the purification column.
- Centrifuge at 16,000 x g for 30 s and discard the flow-through. If the sample volume is greater than 600 μL, apply the rest of the sample to the DNA purification column after the first spin and repeat this step.
- Apply 500 μL of washing buffer to the DNA purification column.
- Centrifuge at 16,000 x g for 30 s.
- Place the column into a new 1.5 mL microcentrifuge tube.
- Add 50 μL of elution buffer and incubate the column at room temperature for 1 min.
- Centrifuge at the maximum speed for 1 min to elute DNA.
- Measure the optical density of DNA at 260 nm on a spectrophotometer using 3 μL of the eluted sample. Use the same volume of elution buffer as a blank.
NOTE: The DNA template can be stored at -20 °C until use.
2. Synthesis of the DENV 3’UTR RNA Standard
NOTE: Maintain the ribonuclease (RNase)-free environment as much as possible to perform the following steps. The set-up of the reagent should be performed inside a clean hood, using nuclease-free pipets, tips, and tubes.
- Mix the in vitro transcription (IVT) reaction (of a total volume of 20 μL) with 100 ng of T7 RNA promoter sequence-fused DENV 3’UTR DNA template (from step 1.6) in a 0.2 mL PCR tube as described in Table 3.
- Incubate the mixture at 37 °C in the thermocycler for 2 h.
- Add 1 μL of DNase to the IVT reaction and continue the incubation at 37 °C for 15 min.
- Purify the in vitro transcribed RNA using an RNA purification kit (Table of Materials).
- Add 80 μL of RNase-free H2O and 350 μL of capture buffer, including 1% β-mercaptoethanol.
- Add 250 μL of 100% ethanol to the IVT reaction and mix it well by pipetting.
- Assemble an RNA purification column with a collection tube and transfer the RNA sample to the purification column.
- Centrifuge it at 8,000 x g for 15 s and discard the flow-through.
- Apply 500 μL of washing buffer to the RNA purification column and centrifuge it at 8,000 x g for 2 min.
- Place the column into a 1.5 mL microcentrifuge tube.
- Add 50 μL of RNase-free H2O and incubate the column at room temperature for 1 min.
- Centrifuge it at the maximum speed for 1 min to elute the RNA.
- Measure the optical density of the RNA at 260 nm on a spectrophotometer, using 3 μL of the eluted sample. Use the same volume of H2O as a blank.
- Determine the copy number of the synthesized DENV 3’UTR RNA. 1 ng of DENV 3’UTR RNA (462 bases, Figure 2) is comparable to 4.05 x 109 copies.
- Store the RNA standard at -80 °C until use.
3. Processing of Virus Samples for RT-qPCR
NOTE: Steps 3.1–3.3 are to be performed inside a biological safety cabinet. In particular, handling of the culture supernatant containing an infectious virus must be conducted under the biosafety level 2 (or higher) environment.
- Mix 199 μL of a processing buffer and 1 μL of nuclease-free proteinase K.
- Serially dilute the synthesized DENV 3’UTR RNA (from step 2.6) 1:10 to obtain 5 x 109 to 5 x 103 copies/μL of RNA standard using cell culture medium (e.g., DMEM supplemented with 10% fetal bovine serum and antibiotics [DMEM/10% FBS]) containing 40 units/mL RNase inhibitor.
- Mix 5 μL of the culture supernatant of DENV-infected cells and the DENV 3’UTR RNA standard with 5 μL of processing buffer/proteinase K solution (from step 3.1) using 8-tube PCR strips or a 96-well PCR plate. As a non-template control (NTC), mix 5 μL of cell culture medium (i.e., no virus is included) with 5 μL of the processing buffer/proteinase K solution.
- After a brief centrifugation, incubate the samples in a thermocycler using the following conditions: 1 cycle (of 25 °C for 10 min and 75 °C for 5 min).
NOTE: Processed samples can be kept at 4 °C if the real-time PCR analysis is done on the same day. For long-term storage, the samples should be stored at -80 °C.
4. Real-time PCR Analysis
NOTE: It is highly recommended to perform steps 4.1–4.4 inside a clean hood to minimize the contamination of any amplicon-related products or RNase.
- Prepare an RT-qPCR master mix with a one-step RT-PCR reagent (Table of Materials) in a 1.5 mL microcentrifuge tube (RNase-free) using DENV 3’UTR-specific primers and a fluorogenic probe (Tables 1). Scale up the volume of each component (Table 4) according to 11% more of the total number of samples, including the standard RNA and NTC reactions.
- Aliquot 8 μL of the master mix into the well to be used in a 96-well real-time PCR plate (Table of Materials).
- Briefly centrifuge the processed samples, including the DENV 3’UTR RNA standard and NTC (from step 3.4) and add 2 μL of the samples to each well of the 96-well real-time PCR plate.
- Seal the PCR plate with optically clear adhesive film.
- Briefly centrifuge the plate at 200 x g to remove air bubbles.
- Place the plate in a real-time PCR instrument and cycle the plate using the following conditions: 1 cycle of the RT stage (25 °C for 10 min), 1 cycle of the polymerase activation stage (95 °C for 2 min), 40 cycles of the amplification stage (95 °C for 10 s and 60 °C for 30 s [single data acquisition at this step]).
- Determine the copy number of the DENV RNA in samples using a real-time PCR-associated software.
- In the Setup window, assign the well of a reaction that is to be analyzed as Unknown sample.
- Assign the well of the serially diluted DENV 3’UTR RNA as Standard and type the expected copy number of RNA standard in each well (e.g., if 5 x 103 copies/μL of RNA standard solution are used, type 5,000). Assign the well as Negative Control for NTC.
- In the Analysis window, click on Analyze and make sure that the correlation coefficient (R2) of the standard curve generated is equivalent to or greater than 0.98.
- Generally, use the default Ct settings (threshold: auto, baseline start cycle: auto, baseline end cycle: auto) for analysis.
NOTE: The copy number of Unknown samples are automatically calculated based on the threshold cycle (Ct) values of the individual reactions. The calculated copy number of each sample is considered as RNA copies per 10 μL RT-qPCR reaction (e.g., if 12,345 is indicated in an Unknown sample, this means that 12,345 copies of DENV RNA are present in the reaction).
Results
For the quantification of DENV RNA by RT-qPCR analysis, a standard of the known copy number, which can be detected by the same primer set, is a prerequisite. In this protocol, the 462 nucleotide-long RNA containing the 3'UTR sequence of the DENV-2 NGC strain was in vitro transcribed from the T7 RNA promoter-fused DENV-2 3'UTR DNA template, which had been amplified by PCR and purified (Figure 2A and 2B). When a serial 10-fold dilution of the standard DENV RNA (from 5 x 109 to 5 x 102 copies in a 10 μL RT-qPCR reaction) was subjected to a direct RT-qPCR analysis using 3'UTR-specific primers and a fluorogenic probe9, a linear curve with a good correlation coefficient (R2 = 0.98726) was obtained (Figure 2C).
Next, this direct RT-qPCR assay was applied to quantify the DENV in the culture supernatant of virus-infected cells. DENV-2 (Singapore isolate EDEN2 329510), which had been propagated in C6/36 mosquito cells and titrated by plaque assay using BHK-21 cells11, was serially diluted (from 8 x 106 to 80 plaque formation units [PFU]/mL). DENV samples were then treated with an equal volume of processing buffer containing proteinase K to deproteinize virions and subjected to a direct RT-qPCR assay targeting the DENV 3'UTR RNA sequence. Again, a good correlation (R2 = 0.99981) between the DENV infectious titer and the Ct, a cycle number that is considered to be the point where the fluorescent signal rises with exponential growth above the background, was obtained (Figure 3A). When Ct values generated from a serial dilution of the DENV stock with known infectious titers were plotted on the standard curve made with in vitro transcribed 3'UTR RNA, all of the plots obtained from 8 x 106 to 80 PFU/mL (equal to 8 x 103 to 8 x 10-2 PFU in a 10 μL RT-qPCR reaction) were within the range of those subjected to standard RNA (5 x 109 to 5 x 103 copies in a 10 μL RT-qPCR reaction, Figure 3B), indicating that DENV samples with a wide range of infectious titers can be analyzed at the same time, using this direct RT-qPCR. In parallel experiments, the effect of processing buffer or proteinase K treatment on the detection of viral RNA by RT-qPCR analysis was investigated. Although treating a log dilution of the DENV sample (8 x 106 to 8 x 102 PFU/mL) with phosphate-buffered saline (PBS) alone prior to RT-qPCR (1:1 dilution of the virus sample with PBS and an incubation of 25 °C for 10 min and 75 °C for 5 min) elicited a regression curve (Figure 3C, black line), and the mean Ct values obtained by the PBS treatment were delayed 2.6–3.2 cycles when compared to the Ct obtained by processing buffer/proteinase K treatment (Figure 3C, dotted line). Additionally, the highest dilution of the virus (8 x 102 PFU/mL [8 x 10-1 PFU per 10 μL RT-qPCR reaction]) could not be detected with this PBS treatment (Figure 3C). In the absence of proteinase K during the processing buffer treatment (Figure 3C, gray line), a similar regression was generated; however, the delay in amplification (0.1–0.8 cycles) was still observed relative to the processing reaction containing proteinase K (Figure 3C, dotted line). These data indicate that, among the conditions tested, treatment of the DENV sample with processing buffer together with proteinase K improves the sensitivity of the RT-qPCR assay.
The direct RT-qPCR assay was further assessed for its applicability to validating antiviral agents against DENV. Mycophenolic acid (MPA), a nonnucleoside inhibitor of inosine monophosphate dehydrogenase that is used as an immunosuppressant in transplantation, has been reported to inhibit DENV in vitro12,13. Although the previous studies employed a conventional plaque assay or a flow cytometry assay to detect viral antigens to demonstrate the inhibitory effect of MPA on DENV infection12,13, in the present study, direct RT-qPCR was applied to evaluate the MPA's antiviral activity. HeLa cells, which had been seeded at a density of 5 x 104 cells/well in a 24-well plate 1 day prior to infection, were exposed to DENV-2 at an MOI of 1 for 1 h and, after washing, cultured with DMEM/10% FBS in the presence of 50–0.016 μg/mL MPA (or 0.1% dimethyl sulfoxide [DMSO]). The culture supernatant was collected 3 days after infection and subjected to the direct RT-qPCR assay using DENV 3'UTR specific primers and a fluorescent probe. Figure 4 shows the DENV RNA copies (per reaction) in the culture supernatants of infected cells treated with increasing concentrations of MPA, which were determined by an in vitro transcribed 3'UTR RNA standard. With a treatment of 50 μg/mL (156 μM) MPA, a reduction to 99.87 ± 0.02% of the DMSO-treated control culture was observed (Figure 4). Importantly, the IC50 (50% inhibitory concentration) value for MPA determined by the data shown in Figure 4 was 0.79 μM, which is similar to the values reported previously12,13. This result, therefore, indicates that this direct RT-qPCR assay is a useful and reliable method for assessing the inhibitory effect of antiviral agents on DENV infection.
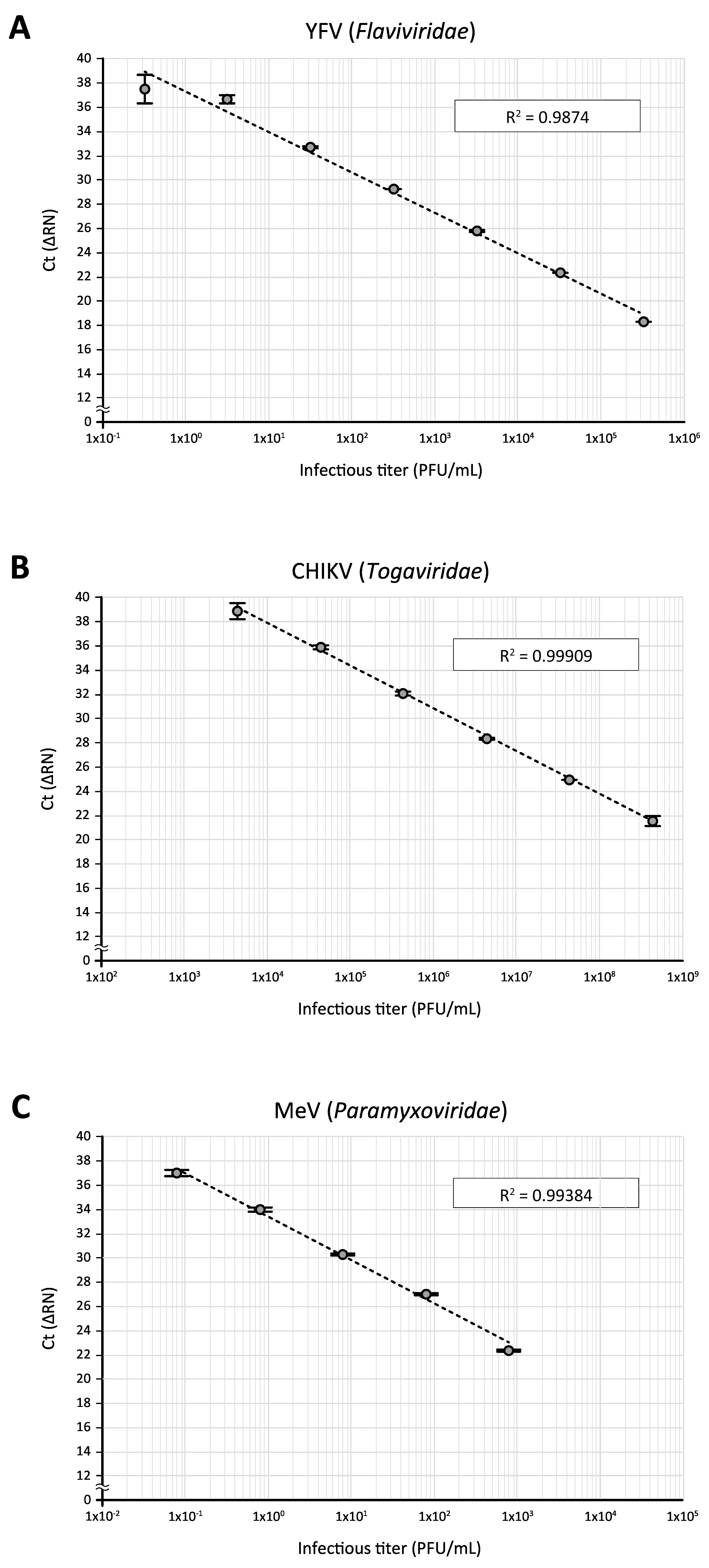
Finally, applications of the direct RT-qPCR assay to other RNA viruses were tested. When a stock of yellow fever virus (YFV) 17D vaccine strain (Flaviviridae), which had been amplified in Vero cells and titrated on BHK-21 cells, was subjected to direct RT-qPCR using YFV-17D-specific primers and a fluorogenic probe14, as described in Table 1, a standard curve with good direct correlation between virus titers and Ct values could be generated with a 10-fold serial dilution of the virus stock (3.2 x 105 to 3.2 PFU/mL, Figure 5A). Likewise, direct RT-qPCR analysis of Chikungunya virus (CHIKV, Togaviridae) Ross strain stock, which had been amplified in Vero cells and titrated on BHK-21 cells, using CHIKV-specific primers and a fluorogenic probe15 (Table 1), gave a good regression between infectious titers (4.4 x 108 to 4.4 x 103 PFU/mL) and Ct values (Figure 5B). This was also the case for the detection of a serial dilution (8 x 102 to 8 x 10-2 PFU/mL, Figure 5C) of the measles virus (MeV, Paramyxoviridae) that had been propagated and titrated with Vero cells, using previously reported primers and a fluorogenic probe16 (Table 1). These data demonstrate the adaptability of the direct RT-qPCR assay for the quantitative detection of various RNA viruses.

Figure 1: Workflow of the direct RT-qPCR assay. The culture supernatant of DENV-infected cells is treated with a processing buffer containing proteinase K to release viral RNA (sample-processing step). The processed sample is then mixed with a one-step RT-PCR reagent and subjected to the real-time PCR assay using DENV 3'UTR-specific primers and a fluorogenic probe. The levels of viral RNA detected in the respective samples can be determined by a serially diluted standard using in vitro transcribed DENV RNA. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: A standard curve generated by DENV 3'UTR RNA. (A) Standard RNA containing 3'UTR of DENV-2 NGC was transcribed in vitro from a T7 RNA promoter sequence-fused PCR fragment. (B) Purified DENV 3'UTR RNA (250 ng) was visualized on a 1% agarose gel (right lane). The left lane shows the molecular weight marker for RNA electrophoresis. (C) A log dilution of the DENV 3'UTR RNA standard was treated with the processing buffer containing proteinase K and subjected to a real-time PCR analysis using a one-step RT-qPCR reagent and DENV 3'UTR-specific primers and a fluorogenic probe set. The mean Ct values (n = 3) obtained in respective dilutions were plotted against the estimated quantity of 3'UTR RNA (5 x 109 to 5 x 102 RNA copies in a 10 μL RT-qPCR reaction). R2 is the correlation coefficient. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: The quantification of DENV RNA in virus stock by direct RT-qPCR. (A) The high-titer DENV-2 stock produced in C6/36 cells was serially diluted 10-fold (8 x 106 to 8 x 101 PFU/mL) with DMEM/10% FBS containing an RNase inhibitor and subjected to the direct RT-qPCR assay using DENV 3'UTR-specific primers/probe. (B) Ct values obtained by RT-qPCR of log-diluted DENV stock (circles) were plotted on a standard curve of the 3'UTR RNA (triangles) transcribed in vitro. The use of 8 x 106 PFU/mL stock in a direct RT-qPCR assay corresponds to 8 x 103 PFU of virus in a 10 μL RT-qPCR reaction. (C) This panel shows the effect of processing buffer and proteinase K treatments on the detection of viral RNA. DENV diluted stock (8 x 106 to 8 x 102 PFU/mL) was incubated with an equal volume of processing buffer containing proteinase K (white circles), processing buffer alone (gray circles), or PBS (black circles) and then subjected to the direct RT-qPCR assay. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Evaluation of the inhibitory effect of MPA by direct RT-qPCR. HeLa cells were infected with DENV-2 at a MOI of 1 and cultured in the presence of increasing concentrations of MPA, a previously reported inhibitor of DENV (or 0.1% DMSO). Three days after infection, culture supernatants of the infected cells were collected and subjected to the direct RT-qPCR assay using DENV 3'UTR-specific primers/probe. The copy number of viral RNA was determined by DENV 3'UTR RNA standard transcribed in vitro. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: The application of direct RT-qPCR for the detection of other RNA viruses. Serially diluted virus stocks of (A) YFV, (B) CHIKV, and (C) MeV were subjected to the direct RT-qPCR assay using PCR primers and fluorogenic probes specific to their respective viral RNA sequences. Please click here to view a larger version of this figure.
{kind=link}
| Purpose | Name | Sequence (5' - 3') | Note |
| T7 promoter-fused DENV-2 3'UTR cDNA amplification | T7-DENV 3’UTR Fwd | TAA TAC GAC TCA CTA TAG GGc gaa tTA GAA GGC AAA ACT AAC ATG AAA | Italic,T7 promoter; lower-case, spacer; bold, DENV-2 NGC nucleotides 10,270-10,292 |
| DENV 3’UTR Rvs | AGA ACC TGT TGA TTC AAC AGC | DENV-2 NGC nucleotides 10,723-10,703 | |
| RT-qPCR analysis of DENV | DENV-2 3'UTR F | AAG GAC TAG AGG TTA GAG GAG ACC C | Reference 9 |
| DENV-2 3'UTR R | GGC GTT CTG TGC CTG GAA TGA TG | ||
| Probe 2 Den-2-4 | 6FAM-AAC AGC ATA TTG ACG CTG GGA AAG ACC-TAMRA | ||
| RT-qPCR analysis of YFV | YFV NS5 F | GAA CAG TGA TCA GGA ACC CTC TCT | Reference 14 |
| YFV NS5 R | GGA TGT TTG GTT CAC AGT AAA TGT G | ||
| YFV NS5 Probe | 6FAM-CTA CGT GTC TGG AGC CCG CAG CAA T-TAMRA | ||
| RT-qPCR analysis of CHIKV | CHIK E1 F | TCG ACG CGC CCT CTT TAA | Reference 15 |
| CHIK E1 R | ATC GAA TGC ACC GCA CAC T | ||
| CHIK E1 P | 6FAM-ACC AGC CTG CAC CCA TTC CTC AGA C-TAMRA | ||
| RT-qPCR analysis of MeV | MeV N F | TGG CAT CTG AAC TCG GTA TCA C | Reference 16 |
| MeV N R | TGT CCT CAG TAG TAT GCA TTG CAA | ||
| MeV N P | 6FAM-CCG AGG ATG CAA GGC TTG TTT CAG A-TAMRA |
Table 1: Oligonucleotide primer and fluorogenic probe sequences used in this study.
| Components | Stock concentration | Volume (μL) | Final concentration |
| PrimeSTAR Max Premix | 2x | 25 | 1x |
| T7-DENV 3’UTR Fwd | 1 μM | 10 | 200 nM |
| DENV 3’UTR Rvs | 1 μM | 10 | 200 nM |
| pEU/DENV 3'UTR | 1 ng/μL | 1 | 20 pg/μL |
| Nuclease-free H2O | 4 | ||
| Total | 50 |
Table 2: Components of a PCR mix for the preparation of DENV 3'UTR template DNA.
| Components | Stock concentration | Volume (μL) | Final concentration |
| T7 sequence-fused DENV 3’UTR DNA template | (variable) | x | 100 ng per reaction |
| ATP solution | 75 mM | 2 | 7.5 mM |
| CTP solution | 75 mM | 2 | 7.5 mM |
| GTP solution | 75 mM | 2 | 7.5 mM |
| UTP solution | 75 mM | 2 | 7.5 mM |
| Reaction Buffer | 10x | 2 | 1x |
| T7 Enzyme Mix | 2 | ||
| Recombinant RNase Inhibitor | 40 units/μL | 1 | 2 units/μL |
| Nuclease-free H2O | 7 - x | ||
| Total | 20 |
Table 3: Components of an IVT mix for synthesizing the DENV 3'UTR RNA standard.
| Components | Stock concentration | Volume (μL) | Final concentration in an RT-qPCR reaction |
| iTaq universal probes reaction mix | 2x | 5.00 | 1x |
| iScript advanced reverse transcriptase | 40x | 0.25 | 100 ng per reaction |
| Primer/probe mix | DENV-2 3'UTR F: 3 μM | 1.00 | 300 nM |
| DENV-2 3'UTR R: 3 μM | 300 nM | ||
| Probe 2 Den-2-4: 2 μM | 200 nM | ||
| Nuclease-free H2O | 1.75 | ||
| Subtotal | 8.00 |
Table 4: Components of a master mix for real-time RT-qPCR analysis of DENV RNA. Note that 2 μL of the processed DENV sample (or standard RNA) should be added to the 8-μL master mix (a total of 10 μL per reaction).
Discussion
Today, viral nucleic acid detection by fluorescent-based real-time PCR is becoming a gold standard for the molecular diagnosis of pathogenic human viruses, due to its sensitivity and rapidity7. This is particularly important for confirming virus infection in the early phase of acute infectious diseases such as dengue fever17. Also, in the field of basic DNEV research, RT-qPCR assay is an indispensable tool for monitoring virus replication in in vitro culture, which will lead to an understanding of the replication biology of DENV and the discovery of antiviral inhibitors. In this study, the fluorescent-based real-time PCR assay was further developed by a combination of a processing buffer and a one-step RT-PCR reagent so that DENV RNA in the culture supernatant of infected cells was able to be quantified by RT-qPCR without purifying the viral RNA genome. When compared to routine assays to detect virus replication, such as plaque assay, ELISA, or conventional RT-qPCR employing RNA purification6,7, a simplified protocol of the RT-qPCR assay resulted in a great reduction of the time and steps necessary to quantify DENV RNA. This is also an important feature that has advantages in minimizing the contamination and loss of genetic materials. Nevertheless, it should be noted that the use of a clean hood is essential in the set-up of the IVT (step 2.1) and RT-qPCR (steps 4.1–4.4) reactions to avoid the cross-contamination of any amplicon-related products or RNase. It is also crucial to handle the culture supernatant, including infectious virus, inside a biosafety cabinet (step 3.3) to reduce a risk of laboratory infection.
Another significance of the direct RT-qPCR assay is that a wide range of viral RNA copies can be analyzed at the same time without dilution or concentration of the sample. When a serially diluted DENV 3'UTR RNA standard was used, the direct RT-qPCR protocol produced a linear standard curve over seven orders of magnitude, and the lower quantification limit was 500 RNA copies (Figure 2). For the detection of DENV in the culture supernatant of infected cells, 8 x 103 to 8 x 10-2 PFU viruses in a 10 μL RT-qPCR were within the range of DENV 3'UTR RNA standard, and the lower detection limit (8 x 10-2 PFU) was calculated to contain 11,400 ± 7,077 RNA copies (Figure 3B). Of note, as reported in several flavivirus studies18,19,20, the larger ratio of viral RNA copies to virus infectious titer (i.e., PFU) in DENV samples was also observed in this study. This overestimation is assumed to be largely due to the presence of defective (i.e., non-infectious) viruses or free viral RNA released from infected and dead cells. Although the ratio of RNA copies:PFU obtained in this study (1.1 x 105 to 1.3 x 105 RNA copies/PFU) was inconsistent with the data shown previously (ratios range from 1 to 3 log)21,20, this may be attributed to the difference in virus strains, cells, or culture conditions employed. Another likely explanation for the discrepancy is that, since no RNA purification process was involved in the direct RT-qPCR assay described herein, more DENV RNA in the culture supernatant could be detected by real-time PCR analysis without the loss of viral genetic materials.
The simplicity of the direct RT-qPCR enhances the ability to handle a large number of samples in multi-well formats, such as a 96-well plate. Therefore, this property will assist the future application of the direct RT-qPCR assay to a high-throughput screening study for the discovery of anti-DENV drugs. Indeed, in this report, application of the direct RT-qPCR assay for the validation of an anti-DENV inhibitor, MPA, was examined, and the result showed that an IC50 value of MPA obtained by the direct RT-qPCR assay (Figure 4) was comparable to that reported in previous studies using plaque assay12,13. This demonstrates that direct RT-qPCR is a useful assay for appropriately evaluating the inhibitory effect of antiviral agents and can be adopted into a drug screening study in future.
In this study, attempts were made to apply the direct RT-qPCR to the detection of other RNA viruses. As shown in Figure 5, when 10-fold dilutions of three other RNA viruses (YFV, CHIKV, and MeV) classified into different genera (Flaviviridae, Togaviridae, and Paramyxoviridae, respectively) were examined using previously reported primers and fluorogenic probes specific to the respective viral RNA sequences. Good correlations between infectious titers and Ct values were obtained by direct RT-qPCR assays, and the correlations were 4-6 linear orders of magnitude. This suggests that the direct RT-qPCR protocol is adaptable to various types of RNA viruses by simply changing the virus-specific primers and fluorogenic probes.
Nevertheless, the sensitivity of detection varied among the viruses tested in this study (Figure 5). One modification of the protocol that can improve the detection of target virus RNA should be to use a new set of primer/probe sequences. Additionally, a key step for the successful direct RT-qPCR assay is thought to be the efficient release of the viral genome during the sample processing step (steps 3.1–3.4). This would be of particular importance for the quantitative detection of stable viruses such as a non-enveloped virus. Although as a preliminary experiment, Coxsackievirus B3 (CVB3), a non-enveloped RNA virus belonging to Picornaviridae, was subjected to the direct RT-qPCR assay using previously described CVB3-specific primers and fluorescent probe22, a positive amplicon signal could not be detected even with a high-titer (1 x 105 PFU/mL) virus stock. This is considered to be largely attributed to the high physical integrity of the non-enveloped virus. So far, some RT-PCR assays that do not require the purification of nucleic acid have been developed to detect several RNA viruses, which employ different protocols in the RNA release step23,24,25,26. Thus, use of the alternative (or additional) treatments, such as an incubation of a virus sample at a high temperature, potentially increases the sensitivity of detection.
In summary, the direct RT-qPCR protocol developed in this study was a simple and rapid but reliable method for quantifying viral RNA in a culture supernatant of infected cells. Because of this simplicity, the direct RT-qPCR assay could process a number of samples in a short time, which holds promise for the high-throughput analysis of virus replication. Furthermore, the application of the direct RT-qPCR protocol to other RNA viruses was also demonstrated. This approach should, therefore, be a useful tool for efficiently monitoring the RNA virus infections in a research laboratory setting and, possibly, in clinical diagnoses.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors thank Subhash G. Vasudevan (Duke-NUS Graduate Medical School, Singapore) for the DENV-2 isolate EDEN2 3295 and Shunro Imura (Kobe Quarantine Station, Japan) for the YFV 17D vaccine strain. The authors are also grateful to the members of the Department of Microbiology and Infection Control for their assistance. This work is supported by MEXT KAKENHI Grant Number JP16737900.
Materials
| Name | Company | Catalog Number | Comments |
| PrimeSTAR Max DNA Polymerase | Takara Bio. Inc | R045A | Comparable PCR reagent kit can be used. |
| SimpliAmp Thermal Cycler | Thermo Fisher Scientific | A24811 | Comparable thermal cycler instrument, but PCR cycle condition needs to be optimized. |
| 6x Gel Loading Dye | New England BioLabs | B7024S | Comparable gel loading dye can be used. |
| 2-Log DNA Ladder | New England BioLabs | N3200 | 0.1-10.0 kilobase pairs. Comparable DNA molecular ladder can be used. |
| Gel Scene Tablet | Astec | GST-33 | Comparable gel imaging system can be used. |
| illustra GFX PCR Purification Kit | GE Healthcare Life Sciences | 28-9034-70 | Comparable gel extraction kit can be used. |
| BioSpectrometer | Eppendorf | 6135000905 | Comparable spectrophotometer can be used. |
| MEGAscrip T7 Transcription Kit | Thermo Fisher Scientific | AM1334 | |
| TURBO DNase | Thermo Fisher Scientific | AM2238 | Included in MEGAscrip T7 Transcription Kit. |
| Recombinant RNase Inhibitor | Takara Bio. Inc | 2313A | 40 units/μL |
| RNeasy Mini Kit | Qiagen | 74104 | Comparable RNA purification kit can be used. |
| CellAmp Direct RNA Prep Kit | Takara Bio. Inc | 3733Q | |
| Proteinase K | Nacalai Tesque | 15679-06 | >600 units/mL. Comparable reagent can be used. |
| iTaq Universal Probes One-Step Kit | Bio-Rad | 1725141 | |
| MicroAmp Fast Optical 96-Well Reaction Plate, 0.1 mL | Thermo Fisher Scientific | 4346907 | Comparable plate or tube can be used dependent on the real-time PCR instrument, but PCR cycle condition needs to be optimized. |
| MicroAmp Optical Adhesive Film | Thermo Fisher Scientific | 4311971 | Comparable film or optical cap can be used dependent on the real-time PCR plate or tube. |
| StepOnePlus Real-Time PCR System | Thermo Fisher Scientific | StepOnePlus-01 | Comparable real-time PCR instrument, but PCR cycle condition needs to be optimized. |
References
- Pastorino, B., Nougairede, A., Wurtz, N., Gould, E., de Lamballerie, X. Role of host cell factors in flavivirus infection: Implications for pathogenesis and development of antiviral drugs. Antiviral Research. 87 (3), 281-294 (2010).
- Fauci, A. S., Morens, D. M. Zika Virus in the Americas--Yet Another Arbovirus Threat. New England Journal of Medicine. 374 (7), 601-604 (2016).
- Wen, Z., Song, H., Ming, G. L. How does Zika virus cause microcephaly?. Genes and Development. 31 (9), 849-861 (2017).
- Guzman, M. G., Harris, E. Dengue. Lancet. 385 (9966), 453-465 (2015).
- Low, J. G., Ooi, E. E., Vasudevan, S. G. Current Status of Dengue Therapeutics Research and Development. Journal of Infectious Diseases. 215 (suppl_2), S96-S102 (2017).
- Sukhavachana, P., Nisalak, A., Halstead, S. B. Tissue culture techniques for the study of dengue viruses. Bulletin of the World Health Organization. 35 (1), 65-66 (1966).
- Tang, K. F., Ooi, E. E. Diagnosis of dengue: an update. Expert Review of Anti-Infective Therapy. 10 (8), 895-907 (2012).
- Suzuki, Y., et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathogens. 12 (1), e1005357 (2016).
- Callahan, J. D., et al. Development and evaluation of serotype- and group-specific fluorogenic reverse transcriptase PCR (TaqMan) assays for dengue virus. Journal of Clinical Microbiology. 39 (11), 4119-4124 (2001).
- Low, J. G., et al. Early Dengue infection and outcome study (EDEN) - study design and preliminary findings. Annals of the Academy of Medicine, Singapore. 35 (11), 783-789 (2006).
- Hishiki, T., et al. Interferon-mediated ISG15 conjugation restricts dengue virus 2 replication. Biochemical and Biophysical Research Communications. 448 (1), 95-100 (2014).
- Diamond, M. S., Zachariah, M., Harris, E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 304 (2), 211-221 (2002).
- Takhampunya, R., Ubol, S., Houng, H. S., Cameron, C. E., Padmanabhan, R. Inhibition of dengue virus replication by mycophenolic acid and ribavirin. Journal of General Virology. 87 (Pt 7), 1947-1952 (2006).
- Akondy, R. S., et al. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proceedings of the National Academy of Sciences of the United States of America. 112 (10), 3050-3055 (2015).
- Edwards, C. J., et al. Molecular diagnosis and analysis of Chikungunya virus. Journal of Clinical Virology. 39 (4), 271-275 (2007).
- Hummel, K. B., Lowe, L., Bellini, W. J., Rota, P. A. Development of quantitative gene-specific real-time RT-PCR assays for the detection of measles virus in clinical specimens. Journal of Virological Methods. 132 (1-2), 166-173 (2006).
- Peeling, R. W., et al. Evaluation of diagnostic tests: dengue. Nature Reviews: Microbiology. 8 (12 Suppl), S30-S38 (2010).
- Bae, H. G., Nitsche, A., Teichmann, A., Biel, S. S., Niedrig, M. Detection of yellow fever virus: a comparison of quantitative real-time PCR and plaque assay. Journal of Virological Methods. 110 (2), 185-191 (2003).
- Colton, L., Biggerstaff, B. J., Johnson, A., Nasci, R. S. Quantification of West Nile virus in vector mosquito saliva. Journal of the American Mosquito Control Association. 21 (1), 49-53 (2005).
- Richardson, J., Molina-Cruz, A., Salazar, M. I., Black, W. Quantitative analysis of dengue-2 virus RNA during the extrinsic incubation period in individual Aedes aegypti. American Journal of Tropical Medicine and Hygiene. 74 (1), 132-141 (2006).
- Wang, W. K., et al. Detection of dengue virus replication in peripheral blood mononuclear cells from dengue virus type 2-infected patients by a reverse transcription-real-time PCR assay. Journal of Clinical Microbiology. 40 (12), 4472-4478 (2002).
- Gnadig, N. F., et al. Coxsackievirus B3 mutator strains are attenuated in vivo. Proceedings of the National Academy of Sciences of the United States of America. 109 (34), E2294-E2303 (2012).
- Rudenko, N., Golovchenko, M., Cihlarova, V., Grubhoffer, L. Tick-borne encephalitis virus-specific RT-PCR--a rapid test for detection of the pathogen without viral RNA purification. Acta Virologica. 48 (3), 167-171 (2004).
- Pastorino, B., et al. Development of a TaqMan RT-PCR assay without RNA extraction step for the detection and quantification of African Chikungunya viruses. Journal of Virological Methods. 124 (1-2), 65-71 (2005).
- Nishimura, N., et al. Detection of noroviruses in fecal specimens by direct RT-PCR without RNA purification. Journal of Virological Methods. 163 (2), 282-286 (2010).
- Kang, K., et al. A direct real-time polymerase chain reaction assay for rapid high-throughput detection of highly pathogenic North American porcine reproductive and respiratory syndrome virus in China without RNA purification. Journal of Animal Science and Biotechnology. 5 (1), 45 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved