
Method Article
Fluorescence Recovery after Photobleaching of Yellow Fluorescent Protein Tagged p62 in Aggresome-like Induced Structures
In This Article
Summary
We describe a comprehensive and practical protocol for fluorescence recovery after photobleaching experiments with live cells. Although the protocol was used to measure the mobility of yellow fluorescent protein-tagged p62 in aggresome-like induced structures, it can be applied to a variety of microscopy systems and fluorescent proteins.
Abstract
Fluorescence recovery after photobleaching (FRAP) is a microscopy technique that can be used to quantify protein mobility in live cells. In a typical FRAP experiment, steady-state fluorescence is observed by repeated imaging with low-intensity laser light. Subsequently, the fluorescent molecules are rapidly and irreversibly impaired via brief exposure to high-intensity laser light. Information about protein mobility is obtained by monitoring the recovery of fluorescence. We used FRAP to determine the mobility of p62 in aggresome-like induced structures (ALIS) in murine macrophages after stimulation with lipopolysaccharide (LPS). Because many existing FRAP protocols are either incomplete or complex, our goal was to provide a comprehensive, practical, and straightforward step-by-step protocol for FRAP experiments with live cells. Here, we describe RAW264.7 macrophage transfection with yellow fluorescent protein-p62 (YFP-p62), induction of ALIS by exposing the cells to LPS, and a step-by-step method for collecting prebleach and postbleach FRAP images and data analysis. Finally, we discuss important factors to consider when conducting a FRAP experiment.
Introduction
Fluorescence recovery after photobleaching (FRAP) is a microscopy technique that can be used to quantify protein dynamics in live cells1,2. The popularity of FRAP has increased because of the widespread commercial availability of laser scanning confocal microscopes with high resolution, speed, and sensitivity, and a "rainbow" of genetically encoded fluorescent proteins, such as green fluorescent protein (GFP) and yellow fluorescent protein (YFP)3. Genetically encoded fluorescent proteins are fused to a protein of interest to allow for the subcellular localization of the protein of interest. In a typical FRAP experiment, the steady-state fluorescence in an acquisition region of interest (ROI) within a cell is observed via repeated imaging of that ROI with low-intensity laser light. Subsequently, the fluorescent molecules are rapidly and irreversibly impaired in a predefined subset of the acquisition ROI, hereafter referred to as the bleach ROI, by brief exposure to high-intensity laser light. As new unbleached proteins replenish bleached proteins over time, the speed and intensity of fluorescence recovery in the bleach ROI provides information about protein mobility (Figure 1)4.
Our interest in determining the mobility of the ubiquitin binding protein p62 (also known as sequestosome-1) in aggresome-like induced structures (ALIS) in murine RAW264.7 macrophages after stimulation with lipopolysaccharide (LPS) led us to review the FRAP literature. Unfortunately, many of the existing FRAP protocols are incomplete or inordinately complex5,6,7,8,9. Some do not provide detailed information about the laser settings, beam path configuration, and image acquisition parameters5,6,7,8,9. Others omitted key details regarding data analysis, such as how to address the issue of bleach ROI drift6,9 or how to calculate important recovery parameters, including the mobile fraction (Mf), immobile fraction (If), and half-time of recovery (t½)5,7. Conversely, others placed too much emphasis on complex mathematical formulas used to calculate Mf, If, and t1/25,6,8,9. Thus, our purpose is to provide a comprehensive, practical, and straightforward step-by-step protocol for FRAP experiments with live cells.
Protocol
1. RAW264.7 Macrophage Transfection
- Culture 100,000 RAW264.7 cells (Table of Materials) in complete culture medium (Dulbecco's Modified Eagle Medium (DMEM)) (Table of Materials) containing 4.5 g/L glucose supplemented with 10% fetal bovine serum (Table of Materials) and penicillin/streptomycin (Table of Materials) onto untreated 35-mm glass-bottom dishes (Table of Materials) and place the dishes in a 37 °C/5% CO2 incubator. On the following day, transfect the cells using 1 mg/mL polyethylenimine (PEI) (Table of Materials).
- Mix 1.5 μg of YFP-p62 with 8 μL of PEI into 166 μL of serum-free DMEM (base medium without fetal bovine serum and penicillin/streptomycin).
- Let the transfection complex mixture sit at room temperature for 15 min prior to adding the mixture into each plate.
- Add the complexes to the existing medium with cells and rock the plate gently.
- Place the plate in a 37 °C /5% CO2 incubator overnight.
- The following morning, aspirate the medium off the plate, then rinse once with complete medium. Add 2 mL of complete medium to the plate and allow the cells to recover until the next day.
2. Induction of ALIS with Lipopolysaccharide (LPS)
- The next day, aspirate the medium from the plates, then add 1 mL of complete medium containing 10 ng/mL LPS (Table of Materials). Allow the plates to incubate for 5 h in a 37 °C/5% CO2 incubator.
- After the LPS treatment, aspirate the medium, then add 1 mL of cold, sterile Tyrode's buffer containing 145 mM NaCl, 5 mM KCl, 10 mM glucose, 1.5 mM CaCl2, 1.0 mM MgCl2, and 10 mM HEPES (pH 7.4) to rinse the plate.
- Aspirate the buffer, then add 1 mL of cold, sterile Tyrode's buffer supplemented with 10 μg/mL nocodazole (Table of Materials). Allow the plates to incubate at 4 °C for 15-20 min prior to FRAP imaging and analysis.
NOTE: The use of Tyrode's buffer containing HEPES as the buffering compound allows for imaging to be performed at room temperature. Nocodazole was used to decrease ALIS movement by microtubules. Culture medium contains bicarbonate as a buffering compound requiring CO2 to buffer. Otherwise, the bicarbonate contained in medium changes pH in the absence of CO2.
3. Set-up of the Confocal Microscope and Selecting the Region of Interest
- Laser Selection and Beam Path Configuration
- Use any suitable confocal microscope (Table of Materials) equipped with AIM or ZEN software (Table of Materials) or any suitable imaging software.
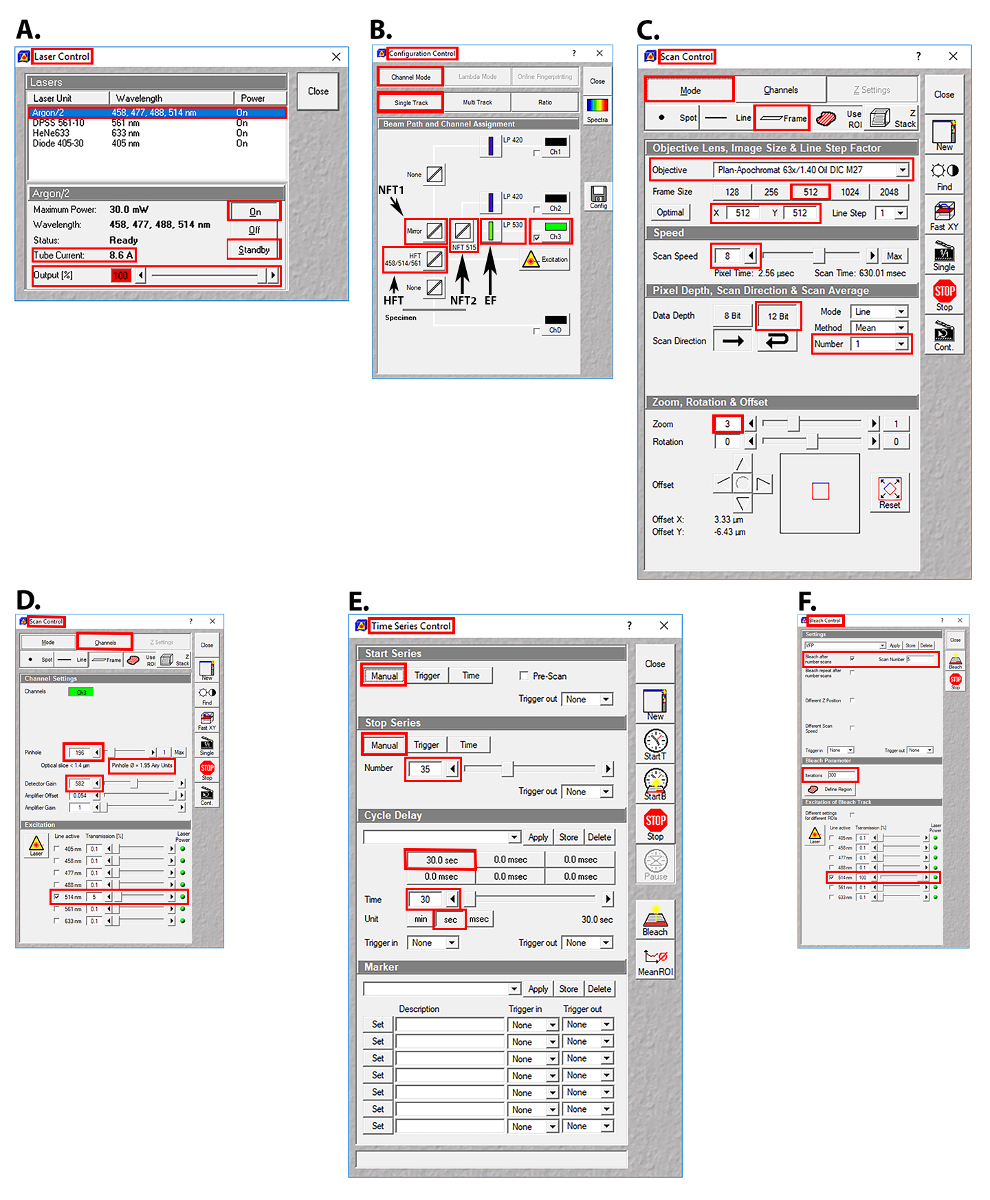
- Select the 514 nm line of the Argon/2 laser, as YFP has peak excitation at 512 nm and peak emission at 527 nm. Click the Acquire button, then click the Laser button. In the Laser Control window, click Argon/2 458, 477, 488, 514 nm, then click the Standby button. After waiting ~3 min for the laser to warm up, click the On button (Figure 2A).
- Set the laser power of the 514 nm Argon/2 laser line to 100% (8.6 A). Click the Acquire button, then click the Laser button. In the Laser Control window, enter 100 in the Output [%] field, then press the Enter button (Figure 2A).
- Set the transmission of the 514 nm Argon/2 laser line to 5%. Click the Acquire button, then the Channels button. In the Scan Control window, click the Channels button, then click the square white box to the left of the text 514 nm in the Line active column to activate the 514 nm laser line. Enter 5 in the Transmission [%] field for the 514 nm laser line (Figure 2D).
- Set the primary dichroic beam splitter (Haupt Farb Teiler (HFT)) to the HFT 458/514/561 position such that these bands are deflected to the specimen for excitation. Click the Acquire button, then the Config button. In the Configuration Control window, click the Channel Mode button, then the Single Track button. Click the HFT button, then select HFT 458/514/561 from the drop-down menu (Figure 2B).
- Set the first secondary dichroic beam splitter (Neben Farb Teiler 1 (NFT1)) to the mirror position, which will deflect 100% of the light to the second secondary dichroic beam splitter (Neben Farb Teiler 2 (NFT2)). Click the Acquire button, then the Config button. In the Configuration Control window, click the Channel Mode button, then the Single Track button. Click the NFT1 button, then select Mirror from the drop-down menu (Figure 2B).
- Set the second secondary dichroic beam splitter (NFT2) to the NFT 515 position to ensure that wavelengths <515 nm will be reflected and wavelengths >515 nm will be transmitted. Click the Acquire button, then the Config button. In the Configuration Control window, click the Channel Mode button, then the Single Track button. Click the NFT2 button, then select NFT 515 from the drop-down menu (Figure 2B).
- Set the long pass (LP) emission filter (EF) to LP 530, so that wavelengths >530 nm will be transmitted to the photomultiplier tube (PMT, detector). Click the Acquire button, then the Config button. In the Configuration Control window, click the Channel Mode button, then the Single Track button. Click the Emission filter button, then select LP 530 from the drop-down menu (Figure 2B).
- Select Channel 3. Click the Acquire button, then the Config button. In the Configuration Control window, click the Channel Mode button, then the Single Track button. Click on the square white box to the left of the Ch3 button.
- Image Acquisition Set-Up
- Select the Plan-Apochromat 63x/1.40 oil objective (Table of Materials). View the specimen through the microscope eyepiece and place the ALIS in the center of the field of view. Click the Acquire button, then the Micro button. In the Microscope Control window, click on Objective, then select the Plan-Apochromat 63x/1.40 oil objective from the drop-down menu.
- Set the frame size to 512 x 512 pixels, the scan speed to 8, and the data depth to 12 bit. Click the Acquire button, then the Scan button. In the Scan Control window, click the Mode button and the Frame button, then click on the 512 button. Enter 8 in the Scan Speed field, press Enter, then click on the 12 Bit button (Figure 2C).
- Set the scan average to 1 and the optical zoom to 3. Click the Acquire button, then click the Scan button. In the Scan Control window, click the down arrow button of the scan average number field, then select 1 from the drop-down menu. Enter 3 in the optical zoom field, then press Enter (Figure 2C).
- Set the pinhole to 1.95 Airy units and detector gain to 582 (just below saturation). Click the Acquire button, then the Scan button. In the Scan Control window, click the Channels button, enter 196 in the Pinhole field, then press Enter. Enter 582 in the Detector Gain field, then press Enter (Figure 2D).
- Ensure that the 514 nm Argon/2 laser line is set to 100% power (8.6 A). Click the Acquire button, then the Laser button. In the Laser Control menu, the value in the Output [%] field should be 100 (Figure 2A).
- Ensure that the 514 nm Argon/2 laser line is set to 5% transmission. Click the Acquire button, then the Scan button. In the Scan Control window, the value in the Transmission [%] field for the 514 nm laser line should be 5 (Figure 2D).
- Create a square-shaped ROI (acquisition ROI) that has the dimensions 150 x 150 pixels (194.6 µm2). Click the Acquire button, then the Edit ROI button. In the Edit ROI menu, click the rectangle icon, then click and drag in the image window to create a square that has the dimensions 150 x 150 pixels (194.6 µm2) (Figure 1A).
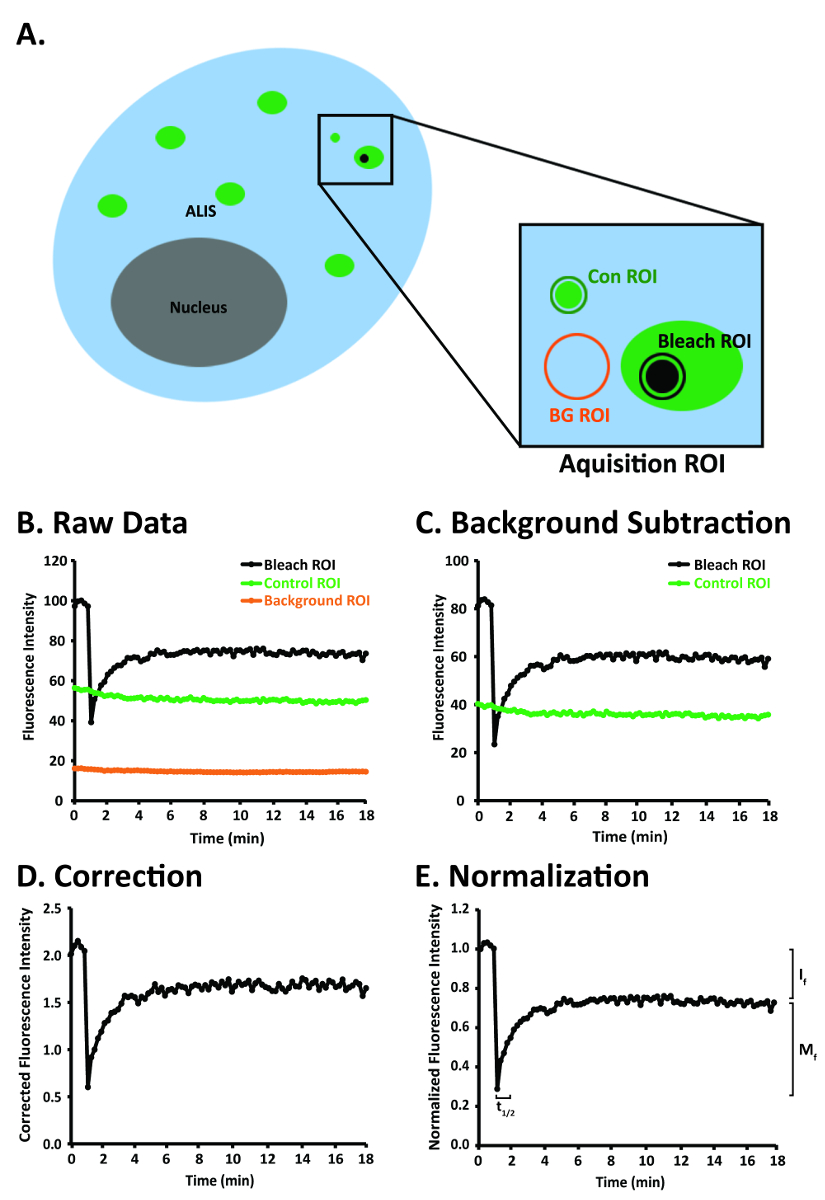
NOTE: The acquisition ROI includes (a) an ALIS of interest, (b) an area of fluorescence that is not an ALIS (control ROI) and is at least 20 x 20 pixels (3.3 µm2), and (c) an area of little to no fluorescence (background ROI) that is at least 20 x 20 pixels (3.3 µm2) (Figure 1A). It is important to include a control ROI in the acquisition ROI so that the photobleaching that may occur with repeated imaging can be controlled. The acquisition ROI that is defined here will be the only area that will be scanned. Repeated laser scanning within an acquisition ROI only will restrict photobleaching to the ROI rather than, for example, photobleaching the entire cell or several cells, and the number of pixels collected at the PMT (detector) will be greater than the number of pixels collected at the PMT (detector) after scanning an entire cell or multiple cells. - Set-up the time series such that the acquisition ROI is scanned 35 times, once every 30 s. Click the Acquire button, then the Time Series button. In the Time Series Control window, click the Manual start series button and the Manual stop series button. Enter 35 in the manual stop series field, press the Enter key, then click the 30.0 sec cycle delay button (Figure 2E).
- Set the bleach control such that the photobleaching will occur after scan number 5, to collect 5 prebleach images of the acquisition ROI. Click the Acquire button, then the Edit Bleach button. In the Bleach Control window, click the white square next to the Bleach after number scans. Enter 5 in the scan number field, then press the Enter key (Figure 2F).
NOTE: Prebleach and postbleach images should be acquired at the same optical depth. - Create a circular-shaped bleach ROI within the ALIS that has a 10-pixel diameter (area = 0.8 µm2). Click Acquire | Edit Bleach | Define Regions. In the Bleach Regions window, click the Circle icon. Click and drag in the image window to create a circle that has a 10-pixel diameter (area = 0.8 µm2) (Figure 2F).
- Set iterations to 300. Click the Acquire button, then the Edit Bleach button. In the Bleach Control window, enter 300 in the Iterations field, then press the Enter key (Figure 2F).
- Set the Argon/2 514-nm laser line to 100% power (8.6 A) and 100% transmission during the bleach. Click the Acquire button, then the Edit Bleach button. In the Bleach Control window, click the white square box to the left of 514 nm. Enter 100 in the Transmission [%] field for the 514 nm laser line, then press the Enter button (Figure 2F).
NOTE: The number of iterations needs to be determined empirically. It will vary depending on the fluorophore, the size of the structure that will be bleached, and the laser.
- Data Collection
- Start the FRAP experiment. Collect the first 5 prebleach images and the first postbleach image, then calculate bleach depth according to the following equation.

If bleach depth is <90, stop the experiment and discard the data, as the bleach depth was not sufficient. When bleach depth is ≥90, collect data for analysis with an image processing program and a spreadsheet program (Table of Materials).
NOTE: When drift of the ALIS is ≥3 µm, stop the experiment and discard the data, as this amount of image drift cannot be corrected for by analysis with image processing software (Table of Materials). When drift of the ALIS is <3 µm, collect the data for data analysis with an image processing program and a spreadsheet program (Table of Materials). - Collect FRAP data from 10 ALIS from 10 cells with less than 3 µm of drift of the ALIS. Next, transfer the AIM .lsm files to a personal computer for data analysis.
- Start the FRAP experiment. Collect the first 5 prebleach images and the first postbleach image, then calculate bleach depth according to the following equation.
- Data Analysis in an Image Processing Program
- Correct for image drift by aligning or matching (i.e., registering) the stack of time-series images of the acquisition ROI. To do so, open each AIM .lsm file with an image processing program (Table of Materials), then select Plugins | Registration | StackReg | Translation. Next, select Plugins | Registration | StackReg | Rigid Body10.
- Set-up ROI Manager to measure the signal intensity in the bleach ROI. Select Analyze | Tools | ROI Manager. Select the Oval tool, then draw a circle in the bleach ROI with a diameter of 10 pixels, then click the Add button.
- Set-up ROI Manager to measure signal intensity in the control ROI. In ROI Manager, select the Rectangle tool, then draw a square of 20 x 20 pixels in the control ROI region, then click the Add button.
- Set-up ROI Manager to measure signal intensity in the background ROI. In ROI Manager, select the Rectangle tool, draw a square of 20 x 20 pixels in the background ROI region, then click the Add button.
- Rename the ROIs. After adding the ROIs to be analyzed in ROI Manager, rename them Bleach ROI, Control ROI, and Background ROI accordingly.
- Measure signal intensity in the ROIs. Select Bleach ROI, Control ROI, and Background ROI, then select More | Multi Measure. Ensure that Measure all 35 slices and One row per slice are selected. In the Set Measurements window, ensure that only Mean gray value is selected.
NOTE: These are the raw FRAP data (Figure 1B). - Paste the results into a spreadsheet program (Table of Materials). Copy the bleach ROI, control ROI, and background ROI signal intensity results then paste them to columns labeled Bleach ROI, Control ROI, and Background ROI, respectively in a spreadsheet program (Table of Materials).
- Data Analysis in a Spreadsheet Program
- Background-correct the signal intensity in the bleach ROI and control ROI (Figure 1A, C). Use the Insert Function tool in a spreadsheet program (Table of Materials) to subtract the values in the column labeled Background ROI from the values in the column labeled Bleach ROI. Subtract the values in the column labeled Background ROI from the values in the column labeled Control ROI. Label these new columns Corrected Bleach ROI and Corrected Control ROI.
- Normalize the signal in the bleach ROI to the background-corrected signal in the control ROI (Figure 1A, D). Use the Insert function in a spreadsheet program (Table of Materials) to divide the values in the column labeled Corrected Bleach ROI by the values in the column labeled Corrected Control ROI. Label this new column Normalized Corrected Bleach ROI.
- Normalize the signal in the Normalized Corrected Bleach ROI column to the average of the 5 prebleach values in the bleach ROI (Figure 1A, E). Calculate the average of the 5 prebleach values in the bleach ROI. Next, divide the values in the column labeled Normalized Corrected Bleach ROI by the average of the 5 prebleach values in the bleach ROI. Label this new column Normalized Corrected Prebleach Average Bleach ROI.
- Mobile Fraction and Half Time of Recovery from Curve-Fitted Data
- Curve-fit the normalized and corrected bleach ROI data using an image processing program (Table of Materials). In the imaging processing program (Table of Materials), select Analyze | Tools | Curve Fitting. Copy the postbleach normalized and corrected bleach ROI values and the corresponding time values, then paste them into the Curve Fitter window. Select Exponential Recovery from the Curve Fitter drop-down menu, then select Fit.
- Calculate the Mf from the parameters of the recovery function, which provides values for: 'a,' a slowly recovering fraction; 'b,' the recovery rate; and 'c,' a rapidly diffusing fraction. Calculate Mf by plugging in the values for 'a' and 'c' into the equation Mf = a + c. Calculate the If using the equation If = 1 - Mf. Calculate t½ by substituting the value for 'b' into the equation
 then solving for t1/2.11
then solving for t1/2.11
Results
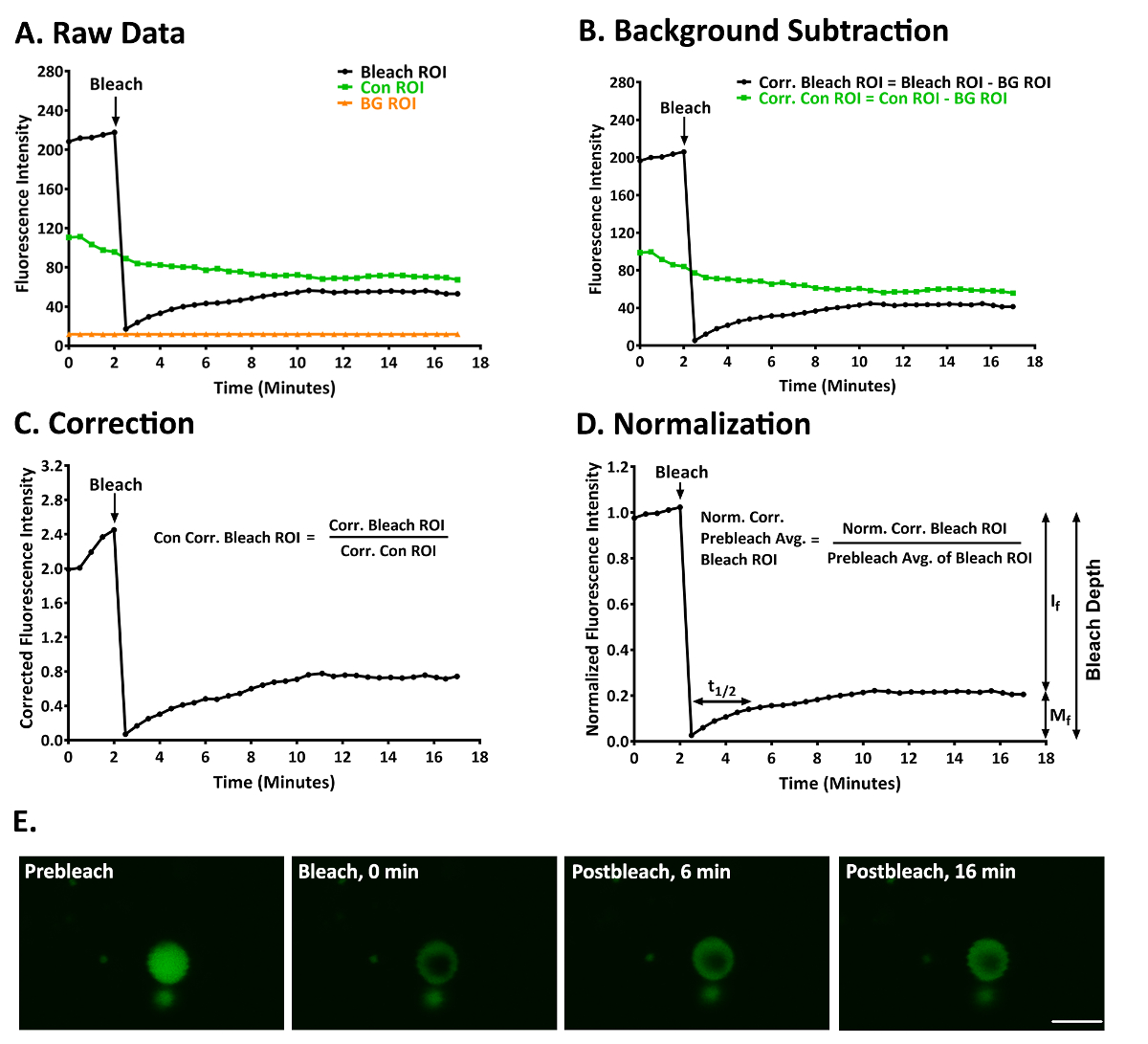
Shown here are the results of a typical experiment in which we used FRAP to examine the degree of mobility of p62 in ALIS in RAW264.7 cells treated with LPS for 3-5 h12. Figure 3A shows the raw data obtained from a bleach, control, and background ROI after the stack of images had been aligned to correct for small amounts (< ~3 µm) of image drift of the ALIS. YFP-p62 fluorescence in this ALIS at prebleach and postbleach is shown in Figure 3E and Supplementary Video 1. Figure 3B shows these data after background-correction. Figure 3C shows these data after correcting for fluorescence in the control ROI. Figure 3D shows these data normalized to the mean fluorescence of the 5 prebleach values in the bleach ROI. The degree of bleaching was sufficient in this experiment (bleach depth = 91.94). YFP-p62 fluorescence within this ALIS recovered slowly, as t1/2 = 128.27 s (2.14 min). YFP-p62 was not a very mobile protein in this experiment, as Mf = 21.97 and If = 78.03.

Figure 1: A diagram of a RAW264.7 cell and steps for image analysis after the FRAP images have been aligned to correct for image drift of the ALIS. (A) Diagram of a RAW264.7 cell depicting several ALIS and the bleach, control, and background ROIs within the acquisition ROI. (B) An idealized depiction of raw FRAP data obtained in the bleach, control, and background ROIs in panel A. (C) An idealized graph of raw FRAP data that have been background-corrected. (D) An idealized graph of background-corrected FRAP data that have been normalized to fluorescence in the control ROI. (E) An idealized graph of normalized and background-corrected FRAP data that have been normalized to the prebleach fluorescence in the bleach ROI. Abbreviations: Con = control; BG = background. This figure was modified from Rabut and Ellenberg4. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Images of the user interface in the AIM software (Table of Materials) for laser selection, beam path configuration, and image acquisition parameters for the FRAP experiments. (A) Laser control screen showing the Argon/2 laser lines, output, and tube current used. (B) Configuration control screen showing the beam splitter and emission filter settings used. (C) Scan control screen showing the setting used for image acquisition. (D) Scan control screen showing the pinhole, detector gain, amplifier offset, amplifier gain, and transmission settings for the Argon/2 514 nm laser line. (E) Time series control screen showing the time series settings used. (F) Bleach control screen showing the prebleach and postbleach parameters used. Abbreviations: HFT = Haupt Farb Teiler (primary dichroic beam splitter); NT1 = Neben Farb Teiler 1 (first secondary dichroic beam splitter); NT2 = Neben Farb Teiler 2 (second secondary dichroic beam splitter); EF = emission filter. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: A representative FRAP experiment to study YFP-p62 dynamics in ALIS in RAW264.7 cells. (A) Raw fluorescence intensity data for a bleach, control, and background ROI. (B) The data given in panel A after the bleach and control ROI had been corrected for fluorescence in the background ROI. (C) The data given in panels A and B after normalizing the fluorescence intensity in the bleach ROI to account for fluorescence in the background-corrected control ROI. (D) The data given in panels A-C after normalizing the fluorescence intensity in the normalized and background-corrected bleach ROI to the average of the first 5 prebleach values in the bleach ROI. Mf = 21.97, If = 78.03, t1/2 = 128.27 s (2.14 min), and bleach depth = 91.94. (E) An image of an ALIS at prebleach (1.5 min) and postbleach (0, 6, and 16 min). Scale bar = 5 µm and is valid for all panels. Abbreviations: Con = control; BG = background; Corr. = corrected; Norm. = normalized; Avg. = average. This figure was modified from Cabe et al.12 Please click here to view a larger version of this figure.
{kind=link}

Supplementary Video 1: A typical ALIS in a RAW264.7 macrophage before and after photobleaching. YFP-p62 fluorescence is slow to recover after the photobleach; hence, p62 is not a very mobile protein. For this experiment, Mf = 25.62, If = 74.38, t1/2 = 442.79 s (7.38 min), and bleach depth = 90.19. Scale bar = 5 µm. This video was modified from Cabe et al.12 Please click here to view this video. (Right-click to download.)
Discussion
We provide a comprehensive, practical, and simple step-by-step protocol for FRAP experiments with live cells. Herein, the protocol was used to measure the mobility of YFP-p62 in ALIS in RAW264.7 macrophages, but it can be applied to many of the laser scanning confocal microscope systems and genetically encoded fluorescent proteins that are now available. For any microscopy system, pilot experiments are critical for determining the optimal FRAP parameters, including acquisition, bleach, and control ROI sizes, laser intensity for photobleaching, and prebleach and postbleach image acquisition. It is reasonable to expect the optimal FRAP parameters to differ for each genetically encoded fluorescent protein and cell line.
Important factors to consider when conducting a FRAP experiment include (a) achieving suitable bleach depth, (b) the use of a brief bleaching step, (c) allowing sufficient time postbleach to observe the full recovery function, (d) photobleaching efficiency, (e) cytotoxicity with repeated FRAP, and (f) the inclusion of a control for fluorescence loss due to repeated imaging. We recommend that bleach depth, which can be calculated according to the equation provided in section 3.3.1, be ≥9013. When bleach depth is <90, the degree of postbleach fluorescence recovery will be underestimated, and the values of If, Mf, and t1/2 will be incorrect. Although the duration and intensity of the bleach-inducing laser pulses may vary between FRAP experiments, it is important that the photobleaching step be brief and substantially faster than the fluorescence recovery function. If it is not, then a significant amount of fluorescence recovery could occur during the bleaching step. With a long bleach time, fluorescence recovery during the bleaching step would not be measured, and it would lead to incorrect measurements of If, Mf, and t1/2. In addition, to obtain correct values for If, Mf, and t1/2, the acquisition ROI should be observed postbleach until the fluorescence level in the bleach ROI has reached a plateau. For example, in our FRAP experiments, there was no difference between the If, Mf, and t1/2 values when we observed YFP-p62 fluorescence in the bleach ROI for 32.2 min postbleach versus when we observed YFP-p62 in the bleach ROI for 15.1 min postbleach; thus, we concluded that the recovery function reached a plateau at 15.1 min postbleach12. With regard to photobleaching efficiency, photobleaching increases with the square of the optical zoom factor14. Thus, the use of high optical zoom lenses is favorable for rapid photobleaching but can result in undesirable photobleaching during acquisition; the latter can be accounted for by imaging a control ROI. Repeated photobleaching is to be avoided as it can lead to the generation of cytotoxic reactive oxygen species (ROS). However, the degree of ROS generation due to exposure to a high-intensity laser is lower for genetically encoded fluorescent proteins than for chemical fluorophores (e.g., fluorescent antibodies)15, and the ROS generated are more likely to react within the genetically encoded fluorescent protein than with other molecules in the cell4. In addition to the increased probability of generating cytotoxic ROS, repeated photobleaching is to be avoided as it is difficult to control. Finally, although low laser transmission is used to acquire all non-bleach images, some photobleaching will invariably occur, which must be controlled. Possible controls for this include monitoring fluorescence in a control ROI within the acquisition ROI, obtaining control images in a neighboring unbleached cell, and performing control experiments with the identical settings for those used in the photobleaching experiments but without the photobleaching event.
Disclosures
The authors have no conflict of interest to disclose.
Acknowledgements
We thank Dr. Seth Robia at Loyola University Chicago for his valuable comments on this manuscript. This work was supported by NIH grant 1R01NS073967-01A1 to Joanna C. Bakowska.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco's Modified Eagle Medium | Hyclone | SH30022.01 | High Glucose (4.5 g/L) |
| Fetal bovine serum | Gemini Bio-Products | 100-106 | |
| Glass bottom dishes | MatTek | P35G-1.5-14-C | 35 mm |
| ImageJ software | National Institutes of Health | N.A. | |
| Lipopolysaccharide (LPS) | Sigma | L4391 | |
| Nocodazole | Sigma | M1404 | |
| Penicillin-streptomycin solution | Corning | 30-002-Cl | 100× |
| Plan-Apochromat 63×/1.40 Oil objective | Carl Zeiss MicroImaging, Inc | 4407629904000000 | |
| Polyethylenimine (PEI) | Polysciences | 23966-1 | linear (MW 25,000) |
| RAW 264.7 murine macrophages | ATCC | TIB-71 | |
| Zeiss confocal microscope | Carl Zeiss MicroImaging, Inc | LSM 510 |
References
- Axelrod, D., Koppel, D. E., Schlessinger, J., Elson, E., Webb, W. W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophysical Journal. 16 (9), 1055-1069 (1976).
- Peters, R., Peters, J., Tews, K. H., Bahr, W. A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochimica et Biophysica Acta. 367 (3), 282-294 (1974).
- Patterson, G., Day, R. N., Piston, D. Fluorescent protein spectra. Journal of Cell Science. 114, 837-838 (2001).
- Rabut, G., Ellenberg, J. Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP, and FLIP. Live Cell Imaging: A Laboratory Manual (1st ed). , 101-126 (2004).
- Fritzsche, M., Charras, G. Dissecting protein reaction dynamics in living cells by fluorescence recovery after photobleaching. Nature Protocols. 10 (5), 660-680 (2015).
- Goodwin, J. S., Kentworthy, A. K. Photobleaching approaches to investigate diffusional mobility and trafficking of Ras in living cells. Methods. 37 (2), 154-164 (2005).
- Hildick, K. L., Gonzàlez-Gonzàlez, I. M., Jaskolski, F., Henley, J. M. Lateral diffusion and exocytosis of membrane proteins in cultured neurons assessed using fluorescence recovery and fluorescence-loss photobleaching. Journal of Visualized Experiments. (60), e3747 (2012).
- Nissim-Rafinia, M., Meshorer, E. Photobleaching assays (FRAP & FLIP) to measure chromatin protein dynamics in living embryonic stem cells. Journal of Visualized Experiments. (52), e2696 (2011).
- Zheng, C., Petralia, R. S., Wang, Y., Kachar, B. Fluorescence recovery after photobleaching (FRAP) of fluorescence tagged proteins in dendritic spines of cultured hippocampal neurons. Journal of Visualized Experiments. (50), e2568 (2011).
- Thevenaz, P., Ruttimann, U. E., Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Transactions on Image Processing. 7 (1), 27-41 (1998).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Cabe, M., Rademacher, D. J., Karlsson, A. B., Cherukuri, S., Bakowska, J. C. PB1 and UBA domains of p62 are essential for aggresome-like induced structure formation. Biochemical and Biophysical Research Communications. 503 (4), 2306-2311 (2018).
- Brown, E. B., Wu, E. S., Zipfel, W., Webb, W. W. Measurement of molecular diffusion in solution by multiphoton fluorescence photobleaching recovery. Biophysical Journal. 77 (5), 2837-2849 (1999).
- Centonze, V., Pawley, J. Tutorial on practical confocal microscopy and the use of the confocal test specimen. Handbook of biological confocal microscopy. , 549-570 (1995).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiological Reviews. 90 (3), 1103-1163 (2010).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved