
Method Article
Predicting In Vivo Payloads Delivery using a Blood-brain Tumor-barrier in a Dish
In This Article
Summary
Drug targeting to central nervous system tumors is a major challenge. Here we describe a protocol to produce an in vitro mimic of the blood-brain tumor-barrier using murine and/or human cells and discuss their relevance for the predictability of central nervous system tumor targeting in vivo.
Abstract
Highly selective by nature, the blood-brain barrier (BBB) is essential for the brain homeostasis in physiological conditions. However, in the context of brain tumors, the molecular selectivity of BBB also shields the neoplastic cells by blocking the delivery of peripherally administered chemotherapies. The development of novel drugs (including nanoparticles) targeting malignant brain tumors ideally requires the use of preclinical animal models to study the drug’s transcytosis and antitumor efficacy. In order to comply with the 3R principle (refine, reduce, and replace) to reduce the number of laboratory animals in experimental setup and perform the high-throughput screening of a large library of antitumor agents, we developed a reproducible in vitro human and murine mimic of the blood-brain tumor-barrier (BBTB) using three-layered cultures of endothelial cells, astrocytes, and patient-derived glioblastoma spheres. For higher scalability and reproducibility, commercial cell lines or immortalized cells have been used in tailored conditions to allow the formation of a barrier resembling the actual BBB. Here we describe a protocol to obtain a BBTB mimic by culturing endothelial cells in contact with astrocytes at specific cell densities on inserts. This BBTB mimic can be used, for instance, for the quantification and confocal imaging of the nanoparticle passage through the endothelial and astrocytic barriers, in addition to the evaluation of the tumor cell targeting within the same assay. Moreover, we show that the obtained data can be used to predict the behavior of nanoparticles in preclinical animal models. In a broader perspective, this in vitro model could be adapted to other neurodegenerative diseases for the determination of the passage of new therapeutic molecules through the BBB and/or be supplemented with brain organoids to directly evaluate the efficacy of drugs.
Introduction
The neurovascular unit is composed of neurons, astrocytes, and the BBB, formed by intricate connections between pericytes, astrocytes, endothelial cells, and the associated basement membrane forming the brain microvasculature1. This tight cellular wall formed by continuous, nonfenestrated vessels finely regulates the movement of ions and molecules (including hormones, nutrients, or drugs) but also of circulating cells1. The notably low transcytosis through the BBB of high-molecular-weight molecules, such as therapeutic antibodies, drug conjugates, or nanocompounds, dramatically restricts the advances in the drug discovery for neurological diseases, including malignant gliomas2. Indeed, orally or intravenously delivered chemotherapies reach the brain parenchyma often at inadequately low concentrations to induce an antitumor effect or are simply unable to cross the BBTB to reach the neoplastic cells3. Several preclinical and clinical studies have not dealt with the issue of BBTB penetration but have attempted to disrupt the BBTB transiently, for instance by using focused ultrasounds4,5, or to circumvent it by the direct in situ delivery of drugs6. However, none of these techniques were able to counteract the inevitable tumor expansion or relapse. Therefore, when developing novel antiglioma therapies, the diffusion through the BBTB should be considered as one of the critical aspects for the successful delivery of the therapeutic agents7.
Due to the very complex nature of the cell interactions within the BBTB, in vivo studies in laboratory animals seem to be the obvious choice when studying the passage of molecules from blood to brain. However, high-scale in vivo methods are relatively complex to establish and, therefore, do not allow the high-throughput screening of molecules in a reasonable time at a reasonable cost. Even more importantly, animal testing has to follow the 3R ethical guideline defined as i) refine, ii) reduce, and, of relevance to the current context, iii) replace by alternative protocols (e.g., in vitro/in silico methods). Therefore, recreating the BBTB in vitro appears as an interesting and attractive possibility, but it also constitutes a complex task challenged by various limitations. Many attempts to recreate this complex compartment with cultured primary cells or cell lines from canine, porcine, murine, and even human origin have been published (as reviewed by Rahman et al.8 and Helms et al.9). These models include three-dimensional microfluidic systems10, BBB-on-a-chip11,12, and multitudes of variants based on the classical co-cultures in inserts systems. Nevertheless, current microfluidic and chip systems are either not suitable for rapid, high-throughput drug-validation studies13,14 or are currently incompatible with studies of drug delivery to brain tumors. In addition, the review of 155 published models using primary cells, inducible pluripotent stem cells (iPSC), or commercial cell lines all co-cultured on inserts showed a trend for interstudy discrepancy in their measurements and/or conclusions8. This lack of interlaboratory reproducibility could be correlated with i) nonnormalized culture conditions, for instance with the optional coating with basement membrane matrix proteins in the cell culture vessel, ii) an increased number of subculture and usage of serum-containing media, both major drivers of genetic and phenotypic modifications of the cell lines15, or iii) the difficulty to reproducibly recreate the right equilibrium between the astroglial and endothelial components in a dish. Although the use of immortalized cells or commercial cell lines to establish an in vitro BBB model lacks some of the properties compared to similar models that only use primary cells, in the described method we show that the right combination of cells exhibits a very comparable performance to published studies in other models of reference16,17. Eventually, the lack of a robust and reproducible model to study the passage of therapeutic compounds targeting brain tumors through the BBTB motivated us to develop the methods described here.
Since the goal was to use the model to predict the in vivo delivery of nanoparticles in preclinical animal models, we first validated the BBTB model by utilizing inserts containing murine endothelial cells in contact with murine astrocytes. In addition to this, we also optimized the model to use certain human cell lines. Once stabilized, the cell barriers are transferred to cultures with patient-derived glioblastoma spheres or commercial glioma cell lines. Thereafter, the transcytosis of nanoparticles and tumor cell targeting can be visualized by confocal microscopy and quantified by collecting samples over time. Importantly, results obtained using the BBTB mimics could reliably predict the behavior of the nanoparticles in vivo, supporting the use of the BBTB mimic prior the preclinical validation.
Protocol
The animal experiments were approved by the Committee for Animal Experiments of the District of Southern Finland (ESAVI/6285/04.10.07/2014).
1. Establishment of the BBTB Mimics
NOTE: Cell culture medium and supplements are detailed in the Table of materials.
- Preparation of astrocytes
NOTE: The following volumes are suitable for a 10 cm Petri dish or a T75 cell culture flask.- Under a sterile cell culture hood, carefully wash the cultured astrocytes with 5 mL of sterile phosphate-buffered saline (PBS). Gently discard the PBS using a vacuum pump and add 2 mL of the cell dissociation reagent for 5 min (at 37 °C, see the Table of Materials) to detach the cells. Check the cell detachment under the microscope. Do not exceed 5 min of incubation to limit the stress on the cells.
- Add 10 mL of sterile complete astrocyte cell culture medium (ABM+) to the vessel to inhibit the activity of the cell dissociation reagent. Use a sterile serological pipet to transfer the detached cells from the vessel to a sterile 15 mL tube. Centrifuge the cell suspension for 3 min at 250 rcf (acceleration: 9 rcf/s, deceleration: 5 rcf/s) at room temperature (RT).
- Meanwhile, prepare the inserts (see the Table of Materials): using sterile forceps, place the inserts with the brain side up (Figure 1A) on the lid of a sterile 6-well plate (Figure 1B). Verify beforehand that the plate can be placed upside down on the inserts without touching or moving the inserts during the process.
NOTE: The proper placement of the inserts allows the entrapment of the astrocyte suspension in-between the membrane and the bottom of the well. - Once centrifuged, carefully discard the supernatant from the cell suspension; resuspend the astrocyte pellet in 1 mL of ABM+ by gently resuspending the pellet on the tube’s wall up to 5x. Avoid excessive pipetting of the cells to limit the stress on the cells. Count the cells and adjust the cell suspension density to 1.5 x 105 cells in 400 µL of ABM+/insert.
- Place the cell suspension in the middle of the brain-side of the insert’s membrane (Figure 1B) and, very carefully, spread it by using capillary force with a sterile pipet tip. Avoid direct contact as the membrane is particularly fragile.
- With the brain side of the inserts still up, place the 6-well plate back on the inserts. This ensures that the cell suspension is trapped between the membrane and the actual bottom of the well (Figure 1C). Avoid air bubbles in the cell suspension, as it will prevent the homogeneous spread of the astrocytes on the membrane.
- Place the plate and inserts, with the brain side up, in the incubator (at 37 °C with 5% CO2) to allow the cell adhesion for a minimum of 2 h (murine immortalized astrocytes) and up to 6 h (human primary astrocytes).
NOTE: As the inserts are maintained upside down, visualization of the cell adhesion is not possible under a microscope. It is, therefore, recommended to seed a separate regular cell culture vessel and control the cell adhesion in the vessel over time. Careful manipulation of the membrane is a must as the results will be unreliable when membranes are damaged. - At the end of the incubation time, verify the absence of cell suspension leaks outside of the seeding area, and discard the inserts if they are leaky. Revert the 6-well plate to its regular position, with inserts that will now have the blood side up (Figure 1A). Add 2.6 mL of ABM+ to each well. Pour 2.5 mL of complete astrocyte medium into each insert and place the plate in the incubator (at 37 °C with 5% CO2).
- Preparation of endothelial cells
NOTE: For the murine brain microvascular endothelial cells (bEND3), the cells must reach 100% confluence to ensure maximal cell-cell contacts triggering the optimal tight junction protein expression on the day of the experiment. This does not apply for the human umbilical vein endothelial cells (HuAR2T) as the presence of astrocytes is required for a tight junction protein expression for these cells.- Proceed as previously described for the astrocytes (steps 1.1.1 and 1.1.2.). Once centrifuged, carefully discard the supernatant; resuspend the endothelial cell pellet in 1 mL of complete endothelial cell culture medium (EBM+) by slowly pipetting the cell suspension on the tube’s wall up to 5x. Avoid excessive pipetting of the cells to limit the stress on the cells. Count the cells and adjust the cell suspension density to 2 x 105 cells in 2.5 mL/insert of endothelial cell culture medium devoid of serum (EBM-) and vascular endothelial growth factor-A (VEGF-A).
- Take out the plate containing the inserts, carefully discard the medium from the blood side, and replace it with 2.5 mL of the endothelial cell suspension. Return the plate to the incubator (at 37 °C with 5% CO2) and leave it overnight for the endothelial cells to adhere to the membrane.
- The next day, prepare a sterile 6-well plate by transferring 3 mL of prewarmed serum-free astrocyte medium (ABM-) to each well. By handling the inserts with sterile forceps, carefully discard the endothelial complete medium from the blood side, place the insert in the new plate containing ABM-, and add 2.5 mL of EBM-.
NOTE: The use of EBM- is critical for the establishment of the endothelial barrier (please refer to the discussion section). - Leave the inserts in the incubator (at 37 °C with 5% CO2) with minimal physical disturbance and temperature variations for 5 days, allowing the production of the endothelial basement membrane, astrocyte contacts with endothelial cells, and eventually, the BBTB mimic formation. Replace the medium on the day of the transfer on glioma cell cultures (please refer to section 1.4).
- Measurement of the BBTB mimic permeability (optional)

- Passive diffusion of the small-molecular-weight fluorescent dye sodium-fluorescein (Na-Fl) over time from the blood to the brain side of the inserts allows the calculation of the permeability values according to the following formula:
Here, dFwell is the fluorescence value measured in the well at a certain time point minus the cell culture medium autofluorescence value, dT is the time in seconds, A is the surface of the barrier in square centimeters, and dFinsert is the fluorescence value measured in the insert at the same time point minus the medium autofluorescence value). - Collect 100 µL of the medium from both the blood and the brain sides of the BBTB mimic and transfer each of them to a separate flat-bottomed, black 96-well plate for subsequent fluorescence measurements. Use the plain media as the blank to correct the autofluorescence.
- Prepare 2.5 mL per well of the Na-Fl (50 µM) in EBM-. Prewarm the Na-Fl solution to 37 °C. Replace the media from the blood side of the inserts with the media containing Na-Fl. Start a timer as soon as the medium is replaced.
- Carefully collect 100 µL of media from both the blood and the brain side of the inserts at 5, 30, 60, and 120 min. Transfer each sample to separate wells of the black 96-well plate.
- Accordingly, replace the collected media from the inserts to maintain the volume balance between both sides. Place the inserts back in the incubator between each sample collection to minimize the temperature variations.
- Quantify the fluorescence from the collected samples, using a plate reader with the filter set on 480/560 nm (excitation and emission, respectively).
NOTE: Fluorescence from the brain side is nearly undetectable at the 5 min time point. High values compared to the blank indicate a leak of/damage to the insert’s membrane or the barrier; therefore, exclude these from further analyses. Expected Na-Fl permeability values for the BBTB should be in the 10-5 to 10-6 cm/s range (Table 1).
- Passive diffusion of the small-molecular-weight fluorescent dye sodium-fluorescein (Na-Fl) over time from the blood to the brain side of the inserts allows the calculation of the permeability values according to the following formula:
- Preparation of glioma cells
NOTE: Although patient-derived glioblastoma spheres are used here, the following protocol can be easily adapted for adherent, commercially available glioblastoma cells such as U-87MG.- Optionally, for immunofluorescence imaging, place up to four round sterile borosilicate coverslips (ø 0.9 cm) per well in a 6-well plate containing 2 mL of poly-D-lysine (0.01%). Incubate at room temperature for 30 min.
- Meanwhile, carefully transfer the tumor spheres from the cell culture vessel to a 15 mL sterile tube using a sterile serological pipet. Centrifuge the tumor spheres for 3 min at 250 rcf.
- Discard the supernatant, gently resuspend the spheres in 1 mL of bFGF/EGF-free (GBM-) glioma cell medium and count the cells. Adjust the cell density to approximately 104 spheres/mL (105 cells/mL) in GBM-.
- Discard the poly-D-lysine from the wells and rinse them 3x with sterile PBS. Seed the plate with 3 mL/well of the tumor spheroid suspension and transfer the inserts with the BBB mimic on the tumor cell suspension.
- Incubate overnight (at 37 °C with 5% CO2) to allow equilibrium between the blood and the brain tumor sides of the assay. On the next day, replace the media in the blood side with EBM- supplemented with the molecules/drugs/nanoparticles of interest. Samples are collected over time for direct quantification as described in the previous section. Cells are fixed at a precise time point for fluorescence imaging (please refer to the sections 2.1 and 2.2).
2. High-resolution Confocal Imaging of the BBTB
NOTE: 4% paraformaldehyde (PFA, pH 7.4, 6 mL per BBTB replicate) is always prepared fresh in PBS. Keep it on ice.
CAUTION: PFA is carcinogenic. Use nitrile gloves to handle PFA and prepare the solution under a chemical fume hood.
- BBTB endothelial expression of tight junction proteins
- Rinse both sides of the membrane with ice-cold PBS (3x for 5 min, 2.5 mL/insert, 3 mL/well). Discard the PBS and add 3 mL and 2.5 mL of ice-cold 4% PFA in the well and in the insert, respectively. Incubate on ice for 10 min. Discard the PFA (according to the institution’s hazardous chemical disposal) and rinse 3x with PBS at RT (2.5 mL/insert, 3 mL/well).
NOTE: Once fixed, samples can be stored in PBS (2.5 mL/insert, 3 mL/well) at 4 °C for a week. - Use a cotton swab to wipe the brain side of the insert and remove the astrocytes. Using a sharp scalpel, carefully cut the membrane into four equal pieces by making two perpendicular cuts, forming a cross. Next, insert the scalpel at the point where the membrane is attached to the insert wall and rotate the insert with the other hand to liberate the four samples. Using fine tweezers, carefully transfer each sample to a 24-well plate containing 200 µL of PBS/well, with the blood side up in each well.
- Block the membranes with 10% fetal bovine serum in PBS (for 30 min at RT, 200 µL/well). Prepare the 1° antibody solution for the immunostaining of the tight junction proteins (Figure 1D) (zonula occludens-1, claudin-5; please refer to the Table of Materials) in 200 µL of blocking solution/well. Optionally, endothelial cell identity is verified by adding an antibody raised against the platelet endothelial cell adhesion molecule (PECAM1 or CD31; please refer to the Table of Materials) to each tight junction antibody solution. Discard the blocking solution and incubate with the primary antibodies’ O/N at 4 °C.
- On the next day, discard the primary antibodies and rinse them with 200 µL of PBS (3x for 5 min at RT). Incubate them with appropriate species-specific fluorophore-conjugated secondary antibodies (1:500 dilution, 200 µL/well, diluted in blocking solution; please refer to the Table of Materials) for 2 h at RT.
- Discard the secondary antibodies, rinse with 200 µL of PBS (3x for 5 min at RT). Remove the PBS and counterstain the cell nuclei by using a 4',6-diamidino-2-phenylindole (DAPI) solution at a final concentration of 1 µg/mL in pure distilled H2O (dH2O; 200 µL/well; please refer to the Table of Materials). Incubate for 7 min at RT. Remove the DAPI and wash the membranes 3x with dH2O (200 µL/well).
- Place a drop of the mounting medium (see the Table of Materials) on a glass microscope slide. Using fine tweezers, carefully take the membrane out of the well, and keeping the orientation, remove the excess of dH2O and place it on the drop of the mounting medium. Add another drop of mounting medium on top of the membrane and carefully cover it with a borosilicate cover glass. Ensure that there are no entrapped air bubbles. Store the samples at 4 °C and away from light until confocal microscopy observations.
NOTE: Astrocyte staining can be performed by placing the pieces of the membranes in the 24-well plate with the brain side up, and with the use of selected astrocyte-specific antibodies (e.g., directed against the glial fibrillary acid protein [GFAP]) (Figure 1E).
- Rinse both sides of the membrane with ice-cold PBS (3x for 5 min, 2.5 mL/insert, 3 mL/well). Discard the PBS and add 3 mL and 2.5 mL of ice-cold 4% PFA in the well and in the insert, respectively. Incubate on ice for 10 min. Discard the PFA (according to the institution’s hazardous chemical disposal) and rinse 3x with PBS at RT (2.5 mL/insert, 3 mL/well).
- BBTB fluorescence staining to detect nanoparticle transcytosis
- Perform live-cell lysosome labeling (e.g., using fluorescent probes [see the Table of Materials]). Dilute the lysosome fluorescent dye at a working concentration of 50 nM in prewarmed EBM- (2.5 mL/insert) or of 75 mM in prewarmed ABM- (3 mL/well) for the lysosome labeling of endothelial cells and astrocytes, respectively. Incubate the cells for 45 min (at 37 °C with 5% CO2); then, rinse 3x with ice-cold PBS (2.5 mL/insert, 3 mL/well).
- Discard the PBS and add 3 mL and 2.5 mL of ice-cold 4% PFA to the well and to the insert, respectively. Incubate them on ice for 10 min. Discard the PFA and rinse the cells 3x with PBS (at RT, 2.5 mL/insert, 3 mL/well).
NOTE: Once fixed, the samples can be stored in PBS (2.5 mL/insert, 3 mL/well) at 4 °C for a week. - Remove the PBS and counterstain the cell nuclei by using a DAPI solution at a final concentration of 1 µg/mL in dH2O (1 mL/insert, 1 mL/well). Incubate them for 7 min at RT. Remove the DAPI and wash the membranes 3x with dH2O (2.5 mL/insert, 3 mL/well).
- Carefully cut the membrane, remove the excess of dH2O, and place it on a drop of mounting medium (see the Table of Materials) on a glass microscope slide. Add another drop of mounting medium on the other side of the membrane and carefully cover it with a borosilicate cover glass. Avoid entrapped air bubbles. Store the samples at 4 °C and keep them protected from light until confocal microscopy imaging.
- Fluorescence staining of tumor cells
- Using fine tweezers, carefully transfer the round coverslips containing the tumor spheres to a 24-well plate filled with ice-cold PBS. Proceed with live-cell lysosome labeling using fluorescent lysosome probes at 75 nM in prewarmed GBM- (200 µL/well). Incubate the samples for 45 min; then, rinse them 3x with ice-cold PBS (200 µL/well).
- Discard the PBS and add 200 µL of ice-cold PFA per well. Incubate on ice for 10 min. Discard the PFA and rinse the samples 3x with PBS (at RT).
NOTE: Once fixed, the samples can be stored in PBS (200 µL) at 4 °C for a week. - Remove the PBS and counterstain the cell nuclei by using a DAPI solution at a final concentration of 1 µg/mL in dH2O (200 µL/well). Incubate them for 7 min at RT. Remove the DAPI and wash the coverslips 3x with dH2O (200 µL/well).
- Using fine tweezers, take out the coverslip, remove the excess of dH2O, and place it on a drop of mounting medium (see the Table of Materials) on a glass microscope slide. Avoid entrapping any air bubbles. Store the samples at 4 °C and keep them protected from light until confocal microscopy observations.
3. In Vivo Comparative Study
- In situ recording of sodium-fluorescein diffusion through the BBB
- Prepare 150 µL of a 50 nM Na-Fl solution in a physiological solution. Keep the solution at 37 °C upon intravenous delivery.
- Anesthetize a mouse with an intraperitoneal injection of a ketamine/xylazine cocktail (300 µL of 100 mg/kg ketamine and 10 mg/kg xylazine in PBS). Once deep anesthesia is established, place the animal on a heating pad to maintain its body temperature.
NOTE: Ten-week-old female Naval Medical Research Institute (NMRI) nude immunocompromised mice have been used to obtain the data presented in Figure 2. However, this protocol is adapted to both immunocompetent and immunocompromised mice. The anesthesia/analgesia method is at the scientist’s discretion. However, inhalation anesthesia such as isoflurane is not recommended as it significantly increases the BBB permeability18. - Place the mouse on a stereotaxic frame (see the Table of Materials) and perform a longitudinal incision of the scalp with fine scissors, followed by gentle dilacerations of the connective tissue, using fine tweezers, to reveal the skull. Using circular movements with a fine microdrill, remove a ø 0.3 mm circular piece of the skull from the left or right parietal bone. Proceed with extreme caution during the drilling and while removing the skull piece to avoid injuring the underlying meningeal tissue and blood vessels.
- Place a drop of physiological solution on the exposed tissue. Using two pairs of fine forceps, carefully remove the meningeal tissue to access the brain cortex. The brain tissue should never be in direct contact with air.
NOTE: Minor hemorrhages from meningeal injury can be stopped using hematologic sponges (please refer to the Table of Materials). - Once the meningeal tissue is removed and the cortex is fully exposed, entrap a drop of physiological solution between the cortex and a ø 0.5 mm borosilicate coverslip. Secure the observation area with a drop of cyanoacrylate glue (please refer to the Table of Materials) spread around the coverslip with a needle. Let the glue dry for 1 min.
- Prepare the implantable catheter for the tail vein injection (Figure 2A). Break the tip of a 25 G needle using Rochester-Ochsner forceps and insert the tip in a 10 cm-long PE20 polyurethane tube (please refer to the Table of Materials) (Figure 2A).
- Insert the catheter into the lateral tail vein of the mouse, using bulldog clamps for the catheter manipulation and insertion (please refer to the Table of Materials) (Figure 2B). Secure the inserted needle with a drop of cyanoacrylate glue. Let the glue dry for 20 s before removing the bulldog clamp. Carefully connect the other end of the catheter to a 25 G needle connected to a syringe containing the Na-Fl solution (Figure 2B).
- NOTE: Do not clamp the tail with the bulldog clamp; it is only used for precise catheter handling. Proper catheter insertion can be confirmed by blood reflux into the transparent tubing.
- Place the animal under the stereomicroscope (see the Table of Materials). Using the low-level autofluorescence in the green channel (480 nm), focus on a region containing relatively large blood vessels (they appear darker due to the hemoglobin absorption of light at this wavelength) and smaller capillaries (Figure 2C). Start the time-lapse acquisition briefly before injecting the fluorescent dye to get a measurement of the background fluorescence.
NOTE: Alternatively, the time-lapse recording can be replaced by snapshot pictures from T0 and from any other predetermined time points. - Inject the solution at a slow and continuous pace, or alternatively, use an automated infusion system. The Na-Fl fluorescence detected in the blood should remain stable (half-life in blood: 286 min), which allows a recording of the BBB diffusion through the cranial window for several minutes. Once the acquisition is complete, carefully remove the catheter and euthanize the animal by cervical dislocation.
- In vivo determination of the BBB permeability
NOTE: Values are obtained from any image-processing software, such as ImageJ, allowing the measurement of the fluorescence signal intensity inside a custom region of interest (ROI).- Using the annotation tool, draw a rectangle-shaped ROI outside a blood vessel, in the brain tissue, at around 5 µm distance from any visible blood vessels filled with Na-Fl. Note the dimensions of the ROI and the measured fluorescence intensity at T0 in that ROI, which is used as blank for the tissue’s autofluorescence. Without displacing the ROI, fast-forward to a postinjection time point (e.g., to the very last recorded frame when the whole solution has been injected to the animal) and note the precise time and fluorescence values measured within the ROI (Figure 2C).
- Move the ROI on a visible blood vessel (Figure 2C) and note the T0 autofluorescence value from the blood. Without displacing the ROI, fast-forward to the same time point as defined in step 3.2.1. and note the fluorescence value measured within the ROI (Figure 2C).

- Use the following formula (adapted from section 1.3) to determine the BBB permeability:
- Here, dFbrain is the fluorescence intensity value minus the T0 blank value in the brain, dT is the acquisition time point in seconds, A is the approximate vessel surface area, taken as the ROI area in square centimeters, and dFblood is the fluorescence intensity value minus the T0 blank value in blood.
NOTE: Expected permeability values for the BBB should be in the 10-6 cm/s range (Table 1).
- Tissue processing for the detection of fluorescent nanoparticles in the murine brain
- Implant patient-derived glioblastoma spheres (5 x 104 cells in 5 µL of sterile PBS) in anesthetized 6-week-old female NMRI nude mice in the corpus callosum. Locate this brain region at the following stereotaxic coordinates, starting from the Bregma: anteroposterior + 0.5 mm, left to right + 2.5 mm, dorsoventral + 3 mm. Allow the brain tumor to grow for 2 weeks.
- Inject the nanoparticles intravenously (100 µg in 100 µL of sterile physiological solution) and allow them to circulate for 8 h. Inject the control mice intravenously with 100 µL of sterile physiological solution.
- Euthanize the mice by cervical dislocation and collect the brains rapidly for snap-freezing in isopentane maintained on dry ice (1 min at -50 °C). Store the brains at -80 °C until tissue processing.
- Cut coronal brain sections with a cryomicrotome. Locate the intracranial implantation by the scar it formed on the cortex and cut 9 µm-thick sections from that area onto the appropriate microscope slides (see the Table of Materials).
- Immerse the brain sections that are immobilized on the slides in ice-cold PBS (2x for 5 min) and, then, fix them in ice-cold 4% PFA (for 5 min). Wash the slides in PBS (3x for 5 min at RT). Place the slides horizontally and pipet the blocking solution containing 10% fetal bovine serum in PBS on the tissue sections covering the entire surface (for 1 h at RT, 500 µL/slide). Prepare the CD31 antibody (please refer to the Table of Materials) in 250 µL of blocking solution/slide. Replace the blocking solution with the antibody and incubate overnight at 4 °C in a humidified chamber.
- The next day, immerse the slides in PBS (3x for 5 min at RT) and incubate them with the corresponding fluorophore-conjugated secondary antibody (1:500 in 250 µL of PBS for 2 h at RT). Rinse 3x in PBS (at RT) and counterstain the cell nuclei by using a DAPI solution at a final concentration of 1 µg/mL in dH2O (250 µL/slide). Incubate the samples for 7 min at RT. Remove the DAPI solution and wash the slides 3x with dH2O.
- On each tissue section, add one drop of mounting medium (please refer to the Table of Materials) and secure the samples with a coverslip. Avoid entrapping air bubbles. Store the samples at 4 °C and keep them protected from light until confocal microscopy observations.
Results
Confocal imaging of the murine BBTB mimic shows the expression and cellular localization of the tight junction proteins zonula occludens-1 (ZO-1) and claudin-5 in bEND3. Contacts between the endothelial cells and astrocytes clearly induced the relocation of the ZO-1 and claudin-5 to the endothelial cell-cell contacts compared to the bEND3 monocultures (Figure 1D). Using immunofluorescence staining to visualize the GFAP-expressing astrocytes at the brain-side of the membrane, it is possible to observe and study the astrocytic processes and end-feet contacting the endothelial cells through the membrane (Figure 1E). The astrocyte-endothelial cell contacts are known to promote and stabilize the tightening of the cellular barrier and are associated with lower permeability values of the BBB19. In accordance with that, we observed a substantial decrease in the permeability of the mouse BBTB mimic for the Na-Fl from 27.63 (± 3.45) x 10-6 cm/s in case of monocultures to 6.74 (± 3.01) x 10-6 cm/s when co-cultured with the hypoxia-inducible factor knock out (HIFko) astrocytes (Table 1). The immortalized HuAR2T form highly permeable cellular barriers (104.92 ± 27.1 x 10-6 cm/s, Table 1). Similar to the murine model, we measured significantly lower permeability of the BBTB to Na-Fl, namely 47.4 (± 14.32) x 10-6 cm/s, when the HuAR2T cells were co-cultured with the human primary astrocytes (Table 1).
In both murine and human BBTB mimics, the presence of the patient-derived glioblastoma spheres induced a slight increase in the permeability values compared to the endothelial cell-astrocyte co-cultures alone (Table 1). This phenomenon is observed with several but not all of the patient glioma sphere models. This may be due to the VEGF-A that is secreted by some of these patient-derived cells.
To compare the permeability values of the in vitro BBTB mimics with the in vivo BBB, we imaged the real-time diffusion of the Na-Fl through a cranial window implanted in nude mice. Using a fluorescence stereomicroscope, Na-Fl diffusion from the blood vessel capillaries deriving from the main pial blood vessels was recorded before, during, and after systemic injection of the probe (Figure 2C). Measurements of the differential fluorescence values from the circulating blood and brain cortical parenchyma over time allowed us to calculate the approximate permeability values of the nude mouse’s BBB for Na-Fl (5.57 ± 2.19 x 10-6 cm/s, Table 1).
To illustrate how this BBTB mimic can be used for visualization of the passage of compounds from to the blood side to the brain side, we compared the transcytosis of ø 110 nm (NP110) and ø 350 nm (NP350) nanoparticles targeting patient-derived glioblastoma spheres. Results obtained in vitro were then compared to the in vivo transcytosis. In the presented example, nanoparticles were surface-coated with the tumor-targeting peptide CooP20 and loaded with the fluorescent dye (FITC) to facilitate the visualization. We labeled the cells using the lysosomal dye and counterstained with DAPI 24 h after the addition of the FITC nanoparticles on the blood-side of the BBTB mimic and acquired confocal micrographs at different levels (e.g., the blood side, the membrane, the brain side, and the patient glioblastoma spheres) (Figure 3A). The NP110-associated fluorescent signal colocalized with the lysosomes in the endothelial cells, astrocytes, and tumor cells. In addition, NP110s were detected in-between the endothelial cells and astrocytes, passing through the membrane pores of the insert (Figure 3A).
The passage of NP110s was quantified by measuring the fluorescence from samples collected from both the blood and brain side. These permeability values were compared to those determined for the nanoparticles of ø 350 nm (NP350). The results show that only NP110 was able to cross the BBTB mimics (Figure 3B). NP350 remained on the blood-side of the BBTB mimic, which resulted in lower permeability values for these nanoparticles.
To highlight the relevance of the BBTB mimics compared to the in vivo models, nude mice were intravenously injected with NP110 or NP350 nanoparticles coated with the tumor-targeting peptide CooP and conjugated with red fluorescent dye (TRITC) for the detection. Tissues collected at several time points revealed that after 8 h, BBB-permeable nanoparticles have extravasated into the brain parenchyma, while the nonpermeable ones that stayed in the circulation were mainly cleared from the systemic circulation in vivo. Therefore, we collected the brains and quantified the number of nanoparticles per square millimeter 8 h postinjection. In accordance with the in vitro findings, NP110, but not NP350, successfully extravasated into the brain parenchyma (Figure 3C). High-magnification imaging of the nanoparticle location in the brain showed that NP110 was homogeneously distributed in the brain parenchyma outside the blood capillaries and successfully homed to the implanted glioblastoma cells (Figure 3D). Despite exhibiting the same tumor targeting moiety (CooP), NP350 was unable to extravasate into the brain parenchyma and was only detected within the luminal side of brain blood vessels (Figure 3E), similar to the results obtained in vitro.

Figure 1: Description of the blood-brain tumor-barrier (BBTB) model. (A) Schematic representation of the locations of different cell types. (B) Illustration of the insert placement on the 6-well plate cover and the seeding technique for the astrocytes on the brain side of the insert’s membrane. (C) Illustration of the 6-well plate placement allowing the astrocyte adhesion. (D) Immunofluorescence micrographs of the tight junction proteins zonula occludens-1 (ZO-1, upper row, red) and claudin-5 (lower row, green). The protein expression is compared to the murine brain microvascular endothelial cells (bEND3) cultured on the blood side of the BBTB alone as a monoculture (left column) or with murine immortalized HIFko astrocytes (right column). Cell nuclei are counterstained with DAPI (blue). (E) Immunofluorescence micrograph showing the glial fibrillary acidic protein (GFAP, red) in HIFko astrocytes cultured at the brain side of the BBTB. The high-magnification image shows astrocyte processes and end-feet (arrows) contacting the endothelial cells through the membrane pores (right panel). The identity of the HIFko astrocytes was verified by immunofluorescence staining of the simian virus 40 large T antigen (SV40 large T, green) used for the immortalization of the cells. Endothelial cells express neither the GFAP nor the SV40 large T and, therefore, can be partially observed through the transparent, opposite side of the membrane as DAPI-only stained cells (dashed lines). Cell nuclei are counterstained with DAPI (blue). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Intravital live determination of the mouse BBB permeability. (A) Preparation of the implantable caudal vein catheter. (1) Tools and equipment are the following: (a) a PE20 polyethylene tube (b) two 25 G needles (c) Rochester-Ochsner forceps, and (d) a small bulldog clamp. (2) A 25 G needle is removed by several torsions using the forceps and (3) carefully inserted in the tube. (4) The other side of the tube is connected to another 25 G needle. (B) Guidance for the catheter implantation and positioning to infuse the sodium-fluorescein solution through the tail vein of a mouse. The circled area indicates the area where a drop of cyanoacrylate glue is placed to secure the catheter. The bulldog clamp is used to handle the catheter and removed once the catheter is secured. (C) Representative imaging and quantification method to determine the sodium-fluorescein permeability values. Prior to the sodium-fluorescein infusion (left column), the autofluorescence/blank is measured within a region of interest (ROI) placed on the brain (top panel, white rectangle) and blood vessel areas (bottom panel, red rectangle). During the sodium-fluorescein infusion (right column), the fluorescence intensity is measured in both ROIs, allowing the calculation of the BBB permeability. Please click here to view a larger version of this figure.
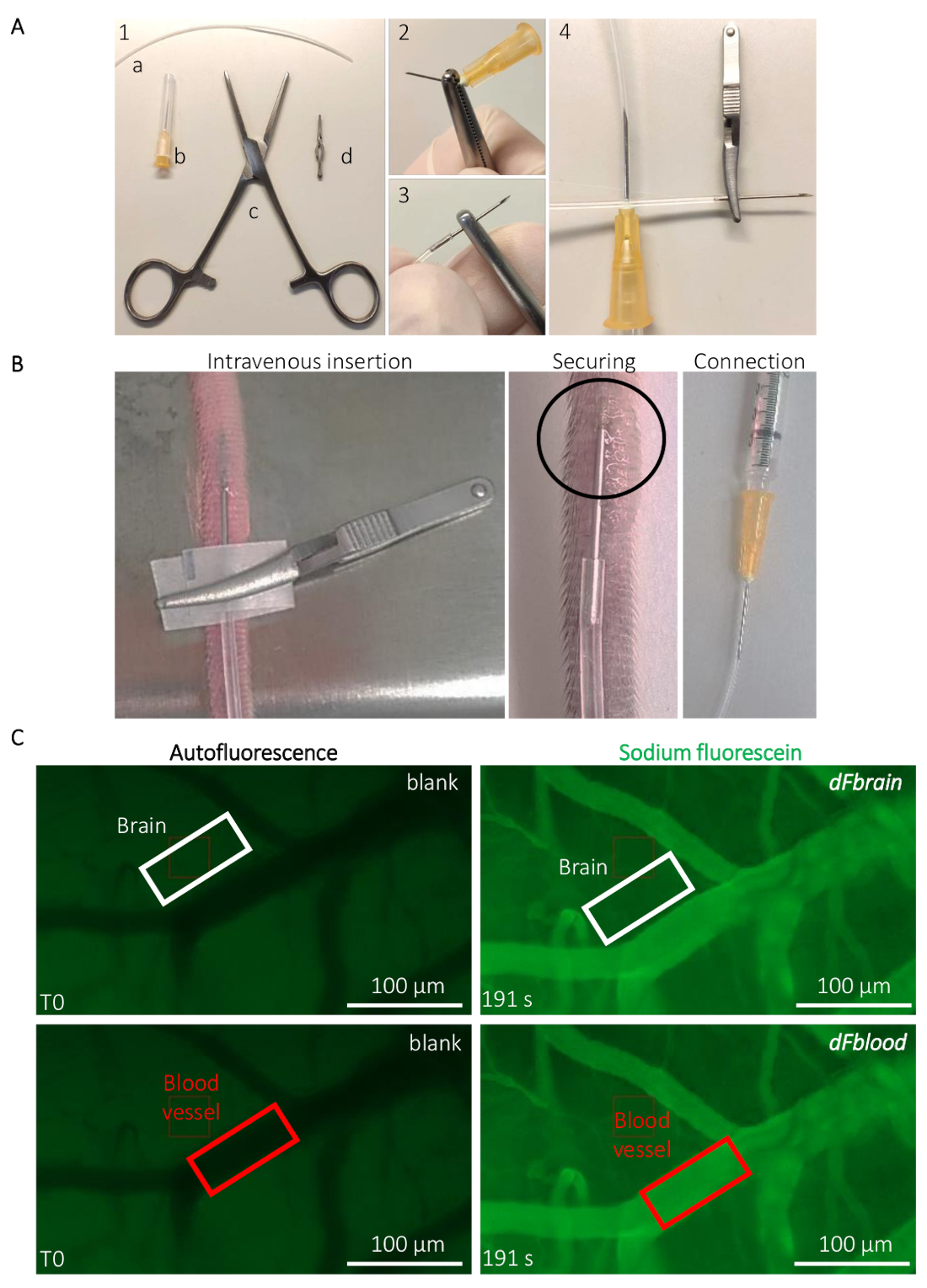{kind=link}

Figure 3: Prediction of the intracerebral transcytosis of nanoparticles through BBB in vivo using the in vitro BBTB model. (A) Graphic representation of the BBTB assay with representative confocal images obtained at the indicated different levels of the murine BBTB model. Nanoparticles of 110 nm in diameter (NP110) and conjugated to FITC (green) were added to the blood side of the BBTB Cells were labelled with the lysosomal probe (LT-99, red). (1) Endothelial cells, (2) Endothelial transcytosis of the nanoparticles through the pores of the membrane (white dashed line), (3) astrocytes, and (4) patient glioblastoma spheres are identified on the graphic and corresponding confocal micrographs (right). Lysosomal encapsulation of the nanoparticles (arrows) suggests active transcytosis through the endothelial and astrocyte layers of the BBTB. (B) Quantification of the indicated nanoparticle permeability through the BBTB in vitro (n = 6). (C) Quantification of the indicated nanoparticle density in the brain tissue sections, 8 h after the caudal vein infusion of the nude mice (n = 3). (D) Confocal micrographs showing the distribution of the ø 110 nm nanoparticles (NP110, red) in murine brain tissue sections labeled with an anti-mouse CD31 antibody (green). The arrow highlights the nanoparticle transcytosis (left panel). Nanoparticles accumulated around the brain tumor cells (tumor, right panel) owing to the CooP-targeting peptide presented on their surface. No significant homing is observed in the brain tissue (Brain). (E) Confocal micrographs show the distribution of the ø 350 nm nanoparticles (NP350, red) in murine brain tissue sections labeled with an anti-mouse CD31 antibody (green). Arrowheads point to the NP350s that were retained in the blood vessel lumen and were unable to cross the BBB, probably due to their larger diameter compared to the NP110s. Cell nuclei are counterstained with DAPI (blue). ** P < 0.01. P-values were calculated using a two-tailed, nonparametric Mann-Whitney U test. The error bars represent the standard deviation. Please click here to view a larger version of this figure.
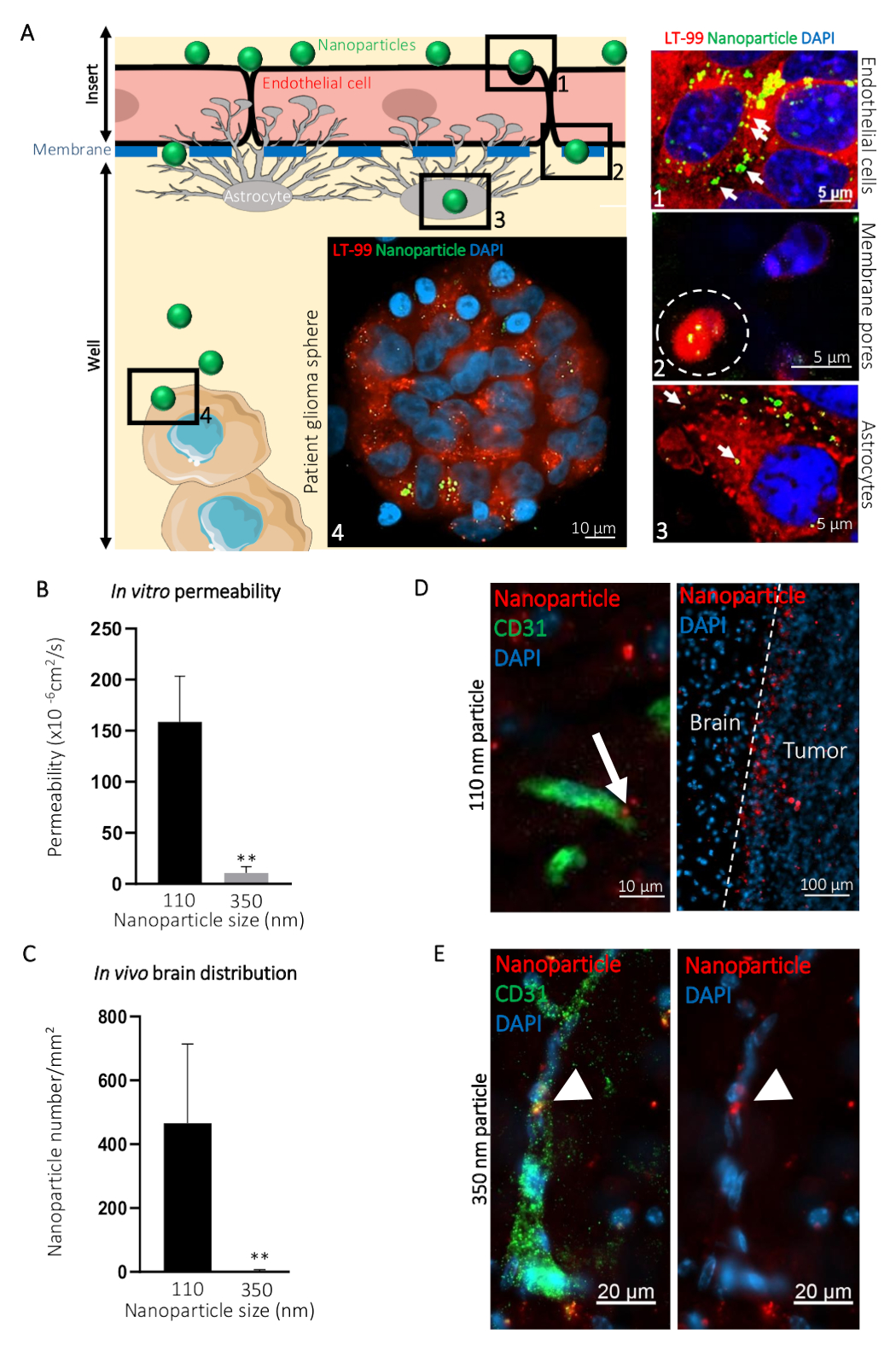{kind=link}
| murine BBTB mimic | bEND3 | bEND3+HIFko As | bEND3+GB | bEND3+HIFko As+GB | In vivo |
| Permeability (10-6 cm/s) | 27.63 | 6.74 | 26.8 | 10.83 | 5.57 |
| SD (10-6 cm/s) | 3.45 | 3.01 | 7.99 | 2.65 | 2.19 |
| human BBTB mimic | HuAR2T | HuAR2T+hIAs | HuAR2T+GB | HuAR2T+hIAs+GB | |
| Permeability (10-6 cm/s) | 104.92 | 47.4 | 89.08 | 48.24 | |
| SD (10-6 cm/s) | 27.1 | 14.32 | 10.21 | 13.07 |
Table 1: Values of the sodium-fluorescein (Na-Fl) permeability (in centimeters per second) determined in vitro in the indicated co-culture systems and in vivo in NMRI nude mice. Data from a representative experiment (n = 3 mice).
Discussion
The rise of the interpatient tumor variability concept rejuvenated the research on personalized cancer medicine21. This variability is also a hallmark of central nervous system neoplasms. Due to the unpredictability of the tumor, response to chemotherapy adds to the sheltering effect of the BBB for drug delivery, and altogether constitutes major challenges in patient care22. In order to develop more effective therapies, it is often necessary to screen large libraries of new molecules. To evaluate the antitumor efficacy and ability of the new therapeutic leads to reach the tumor site, the best option is the preclinical study on patient-derived cells implanted in vivo into murine patient avatars. Due to practical (financial, time, human, and facility resources) and ethical reasons (the 3Rs principle when using laboratory animals), the development of such a large-scale in vivo screening platform is often not possible, and therefore, the cell-based assays remain a model of choice23. The principal reason for selecting established cell lines and avoiding primary cells is to facilitate the reproducibility and reduce the use of laboratory animals, which are the main source for the isolation and establishment of primary murine cultures. The methods presenting here, firmly complying with the 3Rs, could efficiently discard nanoparticles from a further preclinical investigation on the criterium of their inability to cross the model BBTB. As a proof-of-principle, we describe here findings obtained during the development and validation of the BBTB. We were able to confirm in vitro findings in vivo, for instance when measuring the passive diffusion of a 376 Da sodium-fluorescein.
The protocol outlined in this paper describes the preparation of endothelial cells cocultured with astrocytes to form a blood-brain tumor-barrier-like interface in an in vitro setup. Once a physical contact between these two cell types is established, the endothelial cell layer exhibits similarities with the BBB (e.g., a cell surface expression of tight junction proteins and relatively low permeability). Interestingly, the murine BBTB mimic seemed to provide particularly similar Na-Fl permeability values to those obtained with the in vivo mouse BBB permeability measurements24. Therefore, the performance of the BBTB mimics will be directly linked to the choice of the cells used to form the barrier. The bEND3 endothelial cells originate from the brain and are known to be successful in forming barriers when cocultured with astrocytes25. However, we have been using the immortalized HIFko astrocytes26 to generate the BBTB mimic. Due to their lack of hypoxia-inducible factor, these astrocytes do not produce VEGF-A, which is quintessential in the BBTB mimic’s stabilization described here. Astrocytes have been identified as modulators of the BBB permeability, for instance through the release of VEGF-A in response to neuroinflammation27. Activation of the vascular endothelial growth factor receptors (VEGFRs) is a key regulator of the endothelial/vascular permeability both in vitro28 and in vivo29. Therefore, VEGF-A supplements in the medium activate the VEGFR2 on the endothelial cells, which induces the phosphorylation of adherens junction proteins such as VE-cadherin30. The loss of endothelial cell-cell contacts generates highly permeable blood vessels. Similarly, both the strong mitogenic property and unknown composition of the fetal bovine sera used in the cell culture assays cause major issues in the barrier stabilization and assay reproducibility.
Human umbilical vein endothelial cells (HUVECs) are sometimes used to form BBB in vitro31; however, they significantly differ from the brain microvascular endothelial cells, such as the hCMEC/D3 cell line32, in terms of gene expression and barrier-forming properties. However, the relatively higher permeability values obtained with the HuAR2T cells grown alone compared to the bEND3 were greatly reduced by coculturing them with human primary astrocytes. Although the endothelial cells are required to form the cellular wall, it is clear that the astrocytes have an equally important role to play for the BBTB formation and stabilization.
When patient-derived glioma spheres were added to this equation, the mouse BBTB mimic recapitulated some of the features of murine xenografts, such as the drug diffusion through the brain vasculature and the tumor cell targeting. The BBTB mimics discussed here were, for instance, successful in mirroring the in vivo behavior when we screened several nanoparticles with different diameters. To illustrate the parallelism between the in vitro and in vivo models, we used the previously described mesoporous silicate nanoparticles33 with cell-penetrating properties34 conjugated to the tumor-targeting peptide CooP on their surface20. The CooP-targeting peptide homes to invasive tumor cells through the specific binding to the mammary-derived growth inhibitor (MDGI). Several cancers, including gliomas, are overexpressing MDGI compared to the normal tissue35, which makes the CooP a very efficient tumor-targeting moiety capable of increasing the delivery of a payload20. The nanoparticles used here have been previously shown to diffuse into the brain parenchyma (27547955), and when functionalized with Taxol, these nano-cargos have been successful in reducing glioma growth in preclinical models36. The addition of polyethylene glycol (PEG) residues at the surface of the nanoparticles also maintained their static charge to positive values (around + 4 mV), allowing better interaction with the neurovascular unit37 and also increasing their stability in circulation. In the presented data, 3 kDa of PEG was conjugated to NP110s, while NP350s were coated with 10 kDa of PEG. However, increased-molecular-weight PEG also resulted in a significant increase in the nanoparticle diameter, hence their physical capabilities to cross the BBB. Therefore, we checked whether the physical dimensions of the particles prevented their passage through the BBTB and whether these observations could be mirrored in vivo.
In accordance with the previously published observations, we observed that NP110s extravasated through both the BBTB in vitro and the BBB of mice bearing intracranial tumors, while NP350s retained on the luminal side of the BBTB mimic and in the blood vessels of the mice. These similar results strongly suggest that the BBTB model predicted the in vivo ability of nanoparticles to cross the BBB and reach the brain.
The relevance of cellular models of the BBB is frequently discussed, even for the central delivery of nanoparticles38. We show here that primary astrocytes and endothelial cells, both considered as the most relevant in vitro tools, can be replaced by immortalized and/or commercially available cells, ensuring greater scalability and reproducibility. The next generation of the in vitro BBB mimics could be developed by incorporating the microfluidic devices, allowing beautifully formed neurovascular units that structurally resemble the actual BBB12,14. However, such models are currently unsuitable for the high-throughput screening of molecules delivered to gliomas, due to technical limitations in the follow-up of the delivery14. It is indeed difficult to capture the physiological complexity of the BBB in a dish, and the possible lack of some receptors/proteins, known to be expressed by the BBB, could compromise the interpretation of the results. Another argument concerns the great variability in gene expression between in vivo and in vitro conditions, as well as from one cell line to another, especially considering endothelial cells. However, it could also be argued that the neurovascular unit is not a uniform entity within the brain39. Scientific research in biology has reached a humane era where the animal welfare, ethical responsibility, and the cost of using animal lives are always considered before designing an experiment. Therefore, to support the replacement of animals, an increasing number of recent studies show that the awareness of the limitations of the models and careful selection of the cellular models to establish the barrier—with an emphasis on the astrocytes—warrant a match between results obtained in a dish and in animal models40. With the methodology described here, we get one step closer to reducing the number of experimental animals used for screening purposes of BBB transcytosis for potential therapeutics.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was supported by grants from the Finnish Cancer Organizations, Jane & Aatos Erkko Foundation, and Sigrid Juselius Foundation (to P.L. and V.L.J.), the Swiss National Science Foundation (Advanced Postdoc.Mobility grant no: P300PB_164732, to S.K.), the Orion Research Foundation (to S.K.), the Maud Kuistila Memorial Foundation (to S.K.), and the Academy of Finland (TERVA 2017, grant no: 314 498). The Biomedicum Imaging Unit (Helsinki) is acknowledged for providing the microscopy imaging core facility.
Materials
| Name | Company | Catalog Number | Comments |
| Cells | |||
| bEND3 | ATCC | CRL-2299 | Cultured in: DMEM (1g/L glucose) supplemented with 10% FBS, 5 mL L-glutamine and 5 mL penicillin/streptomycin |
| HIFko immortalized mouse astrocytes | Isolated in Dr. Gabriele Bergers Lab | https://doi.org/10.1016/S1535-6108(03)00194-6 | Cultured in: BME-1 supplemented with 5% FBS, 5 mL 1 M HEPES, 5 mL 100 mM sodium pyruvate, 3 g D-glucose and 5 mL penicillin/streptomycin |
| HuAR2T | Isolated in Dr. Dagmar Wirth Lab | https://doi.org/10.1089/ten.tea.2009.0184 | Cultured in: EBM-2 with SupplementMix |
| normal human primary astrocytes | Lonza | CC-2565 | Cultured in: ABM with SingleQuots |
| Material and reagents | |||
| 100 mm x17 mm Dish, Nunclon Delta | ThermoFisher Scientific | 150350 | |
| 10 mL serological pipet | ThermoFisher Scientific | 170361 | |
| 15 mL Conical Sterile Polypropylene Centrifuge Tubes | ThermoFisher Scientific | 339650 | |
| ABM Basal Medium, 500 mL | Lonza | CC-3187 | For primary human astrocytes. ABM+: contains all the additives from the supplement mix. ABM-:all the additives except for rhEGF and FBS |
| Accutase Cell Detachment Solution | Corning | 25-058-CI | |
| AGM SingleQuots Supplements and Growth Factors | Lonza | CC-4123 | |
| B27 supplement | Gibco | 17504-044 | for both GBM + and - medium |
| Basal Medium Eagle | ThermoFisher Scientific | 21010046 | BME-1 |
| Corning Costar TC-Treated 6-Well Plates | Sigma-Aldrich | CLS3506 | |
| Corning Transwell polyester membrane cell culture inserts | Sigma-Aldrich | CLS3452 | |
| D-glucose | Sigma-Aldrich | G8270 | dissolve in 50 mL of BME-1 and sterile filter before adding to the medium |
| Dulbecco's Modified Eagle's Medium/Nutrient F-12 Ham | Gibco | 21331-020 | Specific to the culture of the patient-derived spheres isolated in our lab, may vary for other glioma cell lines |
| EBM-2 growth Medium SupplementMix | PromoCell | c-39216 | EBM+: contains all the additives from the supplement mix. EBM-:all the additives except for VEGF-A and FBS |
| Endothelial Basal Medium 2 (EBM-2) | PromoCell | c-22211 | EBM+: contains all the additives from the supplement mix. EBM-:all the additives except for VEGF-A and FBS |
| Fetal Bovine Serum (FBS), qualified, heat inactivated, E.U.-approved, South America Origin | ThermoFisher Scientific | 10500056 | |
| Fluorescein sodium salt | Sigma-Aldrich | F6377 | |
| Greiner CELLSTAR 96 well plates | Sigma-Aldrich | Greiner 655090 | black polystyrene wells flat bottom (with micro-clear bottom) |
| Menzel-Gläser 0.9 cm round borosilicate Cover Slips | Thermo Scientific | 10313573 | |
| PBS tablets | Medicago | 09-9400-100 | one tablet per liter of dH2O, then sterilize the solution by autoclaving |
| Poly-D-lysine hydrobromide | Sigma-Aldrich | P6407 | |
| Recombinant Human EGF | Peprotech | GMP100-15 | for GBM+ medium |
| Recombinant Human FGF-basic (154 a.a.) | Peprotech | 100-18B | for GBM+ medium |
| Immunofluorescence | |||
| Paraformaldehyde | Sigma-Aldrich | 158127 | |
| 24 mm x 60 mm microscope slide cover glass | ORSAtec | 0224601-D | |
| AlexaFluor 488 and 594 secondary antibodies | ThermoFisher Scientific | dilution: 1/500 | |
| Anti-Claudin-5 antibody | Abcam | ab15106 | dilution: 1/150 |
| Anti-GFAP antibody clone GF5 | Abcam | ab10062 | dilution: 1/150 |
| Anti-Mouse CD31 antibody Clone MEC 13.3 | BD Biosciences | 550274 | dilution 1/800 |
| Anti-SV40 T-antigen antibody | Abcam | ab16879 | dilution: 1/150 |
| Anti-Zonula Occludens-1 | Abcam | ab96587 | dilution: 1/200 |
| DAPI | TOCRIS | 5748 | |
| Fluoromount Aqueous Mounting Medium | Sigma-Aldrich | F4680 | |
| LysoTracker Red DND-99 | ThermoFisher Scientific | L7528 | |
| Animal procedures | |||
| 10 cm curved dissecting scissors | World Precision Instruments | 14394 | |
| BD Microlance 25 G needles | Becton Dickinson | 300600 | |
| Fine Forceps (12.5 cm) | World Precision Instruments | 503283 | for tissue dissociation |
| Intramedic Polyethylene tubing PE20 | Becton Dickinson | 427406 | |
| Ketaminol vet 50 mg/mL | Intervet | Vnr511485 | Ketamine |
| Mains Powered microdrill | World Precision Instruments | 503599 | |
| Menzel-Gläser 0.5 cm round borosilicate Cover Slips | Thermo Scientific | 11888372 | |
| Micro Bulldog clamp | World Precision Instruments | 14119 | |
| Physiological saline solution | Mustela | Sterile single dose vials 20 x 5 mL / 40 x 5 mL - Medical device class | |
| Rochester-Oschner forceps | World Precision Instruments | 501709 | |
| Rompun vet 20 mg/mL | Intervet | Vnr148999 | Xylazine |
| Stereotaxic adapter | World Precision Instruments | 502063 | |
| Sugi Sponge Points | Kettenbach | 31603 | |
| Equipment | |||
| Axio Zoom.V16 fluorescence stereo zoom microscope | Carl Zeiss | ||
| FLUOstar Omega microplate reader | BMG Labtech | ||
| ORCA-Flash 4.0 digital sCMOS camera | Hamamatsu Photonics | ||
| Universal 320 tabletop centrifuge | Hettich | Cat. No. 1401 | |
| ZEISS LSM 880 with Airyscan confocal microscope | Carl Zeiss |
References
- Daneman, R., Prat, A. The blood-brain barrier. Cold Spring Harbor Perspectives in Biology. 7 (1), a020412(2015).
- Quail, D. F., Joyce, J. A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 31 (3), 326-341 (2017).
- Wang, Z., Sun, H., Yakisich, J. S. Overcoming the blood-brain barrier for chemotherapy: limitations, challenges and rising problems. Anticancer Agents in Medicinal Chemistry. 14 (8), 1085-1093 (2014).
- Alkins, R. D., Brodersen, P. M., Sodhi, R. N., Hynynen, K. Enhancing drug delivery for boron neutron capture therapy of brain tumors with focused ultrasound. Neuro Oncology. 15 (9), 1225-1235 (2013).
- Alli, S., et al. Brainstem blood brain barrier disruption using focused ultrasound: A demonstration of feasibility and enhanced doxorubicin delivery. Journal of Controlled Release. 281, 29-41 (2018).
- Ashby, L. S., Smith, K. A., Stea, B. Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: a systematic literature review. World Journal of Surgical Oncology. 14 (1), 225(2016).
- Guishard, A. F., Yakisich, J. S., Azad, N., Iyer, A. K. V. Translational gap in ongoing clinical trials for glioma. Journal of Clinical Neurosciences. 47, 28-42 (2018).
- Rahman, N. A., et al. Immortalized endothelial cell lines for in vitro blood-brain barrier models: A systematic review. Brain Research. 1642, 532-545 (2016).
- Helms, H. C., et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. Journal of Cerebral Blood Flow & Metabolism. 36 (5), 862-890 (2016).
- Wang, J. D., Khafagy, el-S., Khanafer, K., Takayama, S., ElSayed, M. E. Organization of Endothelial Cells, Pericytes, and Astrocytes into a 3D Microfluidic in Vitro Model of the Blood-Brain Barrier. Molecular Pharmaceutics. 13 (3), 895-906 (2016).
- Phan, D. T., et al. Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood-central nervous system interface. Experimental Biology and Medicine (Maywood). 242 (17), 1669-1678 (2017).
- Bang, S., et al. A Low Permeability Microfluidic Blood-Brain Barrier Platform with Direct Contact between Perfusable Vascular Network and Astrocytes. Scientific Reports. 7 (1), 8083(2017).
- Wilhelm, I., Krizbai, I. A. In vitro models of the blood-brain barrier for the study of drug delivery to the brain. Molecular Pharmacology. 11 (7), 1949-1963 (2014).
- Campisi, M., et al. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials. 180, 117-129 (2018).
- Pirsko, V., et al. An Effect of Culture Media on Epithelial Differentiation Markers in Breast Cancer Cell Lines MCF7, MDA-MB-436 and SkBr3. Medicina (Kaunas). 54 (2), (2018).
- Stebbins, M. J., et al. Differentiation and characterization of human pluripotent stem cell-derived brain microvascular endothelial cells. Methods. 101, 93-102 (2016).
- Canfield, S. G., et al. An isogenic blood-brain barrier model comprising brain endothelial cells, astrocytes, and neurons derived from human induced pluripotent stem cells. Journal of Neurochemistry. 140 (6), 874-888 (2017).
- Cao, Y., et al. Hypoxia-inducible factor-1alpha is involved in isoflurane-induced blood-brain barrier disruption in aged rats model of POCD. Behavioural Brain Research. 339, 39-46 (2018).
- Abbott, N. J. Astrocyte-endothelial interactions and blood-brain barrier permeability. Journal of Anatomy. 200 (6), 629-638 (2002).
- Kinnari, P. J., et al. Tumour homing peptide-functionalized porous silicon nanovectors for cancer therapy. Biomaterials. 34 (36), 9134-9141 (2013).
- Levin, V. A. Personalized medicine in neuro-oncology. CNS Oncology. 5 (2), 55-58 (2016).
- Weathers, S. S., Gilbert, M. R. Toward Personalized Targeted Therapeutics: An Overview. Neurotherapeutics. 14 (2), 256-264 (2017).
- O'Duibhir, E., Carragher, N. O., Pollard, S. M. Accelerating glioblastoma drug discovery: Convergence of patient-derived models, genome editing and phenotypic screening. Molecular and Cellular Neuroscience. 80, 198-207 (2017).
- Kaya, M., Ahishali, B. Assessment of permeability in barrier type of endothelium in brain using tracers: Evans blue, sodium fluorescein, and horseradish peroxidase. Methods in Molecular Biology. 763, 369-382 (2011).
- Yang, S., et al. Identification of two immortalized cell lines, ECV304 and bEnd3, for in vitro permeability studies of blood-brain barrier. PLoS One. 12 (10), e0187017(2017).
- Blouw, B., et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell. 4 (2), 133-146 (2003).
- Argaw, A. T., et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. Journal of Clinical Investigation. 122 (7), 2454-2468 (2012).
- Miao, Z., et al. VEGF increases paracellular permeability in brain endothelial cells via upregulation of EphA2. The Anatomical Record (Hoboken). 297 (5), 964-972 (2014).
- Heinolainen, K., et al. VEGFR3 Modulates Vascular Permeability by Controlling VEGF/VEGFR2 Signaling. Circulation Research. 120 (9), 1414-1425 (2017).
- Claesson-Welsh, L. Vascular permeability--the essentials. Upsala Journal of Medical Sciences. 120 (3), 135-143 (2015).
- Adriani, G., Ma, D., Pavesi, A., Goh, E. L., Kamm, R. D. Modeling the Blood-Brain Barrier in a 3D triple co-culture microfluidic system. Conference Proceedings - IEEE Engineering in Medicine and Biology Society. 2015, 338-341 (2015).
- Weksler, B., Romero, I. A., Couraud, P. O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids and Barriers of the CNS. 10 (1), 16(2013).
- Paatero, I., et al. Analyses in zebrafish embryos reveal that nanotoxicity profiles are dependent on surface-functionalization controlled penetrance of biological membranes. Scientific Reports. 7 (1), 8423(2017).
- Prabhakar, N., et al. Stimuli-responsive hybrid nanocarriers developed by controllable integration of hyperbranched PEI with mesoporous silica nanoparticles for sustained intracellular siRNA delivery. International Journal of Nanomedicine. 11, 6591-6608 (2016).
- Hyvonen, M., et al. Novel target for peptide-based imaging and treatment of brain tumors. Molecular Cancer Therapeutics. 13 (4), 996-1007 (2014).
- Feng, X., et al. Mammary-Derived Growth Inhibitor Targeting Peptide-Modified PEG-PLA Nanoparticles for Enhanced Targeted Glioblastoma Therapy. Bioconjugate Chemistry. 26 (8), 1850-1861 (2015).
- Nance, E. A., et al. A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Science Translational Medicine. 4 (149), 149rA119(2012).
- Berg, C. Quantitative analysis of nanoparticle transport through in vitro blood-brain barrier models. Tissue Barriers. 4 (1), e1143545(2016).
- Noumbissi, M. E., Galasso, B., Stins, M. F. Brain vascular heterogeneity: implications for disease pathogenesis and design of in vitro blood-brain barrier models. Fluids and Barriers of the CNS. 15 (1), 12(2018).
- Heymans, M., Sevin, E., Gosselet, F., Lundquist, S., Culot, M. Mimicking brain tissue binding in an in vitro model of the blood-brain barrier illustrates differences between in vitro and in vivo methods for assessing the rate of brain penetration. European Journal of Pharmaceutics and Biopharmaceutics. 127, 453-461 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved