
Method Article
Rescue of Recombinant Zika Virus from a Bacterial Artificial Chromosome cDNA Clone
In This Article
Summary
The recent epidemic of Zika virus highlights the importance of establishing reverse genetic approaches to develop vaccines and/or therapeutic strategies. Here, we describe the protocol to rescue an infectious recombinant Zika virus from a full-length cDNA clone assembled in a bacterial artificial chromosome under the control of the human cytomegalovirus immediate-early promoter.
Abstract
The association of Zika virus (ZIKV) infection with neurological complications during the recent worldwide outbreak and the lack of approved vaccines and/or antivirals have underscored the urgent need to develop ZIKV reverse genetic systems to facilitate the study of ZIKV biology and the development of therapeutic and/or prophylactic approaches. However, like with other flaviviruses, the generation of ZIKV full-length infectious cDNA clones has been hampered due to the toxicity of viral sequences during its amplification in bacteria. To overcome this problem, we have developed a nontraditional approach based on the use of bacterial artificial chromosomes (BACs). Using this approach, the full-length cDNA copy of the ZIKV strain Rio Grande do Norte Natal (ZIKV-RGN) is generated from four synthetic DNA fragments and assembled into the single-copy pBeloBAC11 plasmid under the control of the human cytomegalovirus (CMV) immediate-early promoter. The assembled BAC cDNA clone is stable during propagation in bacteria, and infectious recombinant (r)ZIKV is recovered in Vero cells after transfection of the BAC cDNA clone. The protocol described here provides a powerful technique for the generation of infectious clones of flaviviruses, including ZIKV, and other positive-strand RNA viruses, particularly those with large genomes that have stability problems during bacterial propagation.
Introduction
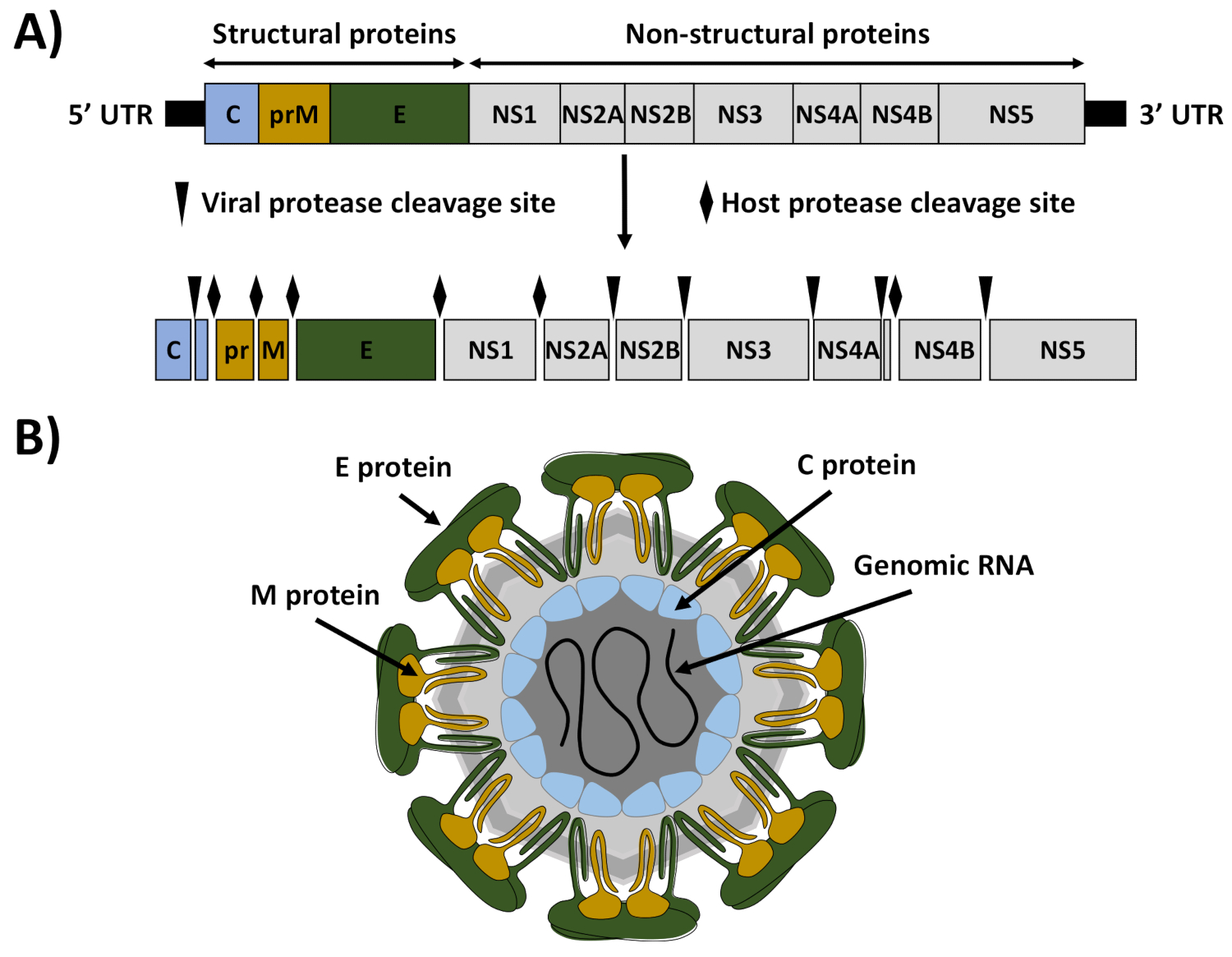
ZIKV is a mosquito-borne member of the Flavivirus genus within the Flaviviridae family that currently constitutes a global public health emergency1. Like other flaviviruses, ZIKV is an enveloped RNA virus with an icosahedral-like structure that contains a positive sense, single-stranded RNA molecule of about 10.8 kb (Figure 1)2. The viral genome encodes a large polyprotein of approximately 3,423 amino acids that is processed by viral and cellular proteases into three structural proteins (capsid [C], premembrane/membrane [prM/M], and envelope [E]) that are involved in the formation of the viral particles and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) that participate in genome replication, virus assembly, and evasion of the host immune response (Figure 1)3.
Historically, ZIKV infection has been associated with a mild febrile illness4,5. However, the explosive recent pandemic of ZIKV infections throughout South and Central America, the South Pacific, and the Caribbean6,7,8, and its association with the occurrence of Guillain-Barré syndrome and microcephaly9,10,11,12,13, have changed the historic perception and potentiated the relevance of ZIKV as an important human pathogen. In this sense, the development of molecular tools, such as infectious cDNA clones, will facilitate the study of viral pathogenesis and the development of genetically defined vaccines and the identification of antiviral drugs for the treatment of ZIKV infection. As described for other flaviviruses, the generation of ZIKV infectious clones is difficult due to the presence of cryptic bacterial promoters in the viral genome14 that allow the leaky expression of toxic viral proteins during the propagation of the cDNA clones in bacteria using standard high-copy-number plasmids15,16,17. To overcome this toxicity problem, several nontraditional approaches have been implemented successfully in the last two years18. These include the use of low-copy-number plasmids19,20, the insertion of introns to disrupt the toxic regions21,22,23, the in vitro ligation of cDNA fragments24,25, mutational silencing of cryptic bacterial promoters present in the viral genome26,27, infectious subgenomic amplicons (ISA)28,29, the Gibson assembly method30, and the use of circular polymerase extension reaction (CPER)31.
Herein, we describe the detailed protocol for the engineering of a full-length cDNA clone of the ZIKV strain ZIKV-RGN13, using a BAC to overcome the toxicity problem, and the rescue of infectious rZIKV by direct transfection of the BAC cDNA clone into Vero cells32, a cell line approved by the Food and Drug Administration (FDA) for the development of human vaccines33. In this system, the full-length cDNA copy of the viral genome is assembled in the BAC plasmid pBeloBAC1134 (Figure 2A), a low-copy-number plasmid (one to two copies per cell) derived from the Escherichia coli F-factor35, which minimizes the toxicity of flavivirus sequences during its propagation in bacteria. The cDNA of the ZIKV genome is assembled in pBeloBAC11 under the control of the human CMV immediate-early promoter, to allow the expression of the viral (v)RNA in the nucleus of transfected cells by cellular RNA polymerase II, and flanked at the 3'-end by the hepatitis delta virus (HDV) ribozyme (RZ), followed by the sequences of the bovine growth hormone (BGH) termination and polyadenylation signals to produce synthetic RNAs bearing authentic 5'- and 3'-ends of the viral genome, respectively (Figure 2B). This cDNA-launched system results in the intracellular expression of capped viral RNA, allowing the recovery of infectious ZIKV without the need for an in vitro transcription step. The BAC approach provides a powerful methodology applicable to constructing stable and fully functional infectious cDNA clones for other flaviviruses, as well as other positive-stranded RNA viruses36,37,38,39,40,41.
Protocol
1. Construction of a ZIKV Infectious cDNA Clone in a BAC
NOTE: The strategy for the assembly of ZIKV in BACs is described for the RGN strain13 (accession number KU527068) (Figure 2).
- Select unique restriction sites appropriately spaced in the viral genome that are absent in the plasmid pBeloBAC11 (Figure 2A).
NOTE: For ZIKV-RGN, the restriction sites Pml I, Afe I, and BstB I (genomic positions 3,347, 5,969, and 9,127, respectively) were selected. In the case that no appropriate restriction sites are available in the viral genome, generate new restriction sites by introducing silent nucleotide mutations in the viral genome during the cDNA fragment design. - Generate by chemical synthesis four cDNA fragments spanning the full-length genome (Z1 to Z4), flanked by the CMV promoter at the 5'-end and the HDV RZ, the BGH termination, and polyadenylation sequences at the 3'-end ( Figure 2B). Each fragment must be flanked by the selected restriction sites (step 1.1).
NOTE: Fragment Z1 is used as a backbone to clone the rest of the fragments in the pBeloBac11. For that end, it must contain the human CMV promoter and must be flanked at the 5'-end by ApaL I (to clone this fragment in pBeloBAC11) and Asc I (absent in the viral genome), and at the 3'-end by the restriction sites selected for the assembly of the infectious clone (Pml I, Afe I, and BstB I) followed by Mlu I (absent in the viral genome) and BamH I (to clone this fragment in pBeloBAC11). Fragment Z4 must contain the genomic region from the last restriction site selected (BstB I) to the end of the viral genome, followed by the HDV RZ, the BGH termination, and polyadenylation sequences, and the Mlu I restriction site (Figure 2B). Alternatively, the cDNA fragments (Z1-Z4) could be generated by a combination of standard reverse transcriptase polymerase chain reactions (RT-PCRs) and overlapping PCRs using specific oligonucleotides. - Assemble the infectious cDNA clone by sequential cloning of fragments Z1 to Z4 into pBeloBAC11 (Figure 2B).
- Digest the pBeloBAC11 plasmid and the Z1 fragment with ApaL I and BamH I. For that, mix 2 µg of pBeloBAC11 plasmid or 1 µg of Z1 fragment with 10 µL of 10x reaction buffer, 20 units of each enzyme, and water to reach a final volume of 100 µL. Incubate at 37°C for 2 h.
- Dephosphorylate the BAC vector using shrimp alkaline phosphatase (SAP). To that end, add 2.5 µL (2.5 units) of SAP to the digested BAC and incubate at 37°C for 1 h. Heat-inactivate the SAP by incubation at 75 °C for 15 min.
- Purify the dephosphorylated BAC vector and fragment Z1 by agarose gel electrophoresis using a commercial gel clean-up kit optimized for the purification of DNA fragments larger than 10 kb (see the Table of Materials).
- Perform the ligation reaction to generate the plasmid (p)BAC-Z1. To that end, mix 150 ng of purified digested BAC vector with the purified insert using a molar ratio of vector-to-insert of 1:3, 1.5 µL of 10x T4 DNA ligase buffer containing 10 mM ATP, one unit of T4 DNA ligase, and water to a final volume of 15 µL.
- Incubate the ligation mixture for 20 h at 16 °C. As control of the ligation reaction, perform in parallel a ligation reaction without insert. Heat-inactivate the ligase by incubation at 65 °C for 15 min.
NOTE: In the case of blunt-ended DNAs, incubate the ligation reaction for 20 h at 14 °C. Due to the large size of the BAC vector (Figure 2A), it is essential to use larger amounts of vector and insert than ligations using conventional plasmids in order to increase the ligation efficiency. - Transform 50 µL of E. coli DH10B electrocompetent cells with 2 µL of the ligation reaction (step 1.3.5) by electroporation (25 µF capacitance, 2.5 kV, and 100 Ω resistance), using electroporation cuvettes fitted with electrodes spaced at intervals of 0.2 cm, following standard protocols.
- Transfer the cells to a polypropylene tube with 1 mL of SOC medium (2% tryptone, 0.5% yeast extract, 0.05% NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose [pH 7.0]) and incubate them at 37°C for 1 h with gentle shaking (200-250 rpm). Plate the cells onto Luria broth (LB) agar plates containing 12.5 µg/mL chloramphenicol and incubate them at 37°C for 16 h.
- Pick 8 to 12 bacterial colonies, make a replica on a fresh LB agar plate containing 12.5 µg/mL chloramphenicol, and test whether they contain the correct insert by direct PCR analysis using specific oligonucleotides.
- Pick a positive colony from the replica plate, grow it in 100 mL of LB containing 12.5 µg/mL of chloramphenicol, and isolate the BAC cDNA by the alkaline lysis method using a commercial plasmid midi kit, following the recommendation for the purification of large low-copy plasmids (see the Table of Materials).
NOTE: BAC cDNA prepared by this method can be contaminated with up to 30% of bacterial genomic DNA, but it is suitable in quality to perform restriction analysis, sequencing, and cloning. Depending on the BAC size, yields of 4-6 µg of the BAC plasmid can be obtained. - Verify the genetic integrity of the cloned cDNA by restriction analysis. Digest 500 ng of the pBAC-Z1 plasmid with Asc I and Pml I at 37 °C for 1 h and confirm the presence of the Z1 fragment by gel electrophoresis. To further confirm that unwanted mutations have not been introduced, sequence the insert with specific oligonucleotides.
- Starting from the plasmid pBAC-Z1 containing the selected restriction sites (Pml I, Afe I, BstB I, and Mlu I), sequentially clone fragments Z2 to Z4 to generate the full-length infectious cDNA clone pBAC-ZIKV (Figure 2B), following similar experimental approaches as described above for fragment Z1 (steps 1.3.1-1.3.10).
2. Preparation of High-purity pBAC-ZIKV for the Rescue of Infectious rZIKV
NOTE: The large-scale preparation of an ultrapure pBAC-ZIKV infectious clone, suitable for the transfection and rescue of infectious viruses, is performed by alkaline lysis with a commercial kit specifically developed for BAC purification (see the Table of Materials). The kit must include an ATP-dependent exonuclease digestion step that removes bacterial genomic DNA contamination, allowing the isolation of BAC cDNA with a grade of purity similar to that obtained with the cesium chloride method.
- Grow a single colony of E. coli DH10B carrying the pBAC-ZIKV infectious clone in 5 mL of LB medium containing 12.5 µg/mL of chloramphenicol at 37°C for 8 h with gentle shaking (200-250 rpm).
- Add 1 mL of bacterial culture (step 2.1) to 500 mL of LB with 12.5 µg/mL of chloramphenicol in a 2 L flask and grow the cells at 37°C for 14-16 h (till an OD600 of 0.6-0.8).
NOTE: Rich broths, such as Terrific Broth (TB), can produce extremely high cell densities, resulting in a lower yield and less purity of the BAC cDNA. - Purify the BAC infectious cDNA clone by alkaline lysis using a commercial kit specifically developed for BAC purification, following the manufacturer's specifications (see the Table of Materials). Keep the purified BAC cDNA at 4 °C. Depending on the BAC size, yields of 30 µg of ultrapure BAC cDNA clone can be obtained.
NOTE: Do not dry the DNA pellet for more than 5 min, as overdrying will make it difficult to dissolve the BAC cDNA. Due to the large size of the BAC cDNA clone, avoid vortexing or pipetting to prevent plasmid shearing.
3. Rescue of Infectious rZIKV from the BAC cDNA Clone by Transfection of Vero Cells
NOTE: Infectious rZIKV is recovered by the transfection of Vero cells with the pBAC-ZIKV cDNA clone, using a commercial cationic lipid transfection reagent (see the Table of Materials; Figure 3).
- One day before transfection, plate on 6-well plates 5 x 105 Vero cells/well in growth medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 5% fetal bovine serum [FBS], 2 mM L-glutamine, and 1% nonessential amino acids) without antibiotics to raise 90% confluent cell monolayers by the time of transfection.
NOTE: We recommend performing the virus rescues in triplicates. - Equilibrate serum-reduced medium (see the Table of Materials) at room temperature and prepare transfection mixtures in sterile microfuge tubes for each transfection sample as follows.
- Add 4 µg of the BAC cDNA clone in 250 µL of serum-reduced medium and mix carefully, avoiding vortexing to prevent plasmid shearing.
- In a separate tube, dilute 12 µL of transfection reagent (1 mg/mL) (see the Table of Materials) in 250 µL of serum-reduced medium, mix by vortexing, and incubate at room temperature for 5 min.
- Combine the diluted BAC cDNA and the transfection reagent (with a 1:3 ratio of DNA:transfection reagent), mix carefully while avoiding vortexing, and incubate at room temperature for 20-30 min.
- During the incubation period of the BAC cDNA/transfection reagentmixture,wash Vero cells with growth medium without antibiotics and leave the cells in 1 mL of fresh medium without antibiotics.
NOTE: The addition of antibiotics during the transfection process may decrease transfection efficiency. - Distribute dropwise the 500 µL of the BAC cDNA/transfection reagent mixture (of step 3.2.3) onto the Vero cells, mix them by rocking the plate back and forth, and incubate the cells in a 5% CO2 humidified incubator at 37 °C for 6 h.
- Remove the transfection medium, add 2 mL of fresh growth medium with antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin), incubate the cells in a 5% CO2 humidified incubator at 37 °C, and check every day for the induction of cytopathic effect (CPE).
NOTE: ZIKV CPE is quite pronounced in Vero cells and it is characterized by the presence of rounded and birefringent cells that detach and float in the tissue culture supernatant. - Collect aliquots (100 µL) of the Vero cell tissue-culture supernatants (step 3.5) every 24 h to determine the efficiency of virus recovery by assessing the presence of ZIKV using a standard plaque assay on fresh Vero cells (Figure 4A).
- After four to six days of transfection, when the CPE is around 50%-75% (Figure 4B), collect the tissue culture supernatants in 15 mL conical tubes and centrifuge at 2,000 x g for 10 min at 4 °C to remove cellular debris.
- Harvest the supernatants containing rZIKV and discard the cell pellets. Aliquot the supernatant in cryotubes and store them at -80 °C to further confirm the presence of rescued rZIKV.
4. Titration of Recovered rZIKV
- A day before titration, seed on 12-well plates 2.5 x 105 Vero cells/well in growth medium to raise 90% confluent cell monolayers by the time of titration.
NOTE: We recommend conducting the titration of the recovered rZIKV in triplicates. - Remove an aliquot of the supernatant from the freezer (step 3.8) and make serial 10-fold dilutions in growth medium without FBS.
- Wash Vero cells 2x with phosphate-buffered saline (PBS) and add 200 µL/well of each virus dilution in triplicate. Place plates in a 5% CO2 humidified incubator at 37 °C and rock every 15 min for a 90 min adsorption period.
- Remove the viral inoculum, overlay the cells with 2 mL of growth medium containing 2% FBS, 1% DEAE-dextran, and 0.6% Agar Noble, and incubate at 37 °C under 5% CO2 for 3-4 days.
NOTE: It is possible to use agarose, methylcellulose, or other overlay media. The time for proper plaque formation will vary depending on the overlay used and the ZIKV strain. - Fix ZIKV-infected cells with 1 mL/well of 4% formaldehyde diluted in PBS at room temperature for 1 h, remove the overlay, and visualize the viral plaques by staining them with 0.1% crystal violet in 20% methanol at room temperature for 15 min.
- Discard the crystal violet, wash 3x with water, allow the plates to dry, and manually count the plaques (Figure 4C, left panel). Viral titer is calculated as plaque forming units per milliliter (PFU/ml).
- Alternatively, viral plaques can be visualized by immunostaining with the pan-flavivirus E protein mouse monoclonal antibody (mAb) 4G2 (Figure 4C, right panel).
- To evaluate ZIKV plaques by immunostaining, after the fixing and removal of the overlay agar as described above (in step 4.5), wash the cells 2x with PBS and permeabilize them by incubation with 200 µL/well of 0.5% Triton-X100 in PBS for 15 min at room temperature.
- Remove the permeabilization solution, wash the cells 3x with PBS, and block them with 200 µL/well of blocking solution (10% FBS in PBS) for 1 h at room temperature.
NOTE: Other standard blocking solutions (e.g., 2.5% BSA in PBS) can be used in this step. - Remove the blocking solution and incubate the cells with 200 µL/well of the pan-flavivirus E protein mAb 4G2 diluted in blocking solution (1 µg/mL) for 1 h at 37 °C.
NOTE: Other mAb or polyclonal antibodies (pAb) can be used instead of 4G2 for the immunostaining and detection of viral plaques. - Wash the cells 3x with PBS, and incubate them with 200 µL of biotinylated anti-mouse secondary antibody diluted (following the manufacturer's recommendation) in blocking solution for 1 h at 37 °C.
- Remove the secondary antibody, wash the cells 4x with PBS, and visualize the viral plaques using an avidin/biotin-based peroxidase kit following the manufacturer's specifications (see the Table of Materials).
5. Confirmation of Successful rZIKV Rescue
NOTE: To further confirm the identity of the rescued virus, ZIKV E protein expression is analyzed by immunofluorescence using the mouse mAb 1176-56 specific for ZIKV E protein (Figure 4D). This mAb is specific for ZIKV E protein, contrary to the situation of the pan-flavivirus E protein mAb 4G2 (step 4.6.3). Alternatively, the virus identity can be confirmed by sequencing.
- A day before immunofluorescence analysis, seed coverslips in 24-well plates containing 1 x 105 Vero cells/well in growth medium to raise 90% confluent cell monolayers by the time of infection.
- Wash the cells 2x with PBS and infect them with a multiplicity of infection (MOI) of 0.5 PFU/cell of the rescued virus in growth medium without FBS (100 µL/well) in triplicate. Incubate the plates at 37 °C for 90 min.
- After viral adsorption, remove the viral inoculum, add 0.5 mL of fresh growth medium with 2% FBS, and incubate the cells in a 5% CO2 humidified incubator at 37 °C for 48 h.
- Remove the tissue culture supernatant, fix the cells with 150 µL/well of 4% paraformaldehyde in PBS for 20 min at room temperature, and permeabilize the cells with 150 µL/well of 0.5% Triton-X100 in PBS for 15 min at room temperature.
- After removing the permeabilization solution and washing the cells 3x with PBS, block the cells with 150 µL/well of blocking solution during 1 h at room temperature.
NOTE: Cell scan be blocked with alternative blocking solutions (e.g., 2.5% BSA in PBS). - Remove the blocking solution and incubate the cells with 100 µL/well of mAb 1176-56, specific for ZIKV E protein, diluted in blocking solution (1 µg/mL) for 1 h at 37 °C.
- Wash the cells 3x with PBS, incubate them at room temperature for 1 h with 100 µL/well of Alexa Fluor 488-conjugated anti-mouse secondary antibody diluted (following the manufacturer's recommendation) in blocking solution, wash the cells extensively with PBS, and incubate them with 150 µL/well of 4',6'-diamidino-2-phenylindole (DAPI; 1 mg/mL) diluted 1:200 in PBS at room temperature for 10 min.
- Wash the cells 3x with PBS, mount the coverslips on an antifade mounting medium (see the Table of Materials), and analyze the samples under a fluorescence microscope.
NOTE: For long-term storage, store samples at 4 °C, protected from light.
6. Amplification and Generation of Viral Stocks
NOTE: Once the identity of the rescued virus is confirmed (section 5), amplify the virus on Vero cells and generate viral stocks for further studies.
- Grow Vero cells in 100 mm x 21 mm plates at 90% confluence and infect them with an MOI of 0.1 PFU/cell as described before.
- When the CPE is around 75% (approximately 48-72 h postinfection), collect the tissue culture supernatant in a 50 mL conical tube and centrifuge at 2,000 x g for 10 min at 4 °C to remove the cellular debris.
- Harvest the supernatant containing rZIKV and discard the cell pellet. Aliquot the supernatant in cryotubes and store them at -80 °C.
- Remove a virus aliquot from the freezer and determine the viral titer by plaque assay as described before (section 4).
Results
The protocol described here allows for the generation of stable ZIKV full-length cDNA infectious clones using a BAC to minimize the toxicity problems associated with several flaviviral sequences. Efficient recovery of infectious rZIKV from the BAC cDNA clone can be easily achieved after the transfection of susceptible Vero cells (Figure 2). Using this protocol, we have generated a stable full-length cDNA clone of the ZIKV strain RGN32 by sequentially cloning four overlapping cDNA fragments into the BAC plasmid pBeloBAC1134 using conventional cloning methods and unique restriction sites present in the viral genome (Figure 2). The full-length cDNA clone was flanked at the 5'-end by the human CMV promoter to allow the expression of vRNA in the nucleus of transfected cells, and at the 3'-end by the HDV RZ followed by the BGH polyadenylation and termination sequences, to produce RNAs containing the exact 3'-end of the viral genome (Figure 2). The stability in bacteria of the generated BAC cDNA clone, together with its easy manipulation using standard recombinant DNA technologies, highlights the potential of the BAC cDNA approach for the rapid and reliable generation of stable full-length cDNA clones of ZIKV and other flaviviruses or positive-stranded RNA viruses with unstable viral genomes.
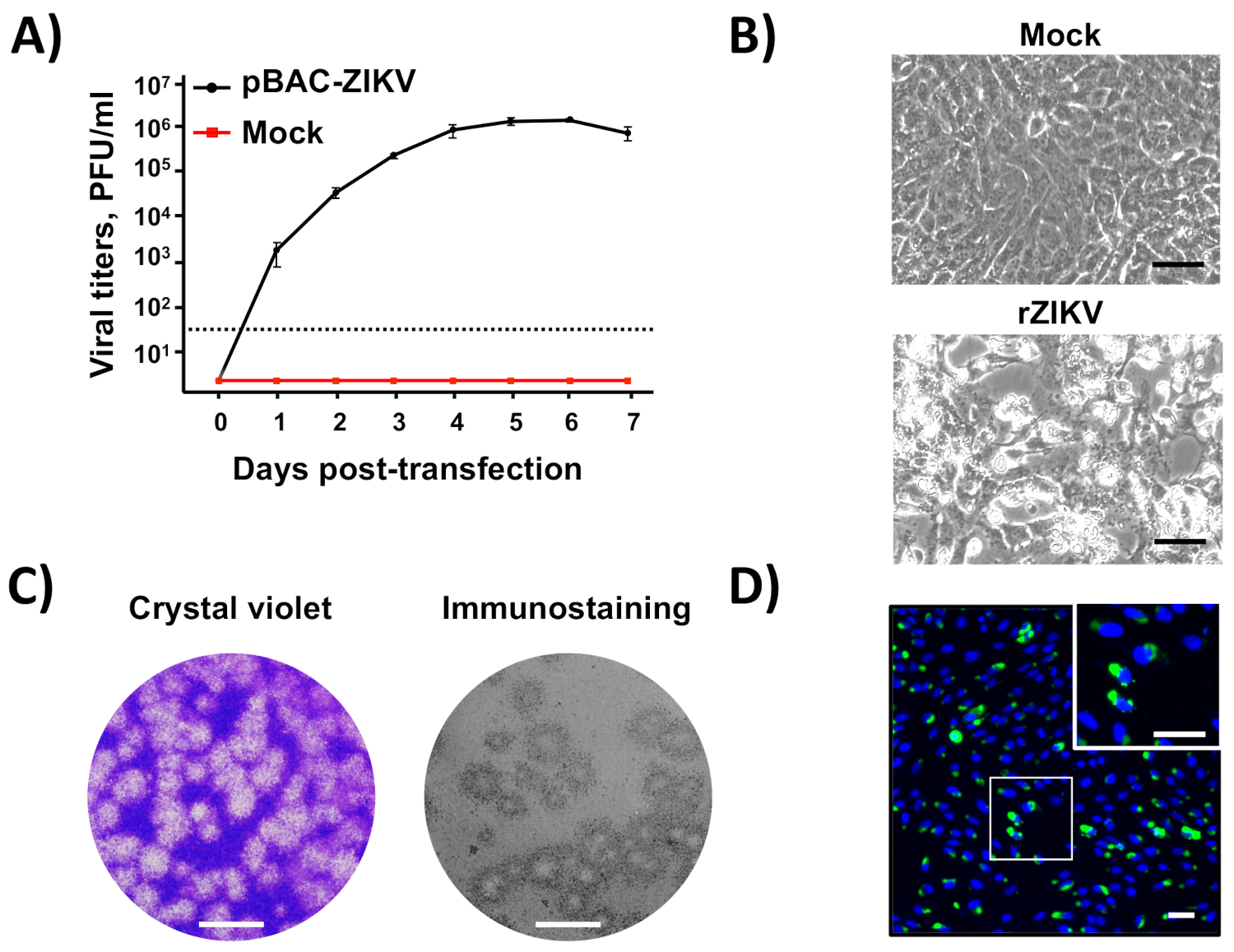
Once the BAC cDNA clone was assembled (Figure 2), infectious virus could be easily recovered after the direct transfection of susceptible Vero cells with the BAC cDNA clone using cationic liposomes(Figure 3). This cDNA-launched system allowed the intracellular expression of the capped vRNA, allowing the recovery of infectious viruses without the need for an in vitro transcription step. Using this approach, we were able to rescue rZIKV-RGN with titers higher than 106 PFU/mL at 5 days posttransfection (Figure 4A). In addition, the rescued virus induced a clear CPE (Figure 4B), generated homogeneous plaques of about 2 mm in size (Figure 4C), and its identity was confirmed by sequencing (data not shown) and immunofluorescence analysis using the mAb specific for ZIKV E protein, 1176-56 (Figure 4D). In vitro data indicate that the recovered rZIKV-RGN replicated efficiently in Vero cells and to levels compared to a natural ZIKV isolate32 (data not shown). Overall, these results demonstrate that infectious rZIKV can be rescued from full-length cDNA clones assembled in a BAC.

Figure 1: ZIKV genome organization and virion structure. (A) Genome organization: ZIKV contains a positive single-stranded RNA that is translated as a single polyprotein. The translated polyprotein was cleaved by viral (arrows) and host (diamonds) proteases to produce the structural proteins capsid (C, blue), matrix (M, brown), and envelop (E, green), and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The 5' and 3' untranslated regions (UTRs) at the end of the viral genome are indicated with black lines. (B) Virion structure: ZIKV virions were decorated with the E and M proteins, anchored in a lipid bilayer with an icosahedral-like structure. Under the viral envelop was the viral nucleocapsid composed of the C protein associated with the viral genomic RNA. This figure has been adapted from Ávila-Pérez et al.18. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Assembly of the ZIKV full-length infectious cDNA clone in a BAC. (A) Schematic representation of the pBeloBAC11 BAC:The regulatory genes parA, parB, parC, and repE,the F-factor replication origin (OriS), the chloramphenicol resistant gene (Cmr), and the lacZ gene are indicated. The relevant restriction sites used to assemble the infectious ZIKV cDNA clone are underlined. (B) Assembly of ZIKV full-length infectious cDNA clone into the pBeloBAC11 BAC:Four overlapping DNA fragments (Z1-Z4), covering the entire ZIKV genome (Figure 1) and flanked by the indicated restriction sites, were generated by chemical synthesis and sequentially cloned into pBeloBAC11 to generate the infectious ZIKV cDNA clone pBAC-ZIKV. The full-length ZIKV infectious cDNA was assembled under the control of the human cytomegalovirus immediate-early promoter (CMV) and flanked at the 3'-end by the HDV ribozyme (RZ) and the bovine growth hormone (BGH) termination and polyadenylation sequences. The acronyms for the viral genes and regulatory elements are as described in Figure 1. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Workflow to generate rZIKV from the BAC cDNA clone. Vero cells at 90% of confluence were transfected in monolayer with the ZIKV full-length infectious cDNA clone pBAC-ZIKV (Figure 2) using cationic liposomes. At 4-6 days posttransfection, when CPE was evident, tissue culture supernatants containing rZIKV were collected and evaluated for the presence of virus (Figure 4) and used for viral amplification in Vero cells. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Recovery and in vitro characterization of rZIKV. (A) Rescue of infectious rZIKV from the BAC cDNA clone: Vero cells at 90% of confluence (6-well plate format, triplicates) were mock-transfected or transfected with 4 µg/well of pBAC-ZIKV (Figure 3), and at the indicated days posttransfection, virus titers in tissue culture supernatants were determined by plaque assay (PFU/mL). The error bars indicate standard deviations from three different transfection experiments.The dotted black line indicates the limit of detection (50 PFU/mL). (B) Viral CPE:Vero cells at 90% confluence (6-well plate format, triplicates) were mock-infected (top) or infected (MOI of 0.5 PFU/cell) with rZIKV, and at 48 h postinfection, the presence of CPE was evaluated by light microscopy. Scale bars = 100 µm. (C) Viral plaque assay and immunostaining: Vero cells at 90% confluence (12-well plate format) were infected with rZIKV, and at 72 h postinfection, viral plaques were visualized by crystal violet staining (left) or by immunostaining (right) using the pan-flavivirus E protein mAb 4G2. Scale bars = 5 mm. (D) Immunofluorescence:Vero cells at 90% confluence (24-well plate format, triplicates) were infected (MOI of 0.5 PFU/cell) with rZIKV and, at 48 h postinfection, analyzed by immunofluorescence using the mAb 1176-56, specific for ZIKV E protein. Cell nuclei were stained with DAPI. A representative merged image of ZIKV-infected Vero cells is shown. The white square in the top right represents an enlarged image of ZIKV-infected Vero cells. Scale bars = 100 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Infectious cDNA clones constitute essential molecular tools for basic research of RNA viruses and the development of vaccines and/or the identification of antiviral strategies. However, for many positive-stranded RNA viruses, including flaviviruses, the generation of infectious cDNA clones are difficult due to the instability of the cloned cDNAs when propagated in bacteria using standard high-copy-number plasmids. In the case of ZIKV and other flaviviruses, this instability is mainly due to the leaky expression of toxic viral proteins from cryptic bacterial promoters present in the viral genome14,15,16,17. Here, we describe an alternative and powerful protocol to generate a stable ZIKV full-length infectious cDNA clone as a single plasmid, based on the use of the BAC plasmid pBeloBAC1134 (Figure 2A) to overcome the toxicity problem, the use of the CMV promoter to allow the expression of the vRNA in the nucleus of transfected cells, and the HDV RZ to generate vRNAs with accurate 3'-ends (Figure 2B). Using this method, we have successfully generated a fully stable infectious clone of the ZIKV strain RGN that allows the efficient and reliable recovery of infectious rZIKV after the direct transfection of susceptible Vero cells (Figure 3 and Figure 4).
A huge effort has been made in the last few years to overcome the instability problems associated with ZIKV infectious cDNA clones, and several approaches have been successfully implemented18, including the in vitro ligation of cDNA fragments24,25, low-copy plasmids19,20, the inactivation of cryptic bacterial promoters by the introduction of silent mutations26,27, intron insertion21,22,23, the Gibson assembly method30, the ISA method28,29, and the use of CPER31. Although these approaches overcome the toxicity problem and are useful to generate ZIKV infectious cDNA clones, some of them are laborious and present several disadvantages, including the need for in vitro ligation and transcription steps that reduce virus recovery efficiency or the introduction of a high number of silent mutations to inactivate cryptic bacterial promoter that could affect viral fitness, among others. The approach described in this protocol presents the following advantages. i) The BAC plasmid pBeloBAC1134 has a strictly controlled replication, keeping one or two copies of plasmid per cell, which minimizes toxicity and allows stable maintenance in bacteria of instable cDNAs. ii) The propagation and modification of BAC plasmids are almost similar to those of conventional plasmids, considering the slight modifications described in this protocol to manipulate large-size BAC-DNA fragments and low-copy plasmids. Notably, the BAC cDNA clone can also be modified into E. coli by homologous recombination using the Red recombination system42,43,44. iii) The use of CMV promoter allows the intracellular expression of capped ZIKV vRNA and the recovery of infectious viruses without requiring an in vitro transcription step. iv) Infectious rZIKV is generated after the direct transfection of susceptible cells (e.g., Vero) with the BAC cDNA clone. Since DNA transfection in mammalian cells is more efficient than RNA transfection, the virus recovery efficiency with the BAC approach is higher than that observed using RNA transcripts, reducing the number of passages in culture cells to generate a viral stock and, consequently, limiting the introduction of unwanted mutations by cell culture adaptation.
Finally, the potential of the BAC approach is supported by the successful use of this method (with slight modifications) to engineer infectious cDNA clones of other flaviviruses, including dengue virus36, and several coronaviruses of high impact in human and animal health, such as transmissible gastroenteritis coronavirus37 (TGEV), feline infectious peritonitis virus38 (FIPV), human coronavirus OC4339 (HCoV-OC43), severe acute respiratory syndrome coronavirus40 (SARS-CoV), and Middle East respiratory syndrome coronavirus41 (MERS-CoV), among others.
In the protocol described here, there are two critical steps that should be considered. One important consideration is identifying appropriate unique restriction sites in the viral genome that are absent in the BAC plasmid. If no adequate restriction sites are available, new restriction sites can be generated during the cloning design by the introduction of silent nucleotide mutations. Another important issue is that the BAC plasmids are present in only one or two copies per cell, and therefore, low yields of BAC plasmids with a high contamination of bacterial genomic DNA are obtained using standard protocols designed for high- and medium-copy-number plasmids. This potential problem is easily overcome using large culture volumes and purifying the BAC plasmid with a commercial kit specifically developed for BAC purification.
In summary, we have developed a powerful ZIKV reverse genetic approach based on the use of a BAC that could be adapted to generate stable and fully functional infectious cDNA clones of other positive-stranded RNA viruses to facilitate the study of the biology of these viruses and the development of vaccines and/or to facilitate the identification of antiviral drugs.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Carla Gómez for her technical assistance in the BAC cDNA clone generation and Snezhana Dimitrova for helping with the video preparation. This work was supported in part by grants from the Spanish Ministry of Economy and Competitiveness (MINECO, grant number BFU2016-79127-R) to F.A.T. and the National Institutes of Health (NIH, grant number 1R21AI120500) to L.M.S. and F.A.T.
Materials
| Name | Company | Catalog Number | Comments |
| 1. Molecular Biology Reagents | |||
| Afe I | New England BioLabs | R0652S | 10,000 Units/mL |
| AmpliTaq DNA Polymerase | ThermoFisher Scientific (Applied Biosystems) | N8080161 | 5,000 Units/mL |
| ApaL I | New England BioLabs | R0507S | 10,000 units/mL |
| Asc I | New England BioLabs | R0558S | 10,000 Units/mL |
| BamH I | New England BioLabs | R0136S | 10,000 Units/mL |
| BstB I | New England BioLabs | R0519S | 20,000 Units/mL |
| Chloramphenicol | Sigma-Aldrich | C0378 | |
| ElectroMAX DH10B Cells | ThermoFisher Scientific (Invitrogen) | 18290015 | Electocompetent DH10B cells |
| Electroporation Cuvettes, 0.2 cm | Bio-Rad | 165-2086 | |
| Ethanol | Merck | 100983 | Flamable |
| Isopropanol | Merck | 109634 | Flamable |
| Large-Construct Kit (10) | QIAGEN | 12462 | For high-purity BAC preparation |
| LB Broth | ThermoFisher Scientific (Invitrogen) | 12780029 | Can be homemade as well |
| LB with Agar | ThermoFisher Scientific (Invitrogen) | 22700041 | Can be homemade as well |
| Methanol | Merck | 106009 | Flamable |
| Mlu I | New England BioLabs | R0198S | 10,000 Units/mL |
| Oligonucleotides | IDT | N/A | |
| Plasmid pBeloBAC11 | New England BioLabs | ER2420S (E4154S) | |
| Plasmid Midi Kit (25) | QIAGEN | 12143 | For midle-scale preparation of BAC plasmids |
| Pml I | New England BioLabs | R0532S | 20,000 Units/mL |
| Polypropylene tubes (10 mL) | DeltaLab | 175724 | Other commercial sources are acceptable |
| QIAEX II Gel Extraction Kit (150) | QIAGEN | 20021 | Gel-clean-up kit optimized for DNA fragments larger than 10 kb |
| Shrimp AlKaline Phosphatase (rSAP) | New England BioLabs | M0371S | 1,000 Units/mL |
| SOC Medium | ThermoFisher Scientific (Invitrogen) | 15544034 | Can be homemade as well |
| Synthesis of cDNA fragments | Bio Basic | N/A | |
| T4 DNA Ligase | Sigma-Aldrich (Roche) | 10481220001 | 1,000 Units/mL |
| 2. Cell Culture Reagents | |||
| 6-Well Plates | ThermoFisher Scientific (Nunc) | 140675 | |
| 12-Well Plates | ThermoFisher Scientific (Nunc) | 150628 | |
| 24-Well Plates | ThermoFisher Scientific (Nunc) | 142485 | |
| Agar Noble | VWR | 214230 | |
| Alexa Fluor 488 Conjugate ant-mouse secondary antibody | Varies | N/A | |
| Biotinylated Anti-Mouse Secondary Antibody | Varies | N/A | |
| Cell Culture Dishes (100 mm x 21 mm) | ThermoFisher Scientific (Nunc) | 172931 | |
| Conical Tubes (15 mL) | VWR | 525-0150 | |
| Conical Tubes (50 mL) | VWR | 525-0155 | |
| Crystal Violet | Sigma-Aldrich | C6158 | |
| DAPI | Sigma-Aldrich | D9542 | Toxic and carcinogenic |
| DEAE-Dextran | Sigma-Aldrich | D9885 | |
| DMEM | ThermoFisher Scientific (Gibco) | 11995065 | |
| Fetal Bovine Serum (FBS) | ThermoFisher Scientific (HyClone)) | SV30160.03 | |
| Formaldehyde | Sigma-Aldrich | F8775 | Toxic and carcinogenic |
| L-Glutamine | ThermoFisher Scientific (Gibco) | 25030081 | |
| Lipofectamine 2000 | ThermoFisher Scientific (Invitrogen) | 11668019 | Transfection reagent |
| Nonessential amino acids | ThermoFisher Scientific (Gibco) | 11140035 | |
| Opti-MEM I Reduced Serum Medium | ThermoFisher Scientific (Gibco) | 31985070 | Transfection medium |
| Pan-flavivirus E protein mAb 4G2 | BEI Resources | NR-50327 | |
| Paraformaldehyde | Electron Microscopy Sciences | 15710-S | Toxic and carcinogenic |
| PBS | ThermoFisher Scientific (Gibco) | 14190144 | |
| Penicillin/Streptomycin | ThermoFisher Scientific (Gibco) | 15140122 | |
| ProLong Gold Antifade Reagent | ThermoFisher Scientific (Invitrogen) | P10144 | |
| Triton-X-100 | Sigma-Aldrich | T8787 | |
| Vectastain ABC Kit | Vector Laboratories Inc | PK-4010 | Avidin/biotin-based peroxidase kit |
| Vero Cells | ATCC | CCL-81 | |
| ZIKV E Protein mAb 1176-56 | BioFront Technologies | BF-1176-56 | |
| 3. Equipment | |||
| Agarose Gel Electrophoresis System | Bio-Rad | 1704468 | Other commercial sources are acceptable |
| Class II Biosafety CO2 Cabinet | Varies | N/A | Other commercial sources are acceptable |
| Desktop Refrigrated Centrifuge | Varies | N/A | |
| Fluorescence Microscope | Varies | N/A | |
| High-speed Refrigrated Centrifuge | Varies | N/A | |
| MicroPulser Electroporator | Bio-Rad | 1652100 | Other machines are acceptable |
| SimpliAmp Thermal Cycler | ThermoFisher Scientific (Applied Biosystems) | A24811 | Other machines are acceptable |
| Vortexer | Varies | N/A |
References
- Friedrich, M. J. WHO Calls Off Global Zika Emergency. JAMA. 317 (3), 246(2017).
- Sirohi, D., et al. The 3.8 Å resolution cryo-EM structure of Zika virus. Science. 352 (6284), 467-470 (2016).
- Lindenbach, B. D., Murray, C. J., Thiel, H. J., Rice, C. M. Flaviviridae. Fields Virology. Knipe, D. M., Howley, P. M. , Wolters Kluwer | Lippincott Williams & Wilkins. 712-748 (2013).
- Boeuf, P., Drummer, H. E., Richards, J. S., Scoullar, M. J., Beeson, J. G. The global threat of Zika virus to pregnancy: epidemiology, clinical perspectives, mechanisms, and impact. BMC Medicine. 14 (1), 112(2016).
- Simpson, D. I. Zika virus infection in man. Transactions of The Royal Society of Tropical Medicine and Hygiene. 58, 335-338 (1964).
- Campos, G. S., Bandeira, A. C., Sardi, S. I. Zika Virus Outbreak, Bahia, Brazil. Emerging Infectious Diseases. 21 (10), 1885-1886 (2015).
- Cao-Lormeau, V. M., et al. Zika virus, French polynesia, South pacific, 2013. Emerging Infectious Diseases. 20 (6), 1085-1086 (2014).
- Faria, N. R., et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science. 352 (6283), 345-349 (2016).
- Costello, A., et al. Defining the syndrome associated with congenital Zika virus infection. Bulletin of the World Health Organization. 94 (6), 406-406A (2016).
- Cugola, F. R., et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 534 (7606), 267-271 (2016).
- do Rosario, M. S., et al. Guillain-Barre Syndrome After Zika Virus Infection in Brazil. American Journal of Tropical Medicine and Hygiene. 95 (5), 1157-1160 (2016).
- Miner, J. J., et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell. 165 (5), 1081-1091 (2016).
- Mlakar, J., et al. Zika Virus Associated with Microcephaly. The New England Journal of Medicine. 374 (10), 951-958 (2016).
- Li, D., Aaskov, J., Lott, W. B. Identification of a cryptic prokaryotic promoter within the cDNA encoding the 5' end of dengue virus RNA genome. PLOS ONE. 6 (3), e18197(2011).
- Aubry, F., Nougairede, A., Gould, E. A., de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: a historical perspective. Antiviral Research. 114, 67-85 (2015).
- Pu, S. Y., et al. Successful propagation of flavivirus infectious cDNAs by a novel method to reduce the cryptic bacterial promoter activity of virus genomes. Journal of Virology. 85 (6), 2927-2941 (2011).
- Ruggli, N., Rice, C. M. Functional cDNA clones of the Flaviviridae: strategies and applications. Advances in Virus Research. 53, 183-207 (1999).
- Avila-Perez, G., Nogales, A., Martin, V., Almazan, F., Martinez-Sobrido, L. Reverse Genetic Approaches for the Generation of Recombinant Zika Virus. Viruses. 10 (11), (2018).
- Annamalai, A. S., et al. Zika Virus Encoding Nonglycosylated Envelope Protein Is Attenuated and Defective in Neuroinvasion. Journal of Virology. 91 (23), (2017).
- Shan, C., et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence Mosquito Transmission, and Antiviral Inhibitors. Cell Host & Microbe. 19 (6), 891-900 (2016).
- Liu, Z. Y., et al. Characterization of cis-Acting RNA Elements of Zika Virus by Using a Self-Splicing Ribozyme-Dependent Infectious Clone. Journal of Virology. 91 (21), (2017).
- Schwarz, M. C., et al. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere. 1 (5), (2016).
- Tsetsarkin, K. A., et al. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. MBio. 7 (4), (2016).
- Deng, C. L., et al. Recovery of the Zika virus through an in vitro ligation approach. Journal of General Virology. 98 (7), 1739-1743 (2017).
- Widman, D. G., et al. A Reverse Genetics Platform That Spans the Zika Virus Family Tree. mBio. 8 (2), (2017).
- Munster, M., et al. A Reverse Genetics System for Zika Virus Based on a Simple Molecular Cloning Strategy. Viruses. 10 (7), (2018).
- Zhao, F., et al. Negligible contribution of M2634V substitution to ZIKV pathogenesis in AG6 mice revealed by a bacterial promoter activity reduced infectious clone. Scientific Reports. 8 (1), 10491(2018).
- Atieh, T., Baronti, C., de Lamballerie, X., Nougairede, A. Simple reverse genetics systems for Asian and African Zika viruses. Scientific Reports. 6, 39384(2016).
- Gadea, G., et al. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology. 497 (Supplement C), 157-162 (2016).
- Weger-Lucarelli, J., et al. Development and Characterization of Recombinant Virus Generated from a New World Zika Virus Infectious Clone. Journal of Virology. 91 (1), (2017).
- Setoh, Y. X., et al. De Novo Generation and Characterization of New Zika Virus Isolate Using Sequence Data from a Microcephaly Case. mSphere. 2 (3), (2017).
- Marquez-Jurado, S., et al. An Alanine-to-Valine Substitution in the Residue 175 of Zika Virus NS2A Protein Affects Viral RNA Synthesis and Attenuates the Virus In Vivo. Viruses. 10 (10), (2018).
- Cheng, B. Y. H., Ortiz-Riaño, E., de la Torre, J. C., Martínez-Sobrido, L. Generation of Recombinant Arenavirus for Vaccine Development in FDA-Approved Vero Cells. Journal of Visualized Experiments. (78), (2013).
- Wang, K., Boysen, C., Shizuya, H., Simon, M. I., Hood, L. Complete nucleotide sequence of two generations of a bacterial artificial chromosome cloning vector. BioTechniques. 23 (6), 992-994 (1997).
- Shizuya, H., et al. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proceedings of the National Academy of Sciences of the United States of America. 89 (18), 8794-8797 (1992).
- Usme-Ciro, J. A., Lopera, J. A., Enjuanes, L., Almazan, F., Gallego-Gomez, J. C. Development of a novel DNA-launched dengue virus type 2 infectious clone assembled in a bacterial artificial chromosome. Virus Research. 180, 12-22 (2014).
- Almazan, F., et al. Engineering the largest RNA virus genome as an infectious bacterial artificial chromosome. Proceedings of the National Academy of Sciences of the United States of America. 97 (10), 5516-5521 (2000).
- Balint, A., et al. Molecular characterization of feline infectious peritonitis virus strain DF-2 and studies of the role of ORF3abc in viral cell tropism. Journal of Virology. 86 (11), 6258-6267 (2012).
- St-Jean, J. R., et al. Recovery of a neurovirulent human coronavirus OC43 from an infectious cDNA clone. Journal of Virology. 80 (7), 3670-3674 (2006).
- Almazan, F., et al. Construction of a severe acute respiratory syndrome coronavirus infectious cDNA clone and a replicon to study coronavirus RNA synthesis. Journal of Virology. 80 (21), 10900-10906 (2006).
- Almazan, F., et al. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio. 4 (5), e00650-e00613 (2013).
- Jamsai, D., et al. Targeted modification of a human beta-globin locus BAC clone using GET Recombination and an I-Scei counterselection cassette. Genomics. 82 (1), 68-77 (2003).
- Lee, E. C., et al. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 73 (1), 56-65 (2001).
- Tischer, B. K., von Einem, J., Kaufer, B., Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. BioTechniques. 40 (2), 191-197 (2006).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved