
Method Article
Detecting and Characterizing Protein Self-Assembly In Vivo by Flow Cytometry
In This Article
Summary
This article describes a FRET-based flow cytometry protocol to quantify protein self-assembly in both S. cerevisiae and HEK293T cells.
Abstract
Protein self-assembly governs protein function and compartmentalizes cellular processes in space and time. Current methods to study it suffer from low-sensitivity, indirect read-outs, limited throughput, and/or population-level rather than single-cell resolution. We designed a flow cytometry-based single methodology that addresses all of these limitations: Distributed Amphifluoric FRET or DAmFRET. DAmFRET detects and quantifies protein self-assemblies by sensitized emission FRET in vivo, enables deployment across model systems-from yeast to human cells-and achieves sensitive, single-cell, high-throughput read-outs irrespective of protein localization or solubility.
Introduction
Assays to study homotypic protein interactions, or "self-assembly" are important because the oligomeric state and solubility of proteins dictate their function. The proteome abounds with homo-multimers1,2,3,4, whereas relatively few proteins function as monomers. Proteins can also assemble aberrantly due to stress, age, or misregulation, leading to pathological changes in activity. Identifying the factors that modulate such events, or even the physical nature of the assemblies, is often exceptionally challenging.
A growing number of proteins are now recognized to self-assemble with extraordinary cooperativity and indeterminate stoichiometry, resulting in their demixing from other cellular constituents, as protein-dense phases. These take the form of disordered condensates, such as droplets and gels, or highly ordered filaments, such as amyloid fibers. The conformational fluctuations associated with the latter render its initial formation, or nucleation, inherently probabilistic at the molecular level5,6. Because the probability of nucleation scales with volume, the formation of such assemblies can be highly stochastic in the spatial confines of living cells7,8. At the extreme of stochastic nucleation-limited phase separation are prions, highly ordered protein assemblies that only rarely nucleate spontaneously, but once formed, template their own growth indefinitely. One such protein, known as ASC, executes a digital prion-like switch in the activity of mammalian innate immune cells. ASC self-assembly is nucleated by its interaction with specific proteins that have themselves oligomerized upon binding pathogen- or danger-associated molecular patterns. The ASC assemblies in turn nucleate procaspase-1 to self-assemble and activate, leading to cytokine maturation and pyroptosis of the cell9,10. The region of ASC responsible for its assembly belongs to the death domain superfamily, which consists of over one-hundred members in the human proteome. Despite the pivotal roles of death domains in innate immunity and programmed cell death, most of them have not yet been characterized with respect to self-assembly. The discovery and characterization of additional proteins with such behavior will be greatly facilitated by a direct, single-cell readout of protein self-assembly.
Classical protein biochemistry approaches to study protein self-assembly, such as size-exclusion chromatography and ultracentrifugation, are largely limited to population level assessments. However, cell-to-cell heterogeneity resulting from nucleation-limited phase transitions cannot be modeled with this level of detail. Single-cell approaches based on fluorescence microscopy regain this capability, but lack the throughput necessary to accurately quantify nucleation or to detect rare assemblies. Moreover, soluble self-assemblies such as most enzymes and pre-amyloid oligomers, are too small and mobile to be resolved by standard light microscopy. They can be detected by more sophisticated approaches such as fluorescence correlation spectroscopy, but these are very limited in cell number and throughput.
Proximity-based assays of protein assembly, such as FRET and split fluorophore complementation, offer a potential solution to these problems. However, they generally require the use of two different constructs expressing the protein of interest fused to complementary tags-the donor and acceptor fluorophores in the case of FRET. This compromises experimental throughput and also reduces sensitivity due to cell-to-cell variation in the relative levels of donor and acceptor. To circumvent this, we designed an assay that employs a photoconvertible fluorophore, mEos3.111, which allows a single construct to express both donor- and acceptor-tagged protein. The emission spectrum of unconverted mEos3.1 (GFP-like donor) sufficiently overlaps with the excitation spectrum of photoconverted mEos3.1 (dsRed-like acceptor) to allow FRET to occur when the molecules are in close proximity (<10 nm). Thus, by exposing cells to an empirically determined dose of 405 nm light, which photoconverts an optimal fraction of total mEos3.1 into the acceptor form, we achieve consistent and reproducible relative levels of donor and acceptor across multiple samples, expression levels, and experiments. We measure the acceptor fluorescence when excited either directly with 561 nm light, or indirectly (by energy transfer from the donor) with 488 nm light (i.e., sensitized emission FRET). We report protein assembly as the ratio of these two values and term it amphifluoric FRET or AmFRET.
In order to calculate protein concentration by flow cytometry, we first calculate the mean fluorescence intensity of Spc42 tagged with mEos3.1. Because yeast cells contain approximately 1000 molecules of Spc42, we then calculate the fluorescence intensity of a single fluorescent green mEos3.1 molecule. By leveraging the even photoconversion at all cellular concentrations (Figure 1E), we then correlate total mEos3.1 fluorescence values for all acceptor intensities following photoconversion. We are then able to divide the total number of moles of fluorescent proteins by the approximate cytosolic volume (as determined using imaging flow cytometry) to obtain the total cytosolic concentration of the protein of interest. For exact calculations, please see the original manuscript8.
By expressing the mEos3.1-fused protein from a 2µ plasmid in yeast, we probe an approximately thousand-fold range of protein concentration in every sample8. We achieve the same in HEK293T cells by virtue of variation in plasmid uptake during transfection, and hence variable copy number.
The resulting distribution of AmFRET, or DAmFRET, for many thousands of cells reveals the concentration-dependence of self-assembly in the cytosol for any protein of interest. Overall, DAmFRET represents an enabling methodology to discover and characterize protein self-assembly with an unprecedented combination of sensitivity, throughput, and reproducibility.
While using an imaging cytometer enables us to obtain in vivo protein concentration measurements, such cytometers are not yet available at most research institutions. Nevertheless, DAmFRET can be run even in a typical non-imaging cytometer to get distributions of protein assembly over the range of protein expression.
Protocol
1. Preparation of Saccharomyces cerevisiae for DAmFRET assay
- Transform experimentally relevant yeast strains with a 2µ galactose-inducible plasmid that expresses the protein of interest8 tagged either at the C or N terminus with mEos3.1 using a standard lithium acetate protocol12 .
- Grow the yeast in liquid culture.
- For every query protein, inoculate yeast colonies (transformed), in triplicate, each into 200 µL of appropriate non-inducing growth media (i.e., media containing 2% dextrose, yeast nitrogen base and amino acids for selection of the plasmid) in a 96-well plate (Figure 1C).
- Incubate cells while shaking on a plate shaker (see the Table of Materials) with 1.5 mm orbit at 1200 rpm at 30 °C for 16 h.
- Induce expression of the gene of interest (representative protocol for 16 h-some proteins may take more or less time).
- Following 16 h of induction, spin the plate at 2200 x g for 2 min at room temperature (RT0 to pellet the samples.
- Remove media by forceful inversion. Resuspend cells with 200 µL of appropriate induction media (i.e., media containing 2% galactose, yeast nitrogen base and amino acids for selection of the plasmid). See Figure 1C.
- Incubate cells while shaking at 30 °C for 12 h.
- Centrifuge (see the Table of Materials) the plate at 2200 x g for 2 min at RT to pellet the samples. Remove media by forceful inversion. Resuspend cells with 200 µL of the induction media.
- Incubate cells while shaking at 1200 rpm at 30 °C for a further 4 h. This resuspension in fresh media should happen 4 h prior to running DAmFRET, in order to reduce autofluorescence.
2. Mammalian plasmid creation
- Construct a mammalian vector that has a constitutive promoter and mEos3.1 with a placeholder to insert the gene of interest and a linker in between them.
NOTE: We constructed a golden gate compatible vector, M1, from a publicly available plasmid (see the Table of Materials) with the following modifications: an existing BsaI site was removed by a point mutation G to A (at position 3719 as per the deposited sequence). mEos3.1 was obtained using site-directed mutagenesis of the fluorophore at I157V. Inverted BsaI sites followed by 4x(EAAAR) linker were inserted, by Gibson assembly, in-frame between CMV promoter and mEos3.1 to produce the final M1 vector. Protein expression is driven by CMV enhancer and promoter; and transcription termination by SV40 polyadenylation signal. - Create a library of inserts, compatible with the constructed vector.
NOTE: We order our inserts for Golden Gate assembly as synthetic linear fragments (see table of materials) flanked by BsaI sites for ligation into M1 between CMV promoter and 4x(EAAAR)-mEos3.1.
3. Mammalian cell culture and transfection
- Culture HEK293T cells in DMEM + 10% FBS + 1x PenStrep media at 37 °C at 5% CO2.
- On the day prior to transfection, seed 5 x 105 cells in a 6-well plate with 2 mL of media.
- Transfect cells with 2 µg of plasmid DNA using a transfection reagent (see the Table of Materials) at a ratio of 3:1 (transfection reagent:DNA). Mix the components in 150 µL of reduced serum media (see the Table of Materials) and incubate it at RT for 15 min, add the mix to each well and incubate for 48 h for protein expression.
- Confirm protein expression after 24 h by epifluorescence microscopy (488 nm excitation, 515 nm emission).
4. Preparation of mammalian cells for DAmFRET assay
- After 48 h of protein expression, remove media from each well and carefully wash cells with 1 mL of 37 °C phosphate-buffered saline (PBS). Following washing, aspirate the PBS.
- Add 0.5 mL of trypsin-ethylenediaminetetraacetic acid (EDTA) (0.25%) and incubate for 5 min at 37 °C.
- Add 0.5 mL of complete DMEM media to each well, resuspend cells and ensure no large clumps are visible.
- Transfer the whole volume of each well into 1.5 mL tubes.
- Spin cells for 5 min at 1000 x g at room temperature (RT).
- Remove the supernatant and resuspend cell pellet in 1 mL of PBS + 10 mM EDTA.
- Spin the cells for 5 min at 1000 x g at RT.
- Remove the supernatant and resuspend the cell pellet in 1 mL 4% paraformaldehyde (PFA) + 10 mM EDTA in PBS.
- Fix cells for 5 min on a shake table with constant movement.
- Spin cells for 5 min at 1000 x g at RT.
- Remove the supernatant and resuspend the cell pellet in 1 mL of PBS + 10 mM EDTA.
- Spin cells for 5 min at 1000 x g at RT.
- Remove the supernatant and resuspend the cells in adequate volume of buffer for the cytometry run (for 1 well of a 96-well plate use 200 µL of PBS + 10 mM EDTA).
- Transfer the resuspended cells to one well of a round bottom 96-well plate.
5. Photoconversion of yeast and mammalian cells for the assay
- Photoconvert samples in a microplate with no cover using a UV lamp (see the Table of Materials) fitted with a 320-500 nm (violet) filter and a beam collimator, positioned 45 cm above the plate, for a duration of 25 min while shaking. These conditions are appropriate when the beam power at the plate is 11.25 mW/cm2, with approximately 17,000 mJ/cm2 of total photon dose8.
6. DAmFRET data collection
- Assay cells using a cytometer with non-collinear 488/561 lasers. The following data are the minimum requirements for calculation of FRET.
- Use a 488 nm excitation/ 515 nm emission channel for the collection of the donor fluorescence.
- Use a 488 nm excitation/ 595 nm emission channel for the collection of the fluorescence of the FRET signal.
- Use a 561 nm excitation/ 595 nm emission channel (non-collinear with the 488/595 channel) for collection of the acceptor fluorescence.
- Use a 405 nm excitation/ 457 nm emission channel for the collection of autofluorescence8.
- Utilize a brightfield image channel for volume calculation.
NOTE:Imaging cytometer settings for S. cerevisiae are as follows:60x objective at low flow rate and high sensitivity; laser powers set to 405 nm at 15 mW, 488 nm at 15 mW, 561 nm at 20 mW.Imaging cytometer settings for HEK293T cells are as follows:40x objective at low flow rate and high sensitivity; laser powers set to 405 nm at 15 mW, 488 nm at 15 mW, 561 nm at 20 mW.Also note that, our imaging cytometer was custom-designed to have spatially separated 488 nm and 561 nm lasers (i.e., on different cameras) so as to obtain clear resolution of FRET from acceptor fluorescence.
- Collect data for 20,000-50,000 single cells per sample within a fluorescence positive gate.
- Gate single cells by a high aspect ratio and a small cell area as opposed to clumped cells or debris which would have low aspect ratio and large or very small cell areas respectively.
NOTE: The equivalent parameters in a non-imaging cytometer are FSC (forward scatter) for approximate cell size and SSC (side scatter) for cell granularity. Additionally, use FSC pulse height versus pulse width as a proxy for single cell gating. Additionally, it is crucial to not collect events that have fluorescence values beyond the upper limit of sensitivity of the cytometer. In our imaging cytometer, we restrict collection of events to a raw maximum pixel value of one less than the saturation limit for every fluorescent channel we collect. Similarly, for conventional flow cytometers, data points in the highest intensity bin (which includes events that are beyond the dynamic range of detection) should not be analyzed.
- Gate single cells by a high aspect ratio and a small cell area as opposed to clumped cells or debris which would have low aspect ratio and large or very small cell areas respectively.
7. Data analysis
- Perform compensation using a non-photoconverted mEos3.1 sample for the pure donor signal and monomeric DsRed2, which has a similar spectrum to the red form of mEos3.1, for the pure acceptor signal13. For more sensitivity, ensure that the FRET detector channel is also considered as a spillover target, in the analysis program.
- Gate samples to select only single cells which are fluorescence positive (as in Figure 2).
- Calculate the AmFRET parameter as the ratio of total FRET signal (488ex/595em) divided by the total FRET acceptor (561ex/595em) signal.
- Visualize data using a flow cytometry software (see the Table of Materials).
NOTE: Cytosolic concentrations for S. cerevisiae in this study were calculated with the settings below as in Khan et al.8. Data analysis was performed using flow cytometry software (see Table of Materials).
Results
Detection of oligomers that do not form puncta
We have previously applied DAmFRET for the characterization of protein phase separation, which typically results in the formation of large protein assemblies that can also be detected by fluorescence microscopy. To demonstrate the applicability of DAmFRET to diffraction-limited protein assemblies, we analyzed a biochemically well-characterized protein that forms discrete homo-heptamers that are far too small to be visualized by light microscopy: the human co-chaperonin, HSPE114. We compared the DAmFRET profiles of yeast cells expressing mEos3.1 alone, or HSPE1-mEos3.1. The latter exhibited uniform positive AmFRET values, while the former exhibited negligible AmFRET (Figure 3A). The images of cells acquired by the imaging flow cytometer (see the Table of Materials) revealed diffuse fluorescence in all channels-donor, FRET and acceptor, for both mEos3.1- and HSPE1-mEos3.1-expressing cells (Figure 3B). To confirm this finding at higher optical resolution, we used confocal microscopy to capture z-stacks for multiple fields of cells. Indeed, the fluorescence was uniformly distributed throughout the cytosol, with no detectable puncta, whether the cells expressed HSPE1-mEos3.1 or the fluorophore alone (Figure 3C). Note that the characterized Kd of HSPE1 is around 3 µM which is slightly below the sensitivity of our system, and hence we do not observe the sigmoidal relationship of FRET to concentration that would be expected for this discrete homo-oligomer. Nevertheless, we conclude that DAmFRET robustly detects soluble homo-oligomerization in vivo at single cell resolution.
Detection of nucleation-limited assemblies
To showcase the ability of DAmFRET to distinguish discrete homo-oligomers from nucleation-limited assemblies such as prions, we analyzed the human inflammasome protein ASC. Whereas WT ASCPYD forms filaments in vivo, the inactive R41E mutant of ASCPYD does not9,10. We expressed in yeast cells either mEos3.1 alone, or mEos3.1 fused to either WT or R41E ASCPYD. Yeast cells expressing the fluorophore alone or the mutant form of ASCPYD exhibited negligible AmFRET over the entire concentration range, indicating an inability to self-interact. In contrast, WT ASCPYD exhibited a DAmFRET profile with two populations: one with negligible AmFRET and the other with high AmFRET (Figure 4A). As confirmed by the fluorescence images (Figure 4B), these populations represent cells that contain only soluble protein or instead contain mostly self-assembled protein, respectively. The discontinuous relationship between the populations, and the fact they occur at overlapping concentrations indicates that a nucleation barrier stabilizes the monomeric form of the protein and can keep it from assembling over the duration of the experiment8. The gap in AmFRET between the two populations indicates that, once nucleation occurs, it near instantaneously templates other monomers to the assembled form and achieves a new steady state level of AmFRET. DAmFRET corroborated prior structural data that the point mutant ASCPYD R41E disrupted nucleation across the concentrations achievable by this expression system (Figure 4A).
Applicability of DAmFRET in mammalian cells
Although yeast cells are ideal host cells for DAmFRET, we desired to extend the applicability of DAmFRET to mammalian cells. In order to avoid cell death caused by functional ASC polymers, we tested DAmFRET in HEK293T cells that lack caspase-1 expression. We expressed the same proteins in HEK293T cells as we did in yeast cells in Figure 4A. The resulting DAmFRET profiles in HEK293T cells qualitatively resemble those in yeast cells (Figures 4A,C). DAmFRET, thus, serves as the most versatile in vivo method to detect and quantify nucleated protein self-assemblies at single cell resolution with high throughput irrespective of both the presence of microscopically visible puncta as well as the cell type.

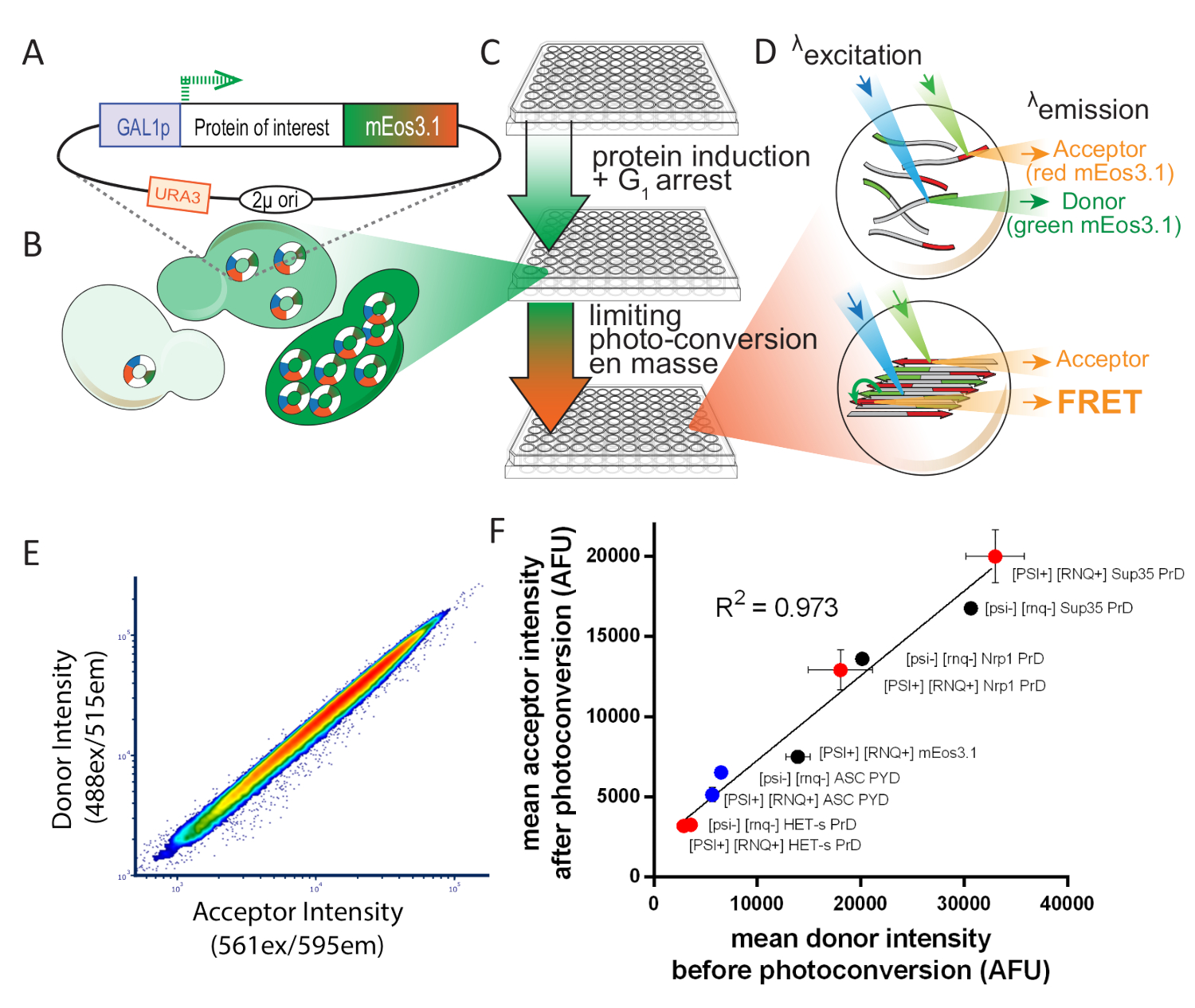
Figure 1: Overview of experimental design for S. cerevisiae. This figure has been adapted from Khan et al.8 with permission. (A) 2µ plasmid map showing the open reading frame for the protein of interest, tagged with photoconvertible mEos3.1 and driven by an inducible promoter. (B) When transformed into yeast cells, the 2µ system of replication leads to high copy number variability among cells. That variation, combined with the transcriptional noise from the GAL1 promoter, results in a broad distribution of protein expression in a population of cells. (C) Experimental overview for yeast DAmFRET assay. To ensure healthy cells for the assay, colonies are first inoculated for proliferation over 16 h in synthetic media containing the non-inducing carbon source, dextrose. Following proliferation, cells are transferred to synthetic media containing the inducing carbon source, galactose, for 16 h. Following induction, cells are partially and uniformly photoconverted by exposure to 405 nm light. (D) Samples are then analyzed using imaging flow cytometry. The spectra and intensities of green and red mEos3.1 render them well-suited for a FRET donor and acceptor, respectively. When in close proximity to one another, as occurs in the polymer depicted in the bottom cell, the red molecules will fluorescence upon excitation of green molecules (FRET). (E) Plot of donor versus acceptor intensities showing a tight linear relationship upon photoconversion, showing that the efficiency of photoconversion is not influenced by expression level. (F) Scatter plot of mean acceptor versus donor intensities from populations of cells expressing a variety of mEos-tagged proteins, in both soluble and amyloid forms, showing that the efficiency of photoconversion is not influenced by the fusion partner or solubility (error bars indicate standard deviation of biological triplicates). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: DAmFRET gating strategy for imaging flow cytometry data. Gating strategy used to analyze only focused, unbudded single cells which have low autofluorescence and express the fluorescent protein. (A) Hierarchy of gates to obtain well-focused, live, single cells for analysis. (B) Histogram of gradient RMS of brightfield channel. This measurement allows for the selection of cells which are properly focused under transmitted light. (C) Density plot of circularity versus area showing gated population of unbudded spherical single cells. (D) Density plot showing autofluorescence (Ch07) versus donor fluorescence (Ch02) intensity showing separate populations of expressing cells (gated) and dark cells. (E) Scatter plot of donor versus acceptor intensities showing a clear loss in donor fluorescence in a subset of cells, resulting from FRET between the donor and acceptor fluorophores. (F) Final DAmFRET plot. Cells containing unassembled protein are centered around zero AmFRET, while cells containing self-assembled protein exhibit a positive AmFRET value. Two overlapping populations in the plot is indicative of a finite barrier to nucleating that protein into higher-order assemblies such as amyloids. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative data of monomeric and heptameric proteins by flow cytometry and microscopy. (A) DAmFRET profile of monomeric mEos3.1 protein (left) and heptameric HSPE1 tagged with mEos3.1 (right) showing increased ratiometric FRET resulting from homotypic assembly. (B) Images from the cytometer, of cells expressing mEos3.1 (left) and HSPE1 (right) shown in all captured channels. (C) Representative images of yeast cells expressing monomeric mEos3.1 (left) and HSPE1 (right). The images are sum projections of confocal slices. At least fifty cells were imaged to corroborate this observation. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: DAmFRET profiles show similar self-assembly behavior of human ASCPYD protein in S. cerevisiae and HEK293T cells. (A) Density plots showing AmFRET versus cytosolic concentration (in µM) of mEos3.1 (left), or mEos3.1 fused to WT (center) or monomerizing mutant of ASCPYD (right) in S. cerevisiae. (B) Images from the cytometer, of cells from the lower and upper gates (left and center panels, respectively) expressing WT ASCPYD; and of cells expressing ASCPYD R41E of the indicated gate (right panel). (C) Density plots showing AmFRET versus acceptor intensity for the same series of proteins as in (A) but expressed in HEK293T cells. Please click here to view a larger version of this figure.
{kind=link}
Discussion
DAmFRET is the most comprehensive method to detect protein self-assembly in vivo. DAmFRET combines direct read-out of homotypic protein-protein interactions over a wide range of concentration, with single cell resolution and high throughput. The direct read-out of DAmFRET and the fact that the fused proteins do not require a specific subcellular localization or insoluble state eliminates false positives and extends its applicability to a broad range of proteins at their native subcellular locations. Notably, by tagging organelles with fluorophores that are spectrally compatible with mEos3.1, such as T-Sapphire and mCardinal, the subcellular localization of soluble and assembled forms of proteins can be determined alongside DAmFRET.
Using the imaging flow cytometer as described here, it takes 8 h to analyze a 96-well plate with approximately 20,000 gated cells per well. However, we also routinely perform DAmFRET with standard (non-imaging) cytometers that achieve much higher throughput (up to 20 samples per minute). In fact, any flow cytometer with spatially separated 488 and 561 nm lasers that is free from log amp artifacts and has the appropriate PMTs and filters for detecting donor, FRET, and acceptor signals is sufficient to perform DAmFRET. At present, this gain in throughput comes at the expense of localization information and volume determination, such that self-assembly must then be analyzed as a function of protein expression rather than concentration. This is not a problem for qualitative analyses. Additionally, it may be possible to estimate cytosolic volume by fusing a spectrally distinct fluorophore to an endogenous "housekeeping" protein whose expression tightly correlates with cytosolic volume.
As we have demonstrated through our deployment of DAmFRET in both yeast and mammalian cells, DAmFRET can be easily adapted to different expression systems. This enables the self-assembly of proteins to be studied in their native cellular contexts. Moreover, it offers the ability to compare protein assembly across widely divergent cell-culture models to study the conservation of mechanisms that govern protein self-assembly.
Disclosures
The authors declare no competing interests.
Acknowledgements
We would like to thank Jeff Lange, Jay Unruh, Jianzheng Wu, Tarique Khan, and Ellen Ketter for their work towards development of the assay. This work was done to fulfill, in part, requirements for PhD thesis research for T.S.K. and A.R.G as students registered with the Open University, UK, and the Stowers Institute for Medical Research Graduate School, USA, respectively. Additional assay-related information can be found at https://doi.org/10.1016/j.molcel.2018.06.016. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-1372. This work was funded by NIH Director’s Early Independence Award DP5-OD009152, March of Dimes Foundation Grant No. 5-FY17-32, and the Stowers Institute for Medical Research.
Author contributions are as follows. Conceptualization: T.S.K., S.V., and R.H.; Methodology: T.S.K., S.V., A.R.G., and A.B; Investigation: S.V., T.S.K., and A.R.G.; Formal Analysis: S.V., and T.S.K. ; Data Curation: T.S.K.; Visualization: T.S.K, and S.V., Writing (Original Draft): S.V., and T.S.K.; Writing (Review, Editing): R.H., S.V., and T.S.K.; and Funding Acquisition: R.H.
Materials
| Name | Company | Catalog Number | Comments |
| 96 Well Plate | Axygen | P96-450R-C-S | |
| ASC 2-92 (mammalian plasmid) | rhm1.0095 | Available on request | |
| ASC 2-92 (R41E) (mammalian plasmid) | rhm1.0096 | Available on request | |
| ASC 2-92 (R41E) (yeast plasmid) | rhx2432 | Available on request | |
| ASC 2-92 (yeast plasmid) | rhx2431 | Available on request | |
| Centrifuge | Eppendorf | 5430R | "centrifuge" in text |
| CSM -Ura | Sunrise Science Products | 1004-100 | |
| Dextrose | EMD Millipore | DX0145-5 | |
| DMEM | Gibco | 11966025 | |
| EDTA | Sigma-Aldrich | EDS-500G | |
| FCS Express 6 | DeNovo | FCS Express 6 Flow | "flow cytometry software" in text |
| Fetal Bovine Serum | VWR Life Science | 45001-108 | |
| Flow Cytometer | BioRad | ZE5 | Non-imaging flow cytometer |
| FUGENE HD | Promega | E2311 | "transfection reagent" in text |
| Galactose | VWR Life Science | 0637-500g | |
| HSPE1 (yeast plasmid) | rhx1531 | Available on request | |
| Imaging Flow Cytometer | EMD Millipore | ImageStream X Mark II | "imaging flow cytometer" in text |
| Mammalian Cells | HEK293T | ||
| mEos3.1 (mammalian plasmid) | rhm1 | Available on request | |
| mEos3.1 (yeast plasmid) | rhx0935 | Available on request | |
| optiMEM | Gibco | 31985062 | "reduced serum media" in text |
| PBS | VWR Life Science | 45000-446 | |
| PenStrep | Gibco | 15070063 | |
| PFA | Sigma-Aldrich | P6148-1KG | |
| Photoconversion Lamp | OmniCure | S1000 | |
| Plasmid #54525 | Addgene | #54525 | "publicly available plasmid" in text |
| Titramax 1000 Plate Shaker | Heidolph Instruments | 1000 | "plate shaker" in text |
| Trypsin-EDTA | Gibco | 25200056 | |
| Yeast Strain | rhy1713 | Available on request- S288c (MATα lyp1Δ can1Δ::STE2pr_SpHIS5 his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 cln3Δ0::GAL1pr_WHI5_hphMX) |
References
- Krissinel, E., Henrick, K. Inference of macromolecular assemblies from crystalline state. Journal of Molecular Biology. 372 (3), 774-797 (2007).
- Kühner, S., van Noort, V., et al. Proteome organization in a genome-reduced bacterium. Science. 326 (5957), 1235-1240 (2009).
- Marianayagam, N. J., Sunde, M., Matthews, J. M. The power of two: protein dimerization in biology. Trends in Biochemical Sciences. 29 (11), 618-625 (2004).
- Matthews, J. M., Sunde, M. Dimers, oligomers, everywhere. Advances in Experimental Medicine and Biology. 747, 1-18 (2012).
- Glover, J. R., Kowal, A. S., Schirmer, E. C., Patino, M. M., Liu, J. J., Lindquist, S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell. 89 (5), 811-819 (1997).
- Tanaka, M., Collins, S. R., Toyama, B. H., Weissman, J. S. The physical basis of how prion conformations determine strain phenotypes. Nature. 442 (7102), 585-589 (2006).
- Michaels, T. C. T., Dear, A. J., Knowles, T. P. J. Stochastic calculus of protein filament formation under spatial confinement. New journal of physics. , (2018).
- Khan, T., Kandola, T. S., et al. Quantifying Nucleation In Reveals the Physical Basis of Prion-like Phase Behavior. Molecular Cell. 71 (1), 155-168 (2018).
- Cai, X., Chen, J., et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 156 (6), 1207-1222 (2014).
- Lu, A., Magupalli, V. G., et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 156 (6), 1193-1206 (2014).
- Zhang, M., Chang, H., et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nature Methods. 9 (7), 727-729 (2012).
- Gietz, D., Jean, A., Woods, R. A., Schiestl, R. H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Research. 20 (6), 1425(1992).
- Nishizawa, K., Kita, Y., Kitayama, M., Ishimoto, M. A red fluorescent protein, DsRed2, as a visual reporter for transient expression and stable transformation in soybean. Plant Cell Reports. 25 (12), 1355-1361 (2006).
- Luke, K., Apiyo, D., Wittung-Stafshede, P. Dissecting homo-heptamer thermodynamics by isothermal titration calorimetry: entropy-driven assembly of co-chaperonin protein 10. Biophysical Journal. 89 (5), 3332-3336 (2005).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved