
Method Article
Genotyping and Quantification of In Situ Hybridization Staining in Zebrafish
In This Article
Summary
Gene editing technologies have enabled researchers to generate zebrafish mutants to investigate gene function with relative ease. Here, we provide a guide to perform parallel embryo genotyping and quantification of in situ hybridization signals in zebrafish. This unbiased approach provides greater accuracy in phenotypical analyses based on in situ hybridization.
Abstract
In situ hybridization (ISH) is an important technique that enables researchers to study mRNA distribution in situ and has been a critical technique in developmental biology for decades. Traditionally, most gene expression studies relied on visual evaluation of the ISH signal, a method that is prone to bias, particularly in cases where sample identities are known a priori. We have previously reported on a method to circumvent this bias and provide a more accurate quantification of ISH signals. Here, we present a simple guide to apply this method to quantify the expression levels of genes of interest in ISH-stained embryos and correlate that with their corresponding genotypes. The method is particularly useful to quantify spatially restricted gene expression signals in samples of mixed genotypes and it provides an unbiased and accurate alternative to the traditional visual scoring methods.
Introduction
The introduction of genome editing technologies (ZFN, TALENs and more recently, CRISPR/Cas9) has led to a massive increase in the number of laboratories around the world that make use of these systems to study the function of specific genes in vivo. Zebrafish in particular are amenable to genetic manipulation and many mutants have been generated in the recent past1,2. For developmental biologists, one of the most common methods to assess the phenotypical consequences of gene mutations in embryonic development is in situ hybridization (ISH). In the absence of obvious morphological defects that separate homozygous mutants from their wild type or heterozygous siblings, it is essential to be able to correctly identify different genotypes accurately.
Classical ISH relies on qualitative analyses of signal intensities to derive conclusions about regulatory interactions between the gene of interest and selected marker genes. Although useful, these analyses suffer from technical variation and may be biased by researcher expectations. Thus, a method was developed to quantify gene expression after imaging ISH-stained embryos, without prior knowledge of the corresponding genotype. This was followed by an efficient DNA extraction and genotyping that allowed us to quantitatively correlate genotype with gene expression3. While the genotyping of embryos post ISH has been used before4,5, image-based quantification of ISH patterns has not been widely used apart from a couple of studies6,7. The most popular alternatives rely on visual scoring or counting of ISH-stained cells8,9,10, both prone to poor reproducibility and researcher bias. This method is particularly useful to study changes in genes with expression patterns that are spatially restricted, such as runx1 or gata2b, both expressed in a restricted subset of aortic floor cells called the haemogenic endothelium11,12.
Here, we aim to provide a practical guide to the implementation of the quantification by image analysis using Fiji13, as well as the DNA extraction and genotyping protocol. This is meant to illustrate visually our previously published method3. Our method allows an accurate representation of the variation in gene expression detected by ISH and an unbiased assignment of gene expression levels to specific genotypes.
Protocol
Procedures involving animal subjects are regulated by the Animals (Scientific Procedures) Act 1986 and have been approved by the Home Office and the local Animal Welfare and Ethical Review Body.
1. Image ISH-stained embryos
- Prepare a glycerol solution (50%–80% in 1x PBS buffer) and mix to homogenize the solution (e.g., leave in a roller for at least 5 min). This solution can be kept for months at room temperature.
- After in situ hybridization14,15,16,17, transfer embryos to the glycerol solution with a 3 mL Pasteur pipette and leave to settle for at least 5 min. If imaging embryos older than 24 hpf (hours post fertilization), they may be bleached as described18.
- Prepare and label enough PCR tubes to transfer ISH-stained embryos after imaging.
- Add 100% glycerol to the bottom of the well in a glass depression slide with a 3 mL Pasteur pipette.
- Using a 3 mL Pasteur pipette, transfer a single ISH-stained embryo to the glass slide and orient as required under a stereomicroscope equipped with a digital camera and bottom and top illumination.
NOTE: Use gel loading pipette tips to position the embryos for imaging, but other tools (e.g., forceps, dissecting needle) will be equally adequate. - Using the first embryo, adjust the illumination and exposure time at the desired magnification. Use these conditions for ALL of the embryos in the same experiment (i.e., if imaging 40 embryos from a heterozygous mutant incross, make sure the illumination, exposure time and magnification are the same for all).
- Image as many embryos as required. Label each image with a unique number. After imaging, transfer the embryo to a PCR tube/plate labelled with the same number.
NOTE: Images should be saved as TIF files, but other formats are also adequate.- If required, remove excess glycerol in the PCR tubes/plates.
NOTE: At this point, the embryos can be stored in the PCR tubes for several weeks at room temperature.
- If required, remove excess glycerol in the PCR tubes/plates.
2. Extract DNA and genotype the ISH-stained embryos
NOTE: Here, use a reliable and inexpensive method to isolate genomic DNA based on the HoTSHOT method19 with a DNA extraction efficiency of 95%-100%3.
- After the imaging is complete, add 40-75 µL of alkaline lysis buffer (e.g., HoTSHOT) to each tube.
- Incubate at 95 °C for approximately 30 min and cool the tubes to 4 °C before adding an equal volume of neutralization buffer. An overnight incubation at 4 °C may improve PCR efficiency.
NOTE: At this point, the genomic DNA can be used for genotyping or stored at -20 °C until it is required. - Genotype samples with an appropriate method (e.g., HRMA, RFLP)3,20,21 as required for the mutation of interest.
- Note the genotype corresponding to each sample (e.g., using a spreadsheet software).
3. Quantify the pixel intensity of ISH-stained embryos (image analysis using Fiji software)
- To quantify the in situ hybridization (ISH) staining signal intensity, convert all images to 8-bit grayscale as described3. If the images were saved as .TIF files, use a Fiji macro for batch conversion3. Alternatively, convert images in other formats (e.g., .JPG) to .TIF using appropriate software and then convert to 8-bit grayscale using Fiji. For convenience, here is the step-by-step procedure that was published previously3, with some alterations.
- Open images in Fiji and invert the image with Edit > Invert. Then change the image type to 8-bit (Image > Type > 8-bit).
- Using the polygon selection tool, draw the region of interest (ROI) manually on the image around the region containing the signal.
- Press t to open the ROI manager. Use the ROI manager's Measure command to measure the intensity of the ROI. Copy the mean value from the Results window to a spreadsheet software.
- Move the same ROI, ensuring the same size and shape as the original region, to a region of the zebrafish not containing any staining. Repeat step 3.4 to measure the background.
- To obtain the mean pixel intensity of the ISH signal, subtract the mean intensity value of the background region from that of the stained region for each embryo.
- Assign each intensity value to a genotype (from step 2.3).
4. Analyze the results with appropriate statistical tests
- Plot all the values on one Q-Q plot to identify any deviations from normal distribution.
NOTE: Normal distribution can also be verified with the Kolmogorov-Smirnov test or the Shapiro-Wilk test. However, for large sample sizes there is a high risk of false positives in these tests. - If there are strong deviations from normal distribution, transform all the values (using ln or sqrt functions) to make sure they are normally distributed before proceeding.
- Analyze the differences between the values (transformed if necessary) assigned to each genotype (wt vs. heterozygote vs. mutant) with 2-tailed ANOVA with 95% confidence levels, accounting for the equality of variances with a Levene's test and Welch correction. For pairwise comparisons between each pair of genotypes, use Tukey's (equal variances) or Games-Howell (unequal variances) post-hoc test.
- If the values are not normally distributed despite the transformation, use a non-parametric test (Kruskall-Wallis) to analyze the differences between ranked values and a post-hoc Dunn's multiple comparisons test with Bonferroni correction for pairwise comparisons.
- Plot the untransformed values (from step 3.6) as dot plots for the best representation of the results.
Results
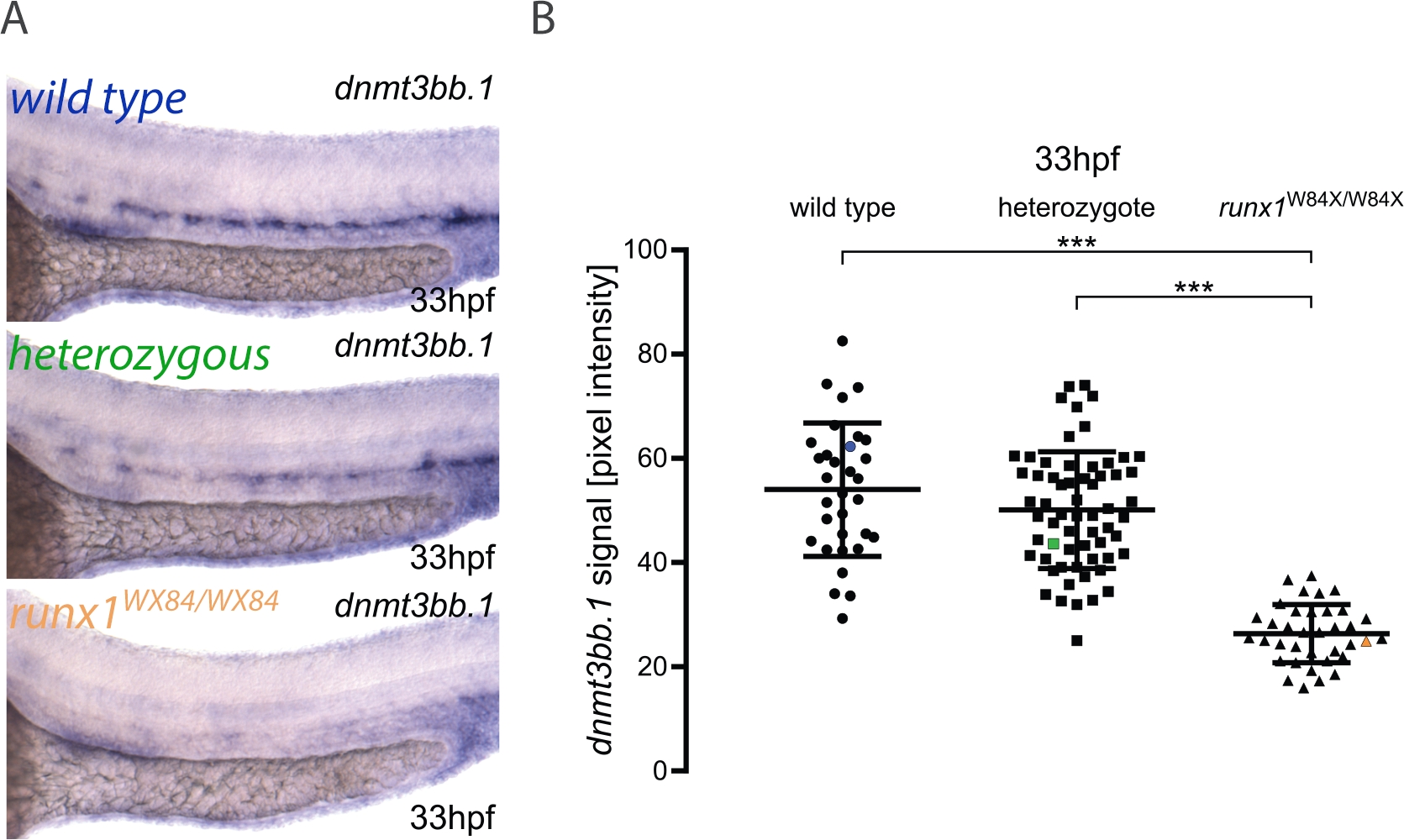
Here, we describe the practical application of the pipeline for image quantitation and embryo genotyping as published elsewhere3. The workflow for the method is shown in Figure 1. To illustrate how to use this method, ISH was performed for dnmt3bb.1 in 33 hpf embryos from a runx1W84X/+ 22 incross (Figure 2). 130 embryos were imaged using the same illumination conditions as detailed in the protocol and labeling them with a unique number. After imaging, each embryo was transferred to a PCR tube for genotyping. At this point, the image analysis was performed to attribute a pixel intensity value to each image. The genotype was then assigned to its corresponding image and the pixel intensity values grouped according to their genotype for statistical analysis. A decrease in dnmt3bb.1 expression was detected in runx1W84X/W84X mutants (Figure 2A,B)3, in agreement with previous observations5. Interestingly, runx1W84X/+ heterozygous embryos showed no significant differences in dnmt3bb.1 expression (Figure 2A,B) compared to its wild type siblings, suggesting that one copy of Runx1 is sufficient to maintain dnmt3bb.1 expression at appropriate levels.
Many zebrafish mutants fail to show an embryonic phenotype that can otherwise be detected using other loss of function technologies like morpholino oligonucleotides (MOs). This discrepancy can be attributed to a number of causes including off-target effects, maternal protein compensation, a hypomorphic allele23 or the recently discovered phenomenon of genetic compensation24,25,26,27. In this example, we asked whether runx1 expression was reduced or lost in lmo4auob100 mutants since previously published data using an lmo4a MO suggested that runx1 is decreased in lmo4a morphants28. Here, the analysis revealed no significant differences in runx1 expression between wild type and lmo4auob100 homozygous mutants3 (Figure 3A,B). Further analysis by single embryo qPCR showed that there was a small but significant decrease in runx1 expression in lmo4auob100 mutants (Figure 3C). Thus, it is possible that image quantification might not be able to detect small differences in expression levels. Alternatively, the lack of difference between genotypes that we detected is real and the qPCR experiments are detecting changes in runx1 expression in other tissues like the telenchephalon where both lmo4a and runx1 are expressed. Researchers should always verify their results with an independent method like qPCR, but ideally enriching for the tissue of interest by flow cytometry, for example.
In rare instances where the ISH has high background (Figure 3D), the pixel intensity value of this area is so high that subtraction from the signal value produces a negative number and in such instances those embryos would be excluded from the analysis. In our experience, this occurred in about 0.4% of the runx1-probed embryos3 but may vary between experiments, probes or batches of reagents. Although that might be a limitation of the method, the low frequency of high background is very unlikely to influence the overall results.
To test the effect of selecting different areas for background corrections, we first measured the pixel intensity of the runx1 ISH signal in 28 hpf embryos, using different regions for background corrections (Figure 4). Four different regions were selected: two in the trunk region (R1, and R2), one in the yolk region (unstained, but likely to accumulate background staining) and a smaller area anterior to the ROI (R4, Figure 4B). Measuring pixel intensity in these regions showed a relatively stable difference in intensity between ROI and either background area (Figure 4C). However, R3 always showed very high values (above those in the ROI). After inversion and conversion to 8-bit, the yolk region appears very bright and thus is not suitable for use as a background correction. R2 was closer to the ROI but contained some ISH signal, and using it for correction decreased the mean pixel intensity when compared with either R1 (located further dorsally, away from the ISH signal) or R4. Thus, either R1 or R4 are appropriate areas that can be used for background correction (despite the area of R4 being smaller than that of R1). Next, we wished to compare how using R1 or R4 affected the outcomes when comparing runx1 expression. For this, we incrossed dll4+/- heterozygotes29 and analyzed runx1 expression in randomly selected wild type and dll4-/-embryos (Figure 4E). Although using R1 or R4 for background correction affected individual values, the mean pixel intensities within the same genotype were not significantly different (Figure 4E). Moreover, comparing runx1 expression still yields similar mean intensity values between genotypes using either R1 or R4 areas as background correction (µR1=16.3 and µR4=18.2, respectively). Taken together, we concluded that although the choice of background area is important, the main criteria is that it does not include yolk regions (prone to accumulation of background staining) and that it should not contain any (specific) staining that might skew the pixel intensity values of the background.

Figure 1: Workflow of the parallel image quantitation and genotyping protocol. Embryos collected from an incross of fish heterozygous for a mutant allele are probed for the measured gene with a standard ISH protocol. After imaging, genomic DNA is extracted using the HotSHOT protocol by adding the lysis buffer directly to the embryo in a 0.2 mL PCR tube, followed by a 30 min incubation at 95 °C. This DNA is used for genotyping of the embryos by PCR, PCR and restriction fragment length polymorphism (RFLP), KASP assays or any other appropriate method. In parallel, the images for each embryo are inverted and converted to 8-bit greyscale. ROIs of identical shape and size containing the ISH signal (yellow) and background (blue) are manually selected and measured. The measurements, assigned to corresponding genotypes, are statistically analyzed. Figure adapted from Dobrzycki et al.3 Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Image quantitation in runx1 mutants reveals reduced levels of dnmt3bb.1 expression by ISH. (A) Example images of ISH in 33 hpf wild type (blue), runx1+/W84X (green) and runx1W84X/W84X (orange) embryos, showing dnmt3bb.1 expression in the dorsal aorta. (B) Pixel intensity values of dnmt3bb.1 mRNA in runx1W84X/W84Xembryos (n=36) are significantly decreased compared to wild types (n=32) and heterozygotes (n=62) (ANOVA, p < 0.001). The coefficients of variation are 24%, 22% and 21% for wild type, heterozygote and mutant groups, respectively. Blue, green and orange data point correspond to the example images from panel A. The bars represent mean ± s.d. ***p<0.001 (Games-Howell post-hoc test). Figure adapted from Dobrzycki et al.3 Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Measuring runx1 expression levels by ISH in lmo4auob100mutants. (A) Representative images of ISH for runx1 in 28 hpf wild type (blue), heterozygous (green) and lmo4auob100/uob100 (orange) embryos, showing the expression in the dorsal aorta. (B) Quantification of the runx1 mRNA signal, detected by ISH, from 28 hpf wild type (n=15), heterozygous lmo4a+/- (het) (n=34) and lmo4auob100/uob100 mutant (n=18) embryos from one clutch shows no significant difference in runx1 pixel intensity among the different genotypes (ANOVA,> p 0.6). Blue, green and orange data point correspond to the example images from panel A. The bars represent mean ± s.d. (C) Boxplots displaying normalized runx1 mRNA levels (2-ΔCt) in single wild type (blue; n=12) and lmo4auob100/uob100 (mut, orange; n=12) embryos, measured by qRT-PCR, showing decreased levels of runx1 in the mutants compared to wild type. *p < 0.05 (t test). (D) Example of an ISH experiment on a 28 hpf embryo (stained for runx1, yellow arrowheads) showing high background. Figure adapted from Dobrzycki et al.3 Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Effect of background intensity correction on measurement outcomes. (A) Representative image of runx1 ISH staining in a wild type embryo at 28 hpf. (B) Same image after inversion and conversion to 8-bit. The region of interest (ROI) is highlighted in green and four different areas used for background correction (R1-R4) are highlighted in yellow. (C) Raw pixel intensity measurements in all regions shown in panel B. Note the intensity in R3 (yolk) is consistently higher than the actual ISH signal in the ROI (n=11). (D) Runx1 expression levels in the ROI using R1, R2 and R4 background areas. Areas for ROI, R1, R2 and R3~28500 pixels; R4~8500 pixels. Note that R3 background was not used for this comparison as the background correction (ROI-R3) consistently yielded negative values. (E) Runx1 expression levels in wild type and dll4-/- mutants using either R1 or R4 for background correction (n=10 for each sample). Statistical analysis in panels D and E was performed using a non-parametric Kruskal-Wallis test, assuming that the pixel intensity values are not normally distributed. Please click here to view a larger version of this figure.
{kind=link}
Discussion
A few factors should be considered when using this method to quantify gene expression. The imaging conditions must be maintained throughout the experiment (for example illumination, exposure times and embryo positioning) to reduce variability between measurements. A critical point is to avoid overstaining of the samples, as differences in staining between samples may be masked. For example, the decrease in VegfA expression in the absence of Eto2 in Xenopus laevis embryos30 could only be detected by carefully monitoring staining over a 24 hour period. Thus, it is good practice to empirically determine adequate staining levels for each gene that best represent its expression, without reaching saturation. Overstaining will also artificially increase the background pixel intensity in the converted 8-bit grayscale images and skew the quantitation results. In extreme cases, the background level in embryonic tissues might be higher than the ISH signal in the selected ROI and these samples should be excluded from the analysis. A similar phenomenon was observed when we tested the suitability of the unstained yolk for background correction (Figure 4). After inversion and conversion to 8-bit the darker pixels in the yolk region become brighter than the ISH signal in the embryo and render the background corrected values negative. Thus, avoid the use of the yolk for background correction. Measuring the background signal in pigmented areas in the embryo (e.g., the eyes or the dorsal part of the trunk from 26/28 hpf onwards) will equally skew the quantitation results and should also be avoided. There are protocols available for bleaching zebrafish embryos, either before or after ISH18 and bleaching embryos older than 24 hpf before imaging is recommended.
Because this method relies on measuring the pixel intensity in a defined area against a background pixel intensity in an equivalent non-stained area, it is not appropriate for quantitation of ubiquitous or near-ubiquitously expressed genes as is. Instead, it is well suited for measuring expression of genes with a spatially restricted distribution where an area for measuring background pixel intensity can be readily identified. Our additional analysis now suggests that using a smaller area (3-4x smaller) for background correction yields similar outcomes to using an equivalent area to that of the ROI. This extends the applicability of the method to genes expressed in wider spatial domains (and thus requiring larger ROIs for intensity measurements), as long as one can use clearly unstained areas of the embryo for background correction.
Finally, we suggest that the genotyping be performed in parallel or after the image quantitation to minimize experimenter bias. Asking a second experimenter to repeat the quantitation on anonymized samples and compare with the first set of measurements will also help reduce experimenter bias. If the images to be quantified are from a comparison between treatments that does not require genotyping (e.g., wild type vs chemical inhibitor or wild type vs. MO knockdown), the experimenter performing the measurements should be blinded to the identity of the sample.
Disclosures
The authors declare they have no competing financial interests.
Acknowledgements
We would like to thank the staff in Biomedical Services in Oxford and Birmingham for excellent zebrafish husbandry. T.D. was funded by a Wellcome Trust Chromosome and Developmental Biology PhD Scholarship (#WT102345/Z/13/Z). R.M. and M.K. were funded by the British Heart Foundation (BHF IBSR Fellowship FS/13/50/30436) and are thankful for their generous support. R.M. acknowledges support from the BHF Centre of Research Excellence (RE/13/1/30181), Oxford.
Materials
| Name | Company | Catalog Number | Comments |
| 0.2 mL PCR tubes (8-strips with lids) | StarLab | A1402-3700 | 96-well plates are equally appropriate for sample handling but beware of cross contamination between samples |
| 3 mL Pasteur pipettes | Alpha Laboratories | LW4114 | |
| Cavity slides | Brand | BR475535-50EA | |
| Digital Camera (Qimaging Micropublisher 5.0) | Qimaging | ||
| Eppendorf Microloader tips | Eppendorf | 10289651 | the tips are used to orient the embryos for imaging in glycerol |
| Excel | Microsoft | ||
| F3000 Fiber Optic Cold Light Source | Photonic | ||
| Fiji | |||
| Glycerol | Sigma | G5516-1L | |
| Graphpad Prism 8.01 | GraphPad Software, Inc. | we prefer to use Graphpad, but other statistics software packages are also suitable (e.g. SigmaPlot or SPSS) | |
| HotSHOT alkaline lysis buffer | 25 mM NaOH, 0.2 mM disodium EDTA, pH 12 | ||
| HotSHOT neutralization buffer | Tris HCl 40 mM, pH 5 | ||
| PBS (10X) pH 7.4 | Thermofisher | 70011044 | |
| Stereomicroscope with illumination stand (Nikon SMZ800N) | Nikon | ||
| Thermocycler | Thermofisher |
References
- Varshney, G. K., et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Research. 25 (7), 1030-1042 (2015).
- Varshney, G. K., et al. CRISPRz: a database of zebrafish validated sgRNAs. Nucleic Acids Research. 44 (D1), D822-D826 (2016).
- Dobrzycki, T., Krecsmarik, M., Bonkhofer, F., Patient, R., Monteiro, R. An optimised pipeline for parallel image-based quantification of gene expression and genotyping after in situ hybridisation. Biology Open. 7 (4), bio031096 (2018).
- Bresciani, E., et al. CBFbeta and RUNX1 are required at 2 different steps during the development of hematopoietic stem cells in zebrafish. Blood. 124 (1), 70-78 (2014).
- Gore, A. V., et al. Epigenetic regulation of hematopoiesis by DNA methylation. Elife. 5, e11813 (2016).
- Fan, Y., et al. Tissue-Specific Gain of RTK Signalling Uncovers Selective Cell Vulnerability during Embryogenesis. PLoS Genetics. 11 (9), e1005533 (2015).
- Wen, B., et al. GATA5 SUMOylation is indispensable for zebrafish cardiac development. Biochimica et Biophysica Acta. 1861 (7), 1691-1701 (2017).
- Espin-Palazon, R., et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 159 (5), 1070-1085 (2014).
- Peterkin, T., Gibson, A., Patient, R. Redundancy and evolution of GATA factor requirements in development of the myocardium. Developmental Biology. 311 (2), 623-635 (2007).
- Genthe, J. R., Clements, W. K. R-spondin 1 is required for specification of hematopoietic stem cells through Wnt16 and Vegfa signaling pathways. Development. 144 (4), 590-600 (2017).
- Kalev-Zylinska, M. L., et al. Runx1 is required for zebrafish blood and vessel development and expression of a human RUNX1-CBF2T1 transgene advances a model for studies of leukemogenesis. Development. 129 (8), 2015-2030 (2002).
- Butko, E., et al. Gata2b is a restricted early regulator of hemogenic endothelium in the zebrafish embryo. Development. 142 (6), 1050-1061 (2015).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Lleras Forero, L., et al. Segmentation of the zebrafish axial skeleton relies on notochord sheath cells and not on the segmentation clock. Elife. 7, (2018).
- Jowett, T., Yan, Y. L. Double fluorescent in situ hybridization to zebrafish embryos. Trends in Genetics. 12 (10), 387-389 (1996).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nature Protocols. 3 (1), 59-69 (2008).
- Narayanan, R., Oates, A. C. Detection of mRNA by Whole Mount in situ Hybridization and DNA Extraction for Genotyping of Zebrafish Embryos. Bio-protocol. , e3193 (2019).
- Monteiro, R., Pouget, C., Patient, R. The gata1/pu.1 lineage fate paradigm varies between blood populations and is modulated by tif1gamma. EMBO JOURNAL. 30 (6), 1093-1103 (2011).
- Truett, G. E., et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 29 (1), 52-54 (2000).
- Wilkinson, R. N., Elworthy, S., Ingham, P. W., van Eeden, F. J. A method for high-throughput PCR-based genotyping of larval zebrafish tail biopsies. Biotechniques. 55 (6), 314-316 (2013).
- Parant, J. M., George, S. A., Pryor, R., Wittwer, C. T., Yost, H. J. A rapid and efficient method of genotyping zebrafish mutants. Developmental Dynamics. 238 (12), 3168-3174 (2009).
- Jin, H., et al. Definitive hematopoietic stem/progenitor cells manifest distinct differentiation output in the zebrafish VDA and PBI. Developement. 136 (4), 647-654 (2009).
- Stainier, D. Y. R., et al. Guidelines for morpholino use in zebrafish. PLoS Genetics. 13 (10), e1007000 (2017).
- El-Brolosy, M. A., Stainier, D. Y. R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genetics. 13 (7), e1006780 (2017).
- Rossi, A., et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 524 (7564), 230-233 (2015).
- Ma, Z., et al. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature. 568 (7751), 259-263 (2019).
- El-Brolosy, M. A., et al. Genetic compensation triggered by mutant mRNA degradation. Nature. 568 (7751), 193-197 (2019).
- Meier, N., et al. Novel binding partners of Ldb1 are required for haematopoietic development. Development. 133 (24), 4913-4923 (2006).
- Kettleborough, R. N., et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 496 (7446), 494-497 (2013).
- Leung, A., et al. Uncoupling VEGFA functions in arteriogenesis and hematopoietic stem cell specification. Developmental Cell. 24 (2), 144-158 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved