Method Article
Flux de spectrométrie de masse d'immunoprécipitation sans étiquette pour le profilage de l'interaction nucléaire à grande échelle
Dans cet article
Résumé
Décrit est un workflow protéomique pour identifier des partenaires d'interaction de protéine d'une fraction subcellulaire nucléaire utilisant l'enrichissement d'immunoaffinité d'une protéine donnée d'intérêt et de la spectrométrie de masse sans étiquette. Le flux de travail comprend la fractionnement subcellulaire, l'immunoprécipitation, la préparation de l'échantillon assisté par filtre, le nettoyage hors ligne, la spectrométrie de masse et un pipeline de bioinformatique en aval.
Résumé
La spectrométrie de masse de purification d'immunoaffinité (IP-MS) est apparue comme méthode quantitative robuste d'identification des interactions protéine-protéine. Cette publication présente un flux de travail protéomique d'interaction complète conçu pour identifier les interactions protéines-protéines à faible abondance du noyau qui pourraient également être appliquées à d'autres compartiments subcellulaires. Ce flux de travail comprend la fractionnement subcellulaire, l'immunoprécipitation, la préparation de l'échantillon, le nettoyage hors ligne, la spectrométrie de masse sans étiquette unique, ainsi que l'analyse de calcul et la visualisation des données en aval. Notre protocole est optimisé pour détecter les interactions compartimentées et à faible abondance qui sont difficiles à identifier à partir de lysates cellulaires entiers (p. ex., interactions des facteurs de transcription dans le noyau) par immunoprécipitation des protéines endogènes compartiments sous-cellulaires fractionnés. Le pipeline de préparation d'échantillons décrit ici fournit des instructions détaillées pour la préparation de l'extrait nucléaire de cellules HeLa, la purification d'immunoaffinité de la protéine endogène d'appât, et l'analyse quantitative de spectrométrie de masse. Nous discutons également des considérations méthodologiques pour effectuer l'immunoprécipitation à grande échelle dans les expériences de profilage d'interaction basées sur la spectrométrie de masse et fournissons des lignes directrices pour évaluer la qualité des données afin de distinguer la véritable protéine positive interactions provenant d'interactions non spécifiques. Cette approche est démontrée ici en étudiant l'interaction nucléaire de la kinase CMGC, DYRK1A, une kinase protéine à faible abondance avec des interactions mal définies dans le noyau.
Introduction
Le protéome humain présente une grande diversité structurelle et biochimique grâce à la formation de complexes multisubunit stables et d'interactions protéinées transitoires. En conséquence, l'identification des partenaires d'interaction pour une protéine d'intérêt est généralement exigée dans les investigations pour démêler le mécanisme moléculaire. Les progrès récents dans les protocoles de purification d'affinité et l'avènement de l'instrumentation de spectrométrie de masse à balayage rapide à haute résolution ont permis une cartographie facile des paysages d'interaction protéique dans une seule expérience impartiale.
Les protocoles d'interaction protéique emploient généralement des systèmes d'expression ectopique avec des constructions de fusion d'affinité pour identifier des interactions de protéine sans exiger des anticorps de haute qualité reconnaissant une protéine d'intérêt1,2. Cependant, les méthodes basées sur les étiquettes épitope ont plusieurs inconvénients. Les interactions physiques avec l'épitope peuvent conduire à la détection de protéines copurifiantes non spécifiques3. En outre, la fusion de ces étiquettes d'épitope au terminal N ou C d'une protéine peut bloquer les interactions protéines-protéines indigènes, ou perturber le pliage des protéines pour favoriser les conformations non physiologiques4. En outre, les systèmes d'expression ectopique surexpriment généralement la protéine d'appât aux concentrations supraphysiologiques, ce qui peut entraîner l'identification d'interactions de protéines artifactual, en particulier pour les gènes sensibles à la posologie5. Pour contourner ces problèmes, la protéine d'appât endogène peut être immunoprécipitée avec les protéines de proies qui interagissent, en supposant la disponibilité d'un anticorps de haute qualité qui reconnaît la protéine indigène.
Fourni ici est un flux de travail protéomique d'interaction pour détecter l'interactome nucléaire d'une protéine endogène utilisant la protéine cmGC kinase DYRK1A comme exemple. La perturbation du numéro de copie de DYRK1A, du niveau d'activité, ou de l'expression peut causer la incapacité intellectuelle grave chez l'homme, et la létalité embryonnaire chez les souris6,7,8,9. DYRK1A présente une régulation spatiotemporelle dynamique10, et compartimenté les interactions protéiques11,12, nécessitant des approches capables de détecter des partenaires d'interaction à faible abondance spécifiques à différents compartiments subcellulaires.
Ce protocole utilise la fractionnement cellulaire des cellules HeLa humaines en fractions de cytosol et de nucléopatme, immunoprécipitation, préparation d'échantillon pour la spectrométrie de masse, et un aperçu d'un pipeline bioinformatique pour évaluer la qualité des données et visualiser les résultats, avec des scripts R fournis pour l'analyse et la visualisation (Figure 1). Les progiciels utilisés dans ce flux de travail sont tous disponibles gratuitement en téléchargement ou peuvent être consultés via une interface web. Pour plus d'informations sur les logiciels et les méthodes de calcul, des tutoriels approfondis et des instructions sont disponibles sur les liens fournis.
Protocole
REMARQUE : Toutes les compositions tampons et les mélanges de protéase sont décrits dans le tableau 1.
1. Préparation des cellules
REMARQUE : Un matériau de départ de 1 à 10 mg de lysate nucléaire par réplique est souhaité pour cette approche de spectrométrie de masse immunoprécipitation (IP-MS). Des quantités de cellules seront données pour 1 mg d'immunoprécipitations nucléaires dans le triplement plus les contrôles de triplicate.
- Si vous utilisez une lignée cellulaire adhérente, développez les cellules à 90 % de consistance dans des plats de 3 x 15 cm par réplique avant la récolte.
REMARQUE : Il est recommandé d'effectuer un minimum de trois immunoprécipitations de répétition pour les conditions d'appât et de contrôle. Ce protocole suppose l'utilisation de contrôles « perles seulement », qui contrôlent les interactions non spécifiques abondantes avec les perles, à partir de la section 4. D'autres types de contrôles peuvent être utiles. Ceux-ci sont décrits en profondeur dans la section de discussion.- Laver les plaques 2x avec du phosphate tamponné saline (PBS) et trypsiniser les cellules en utilisant 3 ml de trypsine de 0,25% par plaque de 15 cm. Faites tourner les cellules à 1 200 x g pendant 5 min et décantez la trypsine.
- Pour les cellules de suspension, atteindre une échelle/densité similaire pour atteindre 70 à 80 mg de protéines totales.
- Cellules de pellet à 1 200 x g et 4 oC pendant 5 min. Décant le support avec soin.
REMARQUE : Les granulés peuvent être combinés au cours de cette étape pour permettre un traitement efficace à l'aide de la fractionnement subcellulaire à grande échelle décrite à la section 2.
- Cellules de pellet à 1 200 x g et 4 oC pendant 5 min. Décant le support avec soin.
- Laver la pastille cellulaire 2x avec pbS 5 mM MgCl2 complété avec des inhibiteurs de la protéase (PI) et des inhibiteurs de la phosphatase (PHIs) (voir tableau 1).
REMARQUE : Les granulés de cellules peuvent être congelés au flash dans de l'azote liquide et stockés à -80 oC jusqu'à ce qu'ils soient prêts à être fractionnements.
2. Préparation de l'extrait nucléaire
REMARQUE : Les inhibiteurs de la protéase et de la phosphatase doivent être ajoutés aux tampons de fractionnement dans les 30 minutes d'utilisation.
- S'ils sont congelés, décongeler les granulés de cellules pendant 15 min dans 1 x de volume de granulés de tampon a froid A et de PI/I.. Placer le granule de cellule sur un écrou à 4 oC pour faciliter la suspension pendant la décongélation. Dans le cas contraire, resuspendre la pastille cellulaire de l'étape 1.3 en 1x volume de granulés Buffer A - PIs /PhIs.
- Pelletà 2 000 x g et 4 oC pendant 10 min. Décant le tampon.
- Suspendre les cellules avec 5x le volume de cellules emballées avec le tampon A et incuber sur la glace pendant 20 min.
- Pellet à 2 000 x g et 4 oC pendant 10 min. Décant le tampon et resuspendre avec 2 x volume cellulaire emballé original Buffer A - PIs/ PhIs et dounce 7x avec "A"/pilon lâche.
- Centrifuger le lysate pendant 10 min à 2 000 x g et 4 oC.
- Soigneusement pipette hors du supernatant et le gel flash avec de l'azote liquide. Conserver le lysate à -80 oC. Le supernatant de cette étape est la fraction subcellulaire cytosolique.
REMARQUE : Le granule nucléaire peut être sauvé au cours de cette étape par congélation éclair avec de l'azote liquide et le stockage à -80 oC - Resuspendre la pastille avec 0,9x volume de granulés de tampon B et PC/Is et mélanger sur un écrou pendant 5 min à 4 oC.
- Pour lyser les noyaux, dounce 20x avec un pilon plus serré "B".
- Mélanger le lysate nucléaire sur un distilateur pendant 30 min à 4 oC afin qu'il soit homogène.
- Centrifuger le lysate nucléaire pendant 30 min à 21 000 x g à 4 oC. Pipette hors du supernatant et sauf comme extrait de protéine nucléaire soluble.
REMARQUE : Le traitement de nucléane du granule nucléaire résultant permet la récupération d'une fraction de protéine chromatine-associée. - Dialyser l'extrait nucléaire soluble contre le tampon C et les PI pendant 3 h à 4 oC.
- Couper une longueur appropriée de 24 mm de dialyse avec un poids moléculaire de 8 kDa coupé. Clamp un côté de la tuyauterie et charger le nucléoplasme dans le tube. Après le chargement du lysate, pincez l'autre extrémité et immerger dans un récipient en verre propre contenant du tampon C et des PI.
- Centrifugelation de l'extrait nucléaire dialysé/nucléoplasme à 21 000 x g à 4 oC pendant 30 min. Aliquot 3x 20 volumes ll d'extrait nucléaire pour validation de fractionnement par tache occidentale. L'extrait nucléaire utilisé pour l'analyse IP-MS peut être aliquoted et flash congelé dans l'azote liquide et stocké à -80 oC, si nécessaire.
3. Validation de la fractionnement subcellulaire
- Effectuez un essai protéique pour déterminer la concentration protéique du lysate nucléaire. Un test de protéine d'acide bicinchoninique fournit la sensibilité suffisante pour l'application en aval.
- Chargez 20 g des fractions cytosoliques et nucléaires sur un gel SDS-PAGE pour l'analyse de tache occidentale comme décrit précédemment13. Passer les voies lors du chargement pour éviter la mauvaise caractérisation d'un échantillon.
- Sonde la tache occidentale pour p84 (THOC1) comme marqueur nucléaire, et GAPDH comme marqueur cytosolique. Déterminer l'étendue de la fractionnement par le rapport de marqueur cytosolique dans la fraction nucléaire et vice versa.
REMARQUE : Des anticorps pour d'autres marqueurs nucléaires et cytosliques peuvent être utilisés.
4. Immunoprécipitation de protéine endogène d'appât nucléaire
REMARQUE: Il est recommandé d'utiliser des tubes à faible rétention à partir de ce point. Cela réduira la liaison non spécifique aux tubes pendant la manipulation de l'échantillon et évitera la perte inutile de l'échantillon. En outre, assurez-vous que LCMS grade H2O est utilisé pour préparer des tampons pour les étapes restantes.
- Préparer un mélange de protéines A/G pour chaque reproduit en combinant 12,5 l de volume de perles pour les protéines A et les protéines-G dans les tubes de microcentrifuge. Conservez les stocks de perles comme une boue contenant 20 % d'éthanol. Déterminer la concentration de perles dans la boue %(v/v) et la pipette le volume nécessaire à l'aide d'une pointe de pipette qui a été coupée sur la pointe pour s'assurer que les perles peuvent entrer dans la pointe.
- Laver le mélange de protéines A/G perles 2x avec 300 l de tampon IP 1. Faire tourner les perles à 1 500 x g à 4 oC pendant 1 min et décanner le tampon.
- Préparer les perles A/G anticorps-protéines : Pour lier l'anticorps aux perles, ajoutez 300 lde de tampon IP 1 et 10 g de l'anticorps désiré. Laisser le mélange perle/anticorps se balancer sur un nutator à 4 oC pendant la nuit. Pour les contrôles perlés seulement, n'ajoutez pas d'anticorps.
REMARQUE : Un total de 10 g d'anticorps par réplique peut être utilisé comme point de départ, mais la quantité exacte devra être optimisée pour chaque anticorps individuel et l'échelle du lysatutilisé utilisé dans l'expérience - Décongeler les lysates nucléaires à partir de l'étape 2.10 dans un bain d'eau et aliquot volumes appropriés dans des tubes microcentrifuge à faible rétention pour 1 mg d'entrée de protéines par réplique.
- Faites tourner le lysate à 16 000 x g pendant 30 min et transférez le supernatant dans un nouveau tube.
- Ajouter 1 l de benzonase (250 unités/L) par 1 mg de lysate nucléaire et de la roche sur un écrou à 4 oC pendant 10 à 15 min.
- Préparer les perles pour prédédouaner le lysate. Ajouter 12,5 L de chaque protéine A et des perles de protéines G à 1,5 ml de tubes à faible rétention comme à l'étape 4.1. Laver 2x avec IP Wash Buffer 1 ' PIs et décanter le tampon.
- Ajouter 1 mg de lysate nucléaire aux perles de l'étape 4.5. Incuber en berçant sur un discausateur pendant 1 h à 4 oC.
- Centrifugeuse lysates prééclaircies à 1 500 x g et 4 oC pendant 1 min.
- Tandis que les lysates nucléaires couvent avec des perles dans l'étape 4.5.1, lavez les perles A/G de protéine d'anticorps 2x avec le tampon IP 1 - PI. Centrifugeuse à 1 500 x g et 4 oC pendant 1 min et décantle le tampon.
- Transférer le lysate nucléaire préautorisé de l'étape 4.6.1 sur les perles A/G anticorps-protéines. Incuber en se balançant sur un écrou à 4 oC pendant 4 h. Centrifugeuse après l'incubation à 1 500 x g et 4 oC pendant 1 min.
- Transférer le supernatant dans des tubes étiquetés comme le flux à travers pour chaque réplique.
- Laver les perles A/G anticorps-protéines avec 1 ml de tampon IP 2 et IP. Centrifugeuse à 1 500 x g et 4 oC pendant 1 min, tampon décantifère, et répéter pour un total de 3x.
- Laver les perles 2x avec 1 ml de tampon IP 1 PIs centrifuging comme dans l'étape précédente. Assurez-vous que tous les tampons sont enlevés après le dernier lavage.
- Elute 2x avec 20 oL de 0,1 M de glycine (pH 2,75) pendant 30 min chacun. Assurez-vous que les tubes basculent pendant l'incubation avec le tampon d'élution. Faire tourner à 750 x g et 4 oC pendant 1 min et pipette hors du supernatant après chaque incubation de 30 min.
REMARQUE : Tandis que la méthode de glycine de bas pH décrite ici élude la plupart des protéines d'appât, quelques interactions d'anticorps-antigènes exigent des conditions plus strictes de mémoire tampon. - Le gel éclair s'élit dans l'azote liquide et se range à -80 oC.
5. Préparation de l'échantillon
REMARQUE : L'insuline épilée dans les échantillons d'élution d'immunoprécipitation aide à la récupération des protéines pendant les précipitations d'acide trichloroacétique (TCA) et le traitement d'échantillon, qui est important pour les protéines endogènes d'appât de faible abondance.
- Décongeler les éluates à température ambiante si elles sont congelées.
- Placer les échantillons sur la glace et ajouter 10 oL d'insuline de 1,0 mg/mL pour chaque 100 l d'éluate. Vortex, puis ajouter immédiatement 10 'L de 1% de désoxycholate de sodium. Vortex l'échantillon à nouveau et ajouter 30 'L de 20% TCA suivie d'un vortex final.
- Incuber les échantillons sur glace pendant 20 min, puis la centrifugeuse à 21 000 x g à 4 oC pendant 30 min.
- Aspirer le supernatant et ajouter 0,5 ml d'acétone qui a été préréfrigéré à -20 oC. Vortex, puis tourner à 21 000 x g et 4 oC pendant 30 min. Répétez cette étape.
- Aspirer le supernatant et sécher à l'air le granule restant dans le fond du tube.
- Préparer l'échantillon pour la spectrométrie de masse à l'aide d'une méthode modifiée de préparation d'échantillons assistés par filtre (FASP), optimisée pour réduire la manipulation de l'échantillon, telle qu'indiquée ci-dessous14.
- Resuspendre la pastille protéique de l'étape 5.1.4 avec 30 L de tampon d'alkylation SDS (voir tableau 1). Incuber l'échantillon sur un bloc de chaleur de 95 oC pendant 5 min. Laisser refroidir à température ambiante pendant 15 minutes avant de passer à l'étape suivante.
- Ajouter 300 l de solution UA et 30 OL de 100 mM de TCEP à chaque échantillon. Chargez cette solution sur un filtre centrifuge de 30 k. Faire tourner le filtre centrifuge à 21 000 x g à température ambiante pendant 10 min.
REMARQUE : La protéine d'appât et ses interactors putatifs devraient être liés au filtre à ce stade. Cependant, le flux à travers peut être maintenu, dans le cas où il ya un problème avec le filtre. - Laver le filtre avec 250 oL d'UA et la centrifugeuse à 21 000 x g pendant 10 min. Décant le flux à travers et répéter pour un total de 3x.
- Laver le filtre avec 100 oL de 100 mM Tris pH 8,5 et centrifugeuse à 21 000 x g pendant 10 min. Décant le débit à travers et répétez pour un total de 3x.
- Ajouter 3 'L de 1 'g/L Lys C resuspendu dans 0.1 M Tris pH 8.5. Remplissez les filtres jusqu'à la marque de 100 l et laissez digérer pendant 1 h à 37 oC tout en se balançant sur un écrou.
- Ajouter 1 'L' de 1 trypsine de catégorie MS. Mélanger délicatement et laisser la trypsine incuber avec l'échantillon pendant la nuit à 37 oC tout en se balançant sur un distilatif.
- Centrifugeuse à 21 000 x g pendant 20 min pour élifier le peptide du filtre.
REMARQUE : Plusieurs tours de centrifugation peuvent être nécessaires pour récupérer toute l'éluate. Si cela n'est pas fait, il y a un risque de perte grave de l'échantillon.
- Saler les peptides à l'aide de colonnes de spin C18. Suivez le protocole fourni par le fabricant.
- Resuspendre le peptide lyophilisé en 7 L de 0,1% TFA en acétonitrile de 5%. Sonicate l'échantillon pendant 3 min pour s'assurer que les peptides ont été suspendus. Faites tourner à 14 000 x g pendant 10 min.
- Transférer le peptide en suspension dans un flacon d'échantillon approprié pour le charger sur le système de spectrométrie de masse (LC/MS) de chromatographie liquide.
6. Adéquation du système LC/MS
REMARQUE : En raison de la petite échelle et de l'abondance généralement plus faible de protéines provenant d'échantillons purifiés par affinité, il est essentiel que la plate-forme LC/MS fonctionne à une sensibilité et une robustesse maximales.
- Ajouter 1 mL d'acide formique de catégorie LC/MS à 1 L d'eau de qualité LC/MS pour la phase A mobile, et ajouter 1 ml d'acide formique de catégorie LC/MS à 1 L d'acétonitrile de catégorie LC/MS pour la phase B mobile.
- Préparer ou installer une colonne capillaire de 75 m de fusion et de silice remplie de résine C18 de la phase inversée de 250 mm de longueur. Les meilleurs résultats seront obtenus avec une injection directe d'échantillons dans la colonne.
- Purger le système de chromatographie liquide Ultra Performance (UPLC) avec des phases mobiles fraîches. Une fois installé une colonne C18, établir un débit stable et un électrospray avec un émetteur approprié (c.-à-d. 20 m id x 360 m od tiré à une pointe de 10 m). Maintenir la colonne à 40 à 60 oC.
- Testez les performances globales du système LC/MS en injectant une norme complexe de contrôle de la qualité, telle que 100 à 200ng d'un digest tryptique de lysate de lysate de cellules entières HeLa. Elute avec un gradient approprié pour un échantillon complexe (c.-à-d., temps d'élution de gradient de 2 à 3 h). Établir une performance du système de base des identifications de peptide et de protéine.
REMARQUE : Pour de meilleurs résultats, 3 000 à 5 000 identifications protéiques ou plus provenant de 20 000 à 35 000 peptides uniques fourniront des performances optimales pour les échantillons expérimentaux. - Pour ce qui est de l'adéquation systématique du système LC/MS, injectez 100 à 200 fmol ou moins d'une seule norme de digestion des protéines, comme l'albumine de sérum bovin (BSA). Elute avec un gradient court (c.-à-d., 20-30 min).
REMARQUE : Les injections multiples d'un digest de protéines aideront à établir les performances de base du système LC/MS, et l'injection répétée après chaque échantillon IP-MS fournit une mesure des performances du système tout au long de l'expérience et permet la détection de la dérive des instruments, ce qui peut biaiser les expériences sans étiquette. Une base de référence des intensités de pointe et des formes de pointe sélectionnées informera sur la MS, le LC et les performances des colonnes. - Pour éviter de surcharger la colonne analytique, chargez une petite partie (15 à 30 % du total) d'un échantillon expérimental sur la colonne et séparez-le à l'aide d'un gradient adapté à des échantillons complexes (c.-à-d. 2 à 3 h). Si le nombre d'identifications protéiques est insatisfaisant, chargez tout l'échantillon sur la colonne.
- Exécutez une norme unique de digestion des protéines entre les échantillons pour surveiller les performances du système LC/MS et le report de l'échantillon. Plusieurs normes peuvent être requises pour réduire le report de l'échantillon en fonction de vos échantillons.
7. Traitement des données
- Téléchargez le logiciel protéomique MaxQuant trouvé à https://www.maxquant.org/.
REMARQUE : Cela sera utilisé pour traiter le fichier de données RAW MS de l'étape 6.6 en tableaux de données des iD protéiques, des noms de gènes et des valeurs quantitatives associées à l'identification de ces données pour l'analyse en aval.- Sélectionnez Charge dans le sous-en-tête des données d'entrée de l'onglet Données brutes. Ouvrez l'emplacement du fichier où les fichiers bruts MS sont stockés et sélectionnez les fichiers bruts pour chaque exécution MS/MS.
- Cliquez sur l'onglet Groupe-Spécifique et sélectionnez Digestion. Dans la liste d'enzymes sélectionnez LysC et cliquez sur la flèche droite pour ajouter cette enzyme dans la liste qui sera utilisée dans la recherche. Ensuite, sélectionnez Instrument et assurez-vous que le bon type d'instrument apparaît dans la liste de déclassement en haut de l'écran. Laissez d'autres paramètres de recherche spécifiques au groupe sur les paramètres standard.
- Cliquez sur l'onglet Paramètres globaux et sélectionnez Séquences. Ajoutez le fichier FASTA approprié pour la taxonomie qui sera utilisé dans cette recherche. Les peptides ne seront pas attribués correctement si cela n'est pas fait. Pour le protéome humain, téléchargez le fichier FASTA d'UniProt à https://www.uniprot.org/help/human_proteome.
- Dans l'onglet Paramètres globaux, cliquez sur Quantificationdes protéines . Dans le menu déroulant Peptides for Quantification, sélectionnez Unique et Razor.
REMARQUE : MaxQuant offre une quantitation alternative des protéines par la quantification absolue basée sur l'intensité (iBAQ) et la quantitation sans étiquette (LFQ). Cependant, l'information sur le nombre de peptides est suffisante pour l'analyse en aval dans ce protocole15. - En bas à gauche de l'interface MaxQuant, sélectionnez le nombre de processeurs à utiliser pour la recherche. Cela affectera directement la durée requise pour la course, alors sélectionnez autant que possible pour cela). Cliquez sur Démarrer en bas à gauche de l'écran pour démarrer la course. Sélectionnez l'onglet Performance en haut de l'écran pour afficher l'évolution de la recherche.
- Lorsque la course est terminée, ouvrez le fichier proteingroups.txt dans Persée, une plate-forme de calcul protéomique, ou tout autre programme de tableur pour afficher les données16.
- Utilisez Persée pour éliminer les contaminants et les coups courants pour inverser les séquences de protéines. Suivez la documentation détaillée de Perséus à http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
REMARQUE : L'ouverture du fichier proteingroups.txt dans un logiciel d'analyse (p. ex., Excel) corrompra automatiquement certains noms de gènes et de protéines.
- Utilisez Persée pour éliminer les contaminants et les coups courants pour inverser les séquences de protéines. Suivez la documentation détaillée de Perséus à http://www.coxdocs.org/doku.php?id=perseus:user:use_cases:interactions.
- Analyser les données expérimentales à l'aide du dépôt de contaminants pour la purification des affinités (CRAPome). Inscrivez-vous un compte à ce dépôt http://crapome.org/ et suivez le tutoriel au besoin17,18.
- Utilisez le flux de travail Analyze Your Data trouvé sur la page d'accueil de CRAPome. Sélectionnez les contrôles externes qui correspondent au système de purification d'affinité utilisé dans cette expérience d'interaction.
REMARQUE : Ces contrôles peuvent être utilisés pour calculer un deuxième enrichissement de changement de pli qui est utile pour détecter les contaminants communs. - Générez un fichier d'entrée à partir de la sortie proteingroups.txt de MaxQuant à l'aide de Persée ou d'une application de feuille de calcul appropriée. Les détails de la mise en forme manuelle peuvent être trouvés à http://crapome.org/?q=fileformatting. Vous pouvez également utiliser le script R fourni « export_CRAPomeSAINT_Input_File.R » pour générer le fichier d'entrée SAINT/CRAPome. Voir README.txt dans les fichiers de codage supplémentaires.
- Exécuter une analyse pour déterminer l'enrichissement par le changement de pli et la probabilité de l'interactome pour chaque protéine d'appât dans l'immunoprécipitation. Assurez-vous que les « contrôlesd'utilisateur » sont sélectionnés dans le menu déroulant sous FC-A, « Contrôles CRAPome » ou « Tous les contrôles » sont sélectionnés dans le drop-down FC-B et le score de probabilité est sélectionné pour générer des probabilités SAINT. Lorsque la course est terminée, affichez la sortie disponible sous la note de ' Résultats d'analyse' avec un ID d'emploi. Téléchargez la matrice de données à partir des « Résultats d'analyse » pour la planification future et la visualisation des données.
- Utilisez le flux de travail Analyze Your Data trouvé sur la page d'accueil de CRAPome. Sélectionnez les contrôles externes qui correspondent au système de purification d'affinité utilisé dans cette expérience d'interaction.
- Tracer les protéines en fonction de FC-A (IP vs contrôles de l'utilisateur) et la probabilité SAINT en suivant les R-Scripts comme prévu dans les fichiers de codage supplémentaires.
REMARQUE : Un ensemble de scripts R est fourni pour produire des parcelles de FC-A vs SAINT probabilité et iBAQ vs log2 (abondance de protéines), coloré par la gamme de valeur p ajustée de l'analyse empirique Bayes des intensités sans étiquette. Les détails de l'analyse statistique différentielle et de l'intrigue se trouvent dans README.txt et le script R "main_differential_analysis. R" dans les fichiers de codage supplémentaires. - Évaluer où les protéines interagissant connues de la protéine d'appât sont classées par FC-A et SAINT. Faire une coupure de FC-A 'gt; 3.00 et SAINT 'gt; 0.7 pour les expériences d'appât unique dans triplicate comme point de départ.
REMARQUE : La sélection des seuils pour un interagiteur « à haute confiance » et un interactionur « à faible confiance » doit être éclairée par des renseignements biologiques.
8. Visualisation des données
REMARQUE : De nombreux programmes peuvent visualiser efficacement les données protéomiques (p. ex., R, Perseus, Cytoscape, STRING-DB). L'analyse de la connectivité entre les coups de confiance et l'enrichissement fonctionnel de ces interacteurs peut être une stratégie utile pour hiérarchiser les hits pour une validation et une caractérisation fonctionnelle supplémentaires.
- Téléchargez Cytoscape, un outil de visualisation réseau open source au19https://cytoscape.org/download.html .
- Préparer un fichier d'entrée pour les données d'interaction sous forme de fichier délimité formaté avec trois colonnes : appât (nœud source), proie (noeud cible), type d'interaction (type de bord). Cela peut être fait dans Perséus ou n'importe quel programme de feuille de calcul de votre choix.
- Sélectionnez la table d'importation de l'icône De fichier vers la partie supérieure gauche du programme (désignée par une flèche vers le bas et une matrice dans l'icône). Cytoscape remplira auto-remplira les données d'interaction dans un réseau prêt pour le formatage et la conception personnalisés.
- Sélectionnez l'onglet Style sur le panneau de contrôle de Cytoscape et cliquez sur les carrés de la colonne Def pour ajuster l'attribut pour l'ensemble du réseau. Sélectionnez des nœuds ou des bords spécifiques dans le réseau, puis sélectionnez le carré dans la colonne Byp. du menu de style pour contourner les paramètres par défaut et ajuster uniquement les objets sélectionnés. Vous pouvez également cliquer sur le menu déroulant en haut du menu de style pour afficher les formats réseau préréglés.
REMARQUE : Les données d'interaction protéine-protéine de STRING-db peuvent être intégrées dans ce réseau à ce moment-là manuellement par le fichier d'entrée ou par divers outils d'enrichissement disponibles en tant que plug-ins dans Cytoscape, http://apps.cytoscape.org/20. Un plug-in cytoscape recommandé pour l'analyse d'enrichissement est trouvé à http://apps.cytoscape.org/apps/cluego21. - Pour accroître la confiance dans l'ensemble de données généré dans ce flux de travail, effectuez des expériences réciproques IP-MS ou IP-western qui ciblent les protéines de proies d'intérêt comme appât.
Résultats
La majorité de la masse protéique identifiée dans une expérience IP-MS se compose de protéines non spécifiques. Ainsi, l'un des principaux défis d'une expérience IP-MS est l'interprétation des protéines qui sont des interacteurs de confiance par rapport aux interacteurs non spécifiques. Pour démontrer les paramètres cruciaux utilisés dans l'évaluation de la qualité des données, l'étude a analysé les immunoprécipitations triplicate de 5 mg d'extrait nucléaire HeLa en utilisant un contrôle de perle seulement. La première vérification interne pour s'assurer qu'une expérience IP-MS est fiable est de savoir si la protéine d'appât se classe comme l'une des protéines enrichies les plus élevées identifiées par le changement de pli sur le contrôle et la probabilité DE SAINT. Dans ce cas, l'appât DYRK1A classé parmi les trois premières protéines enrichies sur le contrôle (Figure 2A,B)). Dans une étude d'interaction nucléaire de DYRK1A utilisant quatre anticorps indépendants, une coupure de FC-A de la limite de probabilité de 'gt;3.00 et DE SAINT 'gt;0.7 a fourni une coupure rigoureuse pour l'identification des interactors nouveaux et précédemment validés22. Lorsqu'il est appliqué à cette expérience, une séparation claire pourrait être observée entre les acteurs à haute confiance et les protéines copurifiées identifiées comme non spécifiques (Figure 2A,B). L'application d'un seuil d'enrichissement et de probabilité de changement de pli augmente la rigueur en exigeant un enrichissement constamment élevé des ID de protéine à travers des répliques biologiques.
En plus de la notation statistique, le workflow d'analyse de CRAPome cartographie également les interactions précédemment signalées sur les données appât-proie23. Bien que cette cartographie puisse être utile pour le seuil des interactions à haute et à faible confiance, les interactions précédemment signalées peuvent obtenir de mauvais résultats par les probabilités FC-A et SAINT, ce qui peut indiquer que de nombreuses interactions connues d'un appât donné peuvent n'exister que dans des types de cellules, des contextes ou des organites spécifiques. Pour l'exemple du jeu de données DYRK1A, les valeurs IREF interactor FC-A étaient aussi basses que 0,45, ce qui représente un très faible enrichissement sur le contrôle (figure 2C). Pour éviter l'inflation des faux positifs, le seuil statistique doit être effectué d'une manière qui donne la priorité à la rigueur plutôt qu'à la réduction des faux négatifs. Il convient de noter que la détection de ces interactions était indépendante de l'abondance des protéines (Figure 2C). Le numéro de copie absolu calculé de chaque interaction iREF au sein des cellules HeLa n'a montré aucune corrélation avec les niveaux de détection d'un partenaire d'interaction par IP-MS24.
Cytoscape sert d'outil efficace pour visualiser plusieurs couches de données d'interaction19. Dans l'expérience d'immunoprécipitation DYRK1A décrite ici, l'utilisation combinée de FC-A et de SAINT -gt; 0.9 a réduit la liste des interactors de haute confiance à six protéines (Figure 2D). Cependant, lors de l'application d'une coupure FC-A de 3,0 dans l'isolement, huit protéines supplémentaires ont été ajoutées au réseau. Ces interacteurs protéiques supplémentaires ont une connectivité élevée avec les acteurs déjà dans le réseau, ce qui suggère qu'ils sont associés à des complexes similaires ou des rôles fonctionnels. À cette fin, les preuves de la STRING-DB des interactions protéines-protéines ont été intégrées dans ce réseau sous forme de lignes pointilléesbleues 20. Bien que cette expérience à appât unique et triplicate fournisse un échantillon limité du réseau d'interaction DYRK1A, l'utilisation d'appâts, de répliques et d'intégration supplémentaires de grands ensembles de données publiques peut être utilisée pour élargir le réseau d'interactions à haute confiance. Les seuils statistiques seront donc spécifiques à chaque expérience individuelle et devront être évalués en profondeur.

Figure 1 : Flux de travail représentatif protéomique pour ip-MS subcellulaire. Les cellules sont cultivées dans des flacons de fond ronds de 4 L ou des plats de culture tissulaire de 15 cm et récoltées en même temps pour la fractionnement subcellulaire. Les cellules sont fractionnées en une pastille cytosolique, nucléaire et nucléaire, et les immunoprécipitations sont faites à partir de 1 à 10 mg de lysate nucléaire à l'aide d'un ou plusieurs anticorps reconnaissant le même appât. Le nettoyage de l'échantillon assisté par filtre (FASP) et hors ligne sont effectués avant la spectrométrie de masse à tir unique. Un pipeline de calcul en aval est utilisé pour traiter les données en données d'interaction interprétables. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
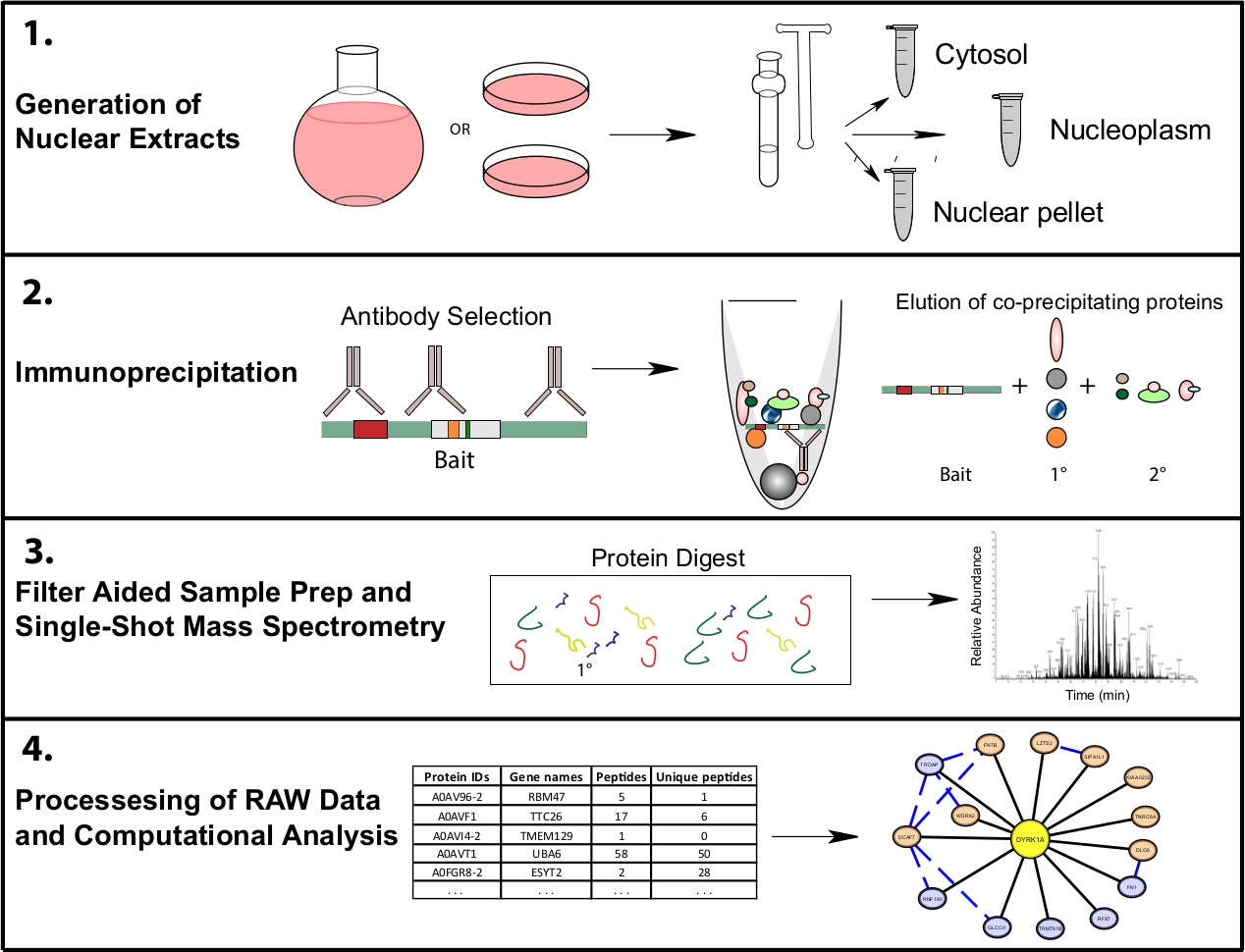{kind=link}

Figure 2 : Données représentatives pour une expérience IP-MS à un seul appât. (A) Sortie de probabilité FC-A et SAINT du flux de travail d'analyse CRAPome pour une expérience optimale utilisant un seul anticorps pour la kinase DYRK1A (n ' 3). Les contrôles perlés seulement ont été utilisés pour la comparaison. Les lignes solides rouges représentent des seuils fixés à FC-A 'gt; 3.00 et SAINT 'gt; 0.7. (B) Estimations de l'abondance des protéines MaxQuant (iBAQ) de la production par rapport au rapport log2 de l'abondance des protéines dans la propriété intellectuelle DYRK1A à la lutte, colorée par la gamme de valeur p ajustée de l'analyse empirique bayes des intensités sans étiquette. (C) FC-A et nombre estimatif de copies de protéines répertoriées comme protéines en interaction dans la base de données iRef23,24. (D) Visualisation du réseau Cytoscape des interactors DYRK1A. Noeuds bleus - FC-A -gt; 3.00, SAINT -gt; 0.7. Noeuds oranges - FC-A - 3.00. Bords noirs et protéines identifiées comme interacteurs dans l'expérience IPMS. Bord pointillé bleu - interaction SAINT entre la protéine de proie (confiance 'gt; .150). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
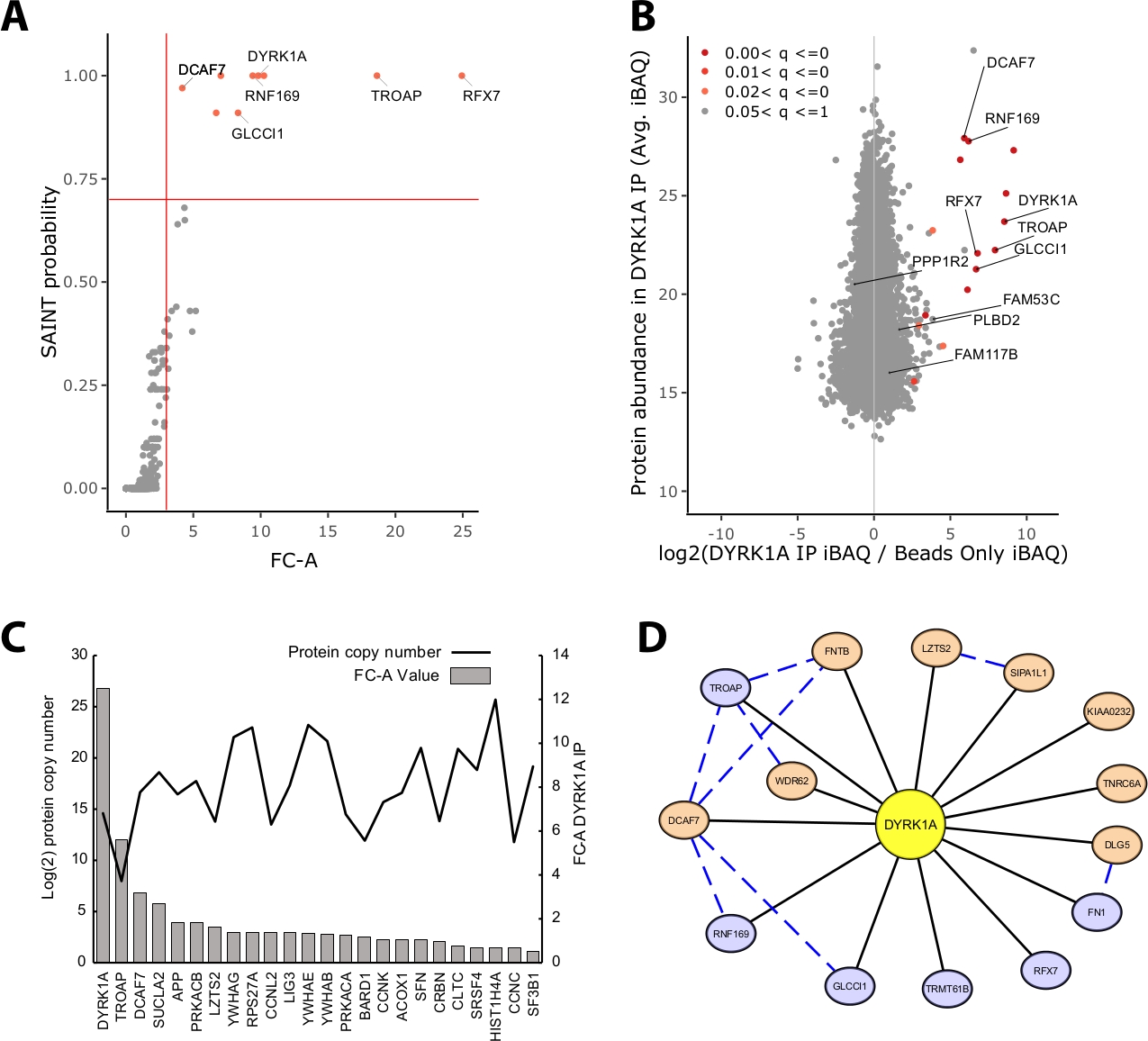{kind=link}
| Mélange d'inhibiteur de la protéase (PI) | |
| Réactif | Concentration finale |
| Sodium Metabisulfite | 1 mM |
| Benzamidine Benzamidine | 1 mM |
| Dithiothreitol (TNT) | 1 mM |
| Fluorure de phénylmethanesulfonyl (PMSF) | 0,25 mm |
| Mélange d'inhibiteur de la phosphatase (PhI) | |
| Réactif | Concentration finale |
| MicrocystinE LR | 1 M |
| Sodium Orthovanadate | 0,1 mm |
| Fluorure de sodium | 5 mM |
| Tampons de fractionnement subcellulaires : | |
| Tampon A pH 7.9 | |
| Réactif | Concentration finale |
| Hepes | 10 mM |
| MgCl2 (en) | 1,5 mm |
| Kcl | 10 mM |
| Tampon B pH 7,9 | |
| Réactif | Concentration finale |
| Hepes | 20 mM |
| MgCl2 (en) | 1,5 mm |
| Nacl | 420 mM |
| Acide éthylènediaminetetraacetic (EDTA) | 0,4 mm |
| Glycérol | 25% (v/v) |
| Tampon C pH 7,9 | |
| Réactif | Concentration finale |
| Hepes | 20 mM |
| MgCl2 (en) | 2 mM |
| Kcl | 100 mm |
| Acide éthylènediaminetetraacetic (EDTA) | 0,4 mm |
| Glycérol | 20% (v/v) |
| Tampons d'immunoprécipitation : | |
| Tampon IP 1 | |
| Réactif | Concentration finale |
| Hepes | 20 mM |
| Kcl | 150 mm |
| Edta | 0,1 mm |
| NP-40 | 0,1 % (v/v) |
| Glycérol | 10% (v/v) |
| Tampon IP 2 | |
| Réactif | Concentration finale |
| Hepes | 20 mM |
| Kcl | 500 mm |
| Edta | 0,1 mm |
| NP-40 | 0,1 % (v/v) |
| Glycérol | 10% (v/v) |
| SDS Alkylation Buffer pH 8.5 | |
| Réactif | Concentration finale |
| Sds | 4 % (v/v) |
| Chloroacétamide | 40 mM |
| Ptce | 10 mM |
| Tris | 100 mm |
| UA pH 8,5 | |
| Réactif | Concentration finale |
| Urée | 8 M |
| Tris | 0,1 M |
| - utiliser HPLC grade H2O | |
Tableau 1 : Compositions tampons
Fichiers de codage supplémentaires. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Le workflow protéomique décrit ici fournit une méthode efficace pour identifier les acteurs de protéines à haute confiance pour une protéine d'intérêt. Cette approche diminue la complexité de l'échantillon par fraction subcellulaire et se concentre sur l'augmentation des partenaires d'interaction d'identification grâce à une préparation robuste de l'échantillon, le nettoyage de l'échantillon hors ligne et un contrôle rigoureux de la qualité du système LC-MS. L'analyse des données en aval décrite ici permet une simple évaluation statistique des protéines identifiées comme copurifiantes avec l'appât. Cependant, en raison d'un grand nombre de variables expérimentales (échelle, lignée cellulaire, choix d'anticorps), chaque expérience nécessite des seuils et des considérations différents en ce qui concerne la visualisation et l'enrichissement des données.
La première considération de conception dans une expérience IP-MS est la sélection d'anticorps qui seront utilisés pour la copurification de la protéine d'intérêt avec ses partenaires en interaction. Bien que la disponibilité d'anticorps commerciaux se soit étendue pour couvrir de plus grandes parties du protéome humain au cours des dernières décennies, il existe encore de nombreuses protéines pour lesquelles les réactifs sont limités. En outre, les anticorps qui ont été validés pour des applications telles que la détection de taches occidentales peuvent être incapables d'enrichissement sélectif de la protéine cible dans une expérience d'immunoprécipitation. Avant de mener une expérience protéomique d'interaction à grande échelle, il est suggéré de compléter une propriété intellectuelle à partir d'un plat de 10 cm confluent à 90 %, ou numéro de cellule équivalent, et de sonder la protéine cible d'intérêt par le ballonnement occidental. Si plus d'un anticorps unique est disponible pour l'immunoprécipitation, il est en outre suggéré de sélectionner plusieurs anticorps reconnaissant les épitopes dans différentes parties de la protéine. La liaison d'un anticorps à une protéine d'appât peut obstruer l'interface de liaison nécessaire pour les partenaires en interaction putative. La sélection d'un épitope secondaire pour la protéine d'appât augmentera la couverture du profil d'interaction identifié par une expérience basée sur la spectrométrie de masse.
Une deuxième considération importante réside dans le choix du contrôle approprié pour distinguer les interactions à haute confiance des interactions à faible confiance ou non spécifiques de celles identifiées comme copurifiantes avec l'appât. Le contrôle le plus strict pour une expérience IP-MS est de compléter l'immunoprécipitation à partir d'une lignée cellulaire CRISPR KO de l'appât. Un tel contrôle permet d'identifier et de filtrer les protéines non spécifiques qui se lient directement à l'anticorps plutôt qu'à la protéine d'appât. Dans les cas où la génération d'une lignée cellulaire CRISPR KO de chaque protéine d'appât n'est pas faisable, un contrôle de perles d'IgG du même isotype de l'anticorps d'appât peut être employé. Dans les expériences utilisant un panel d'anticorps représentant plusieurs espèces, l'utilisation d'un contrôle des perles seulement peut être appropriée, mais augmentera le taux de faux positifs identifiés comme des interacteurs à haute confiance.
La sélection de la lignée cellulaire utilisée dans une expérience IP-MS dépend de plusieurs facteurs clés. L'expression et la localisation des protéines dépendent en grande partie du type cellulaire. Tandis que des estimations d'expression d'ARN peuvent être trouvées pour la plupart des gènes dans beaucoup de lignes fréquemment utilisées de cellules, l'expression de protéine est mal corrélée avec l'expression d'ARN et doit être déterminée expérimentalement25. Les lignées cellulaires dans lesquelles une protéine d'appât est exprimée en très faible nombre de copies devraient être évitées pour contourner les problèmes associés à l'augmentation drastique de l'échelle de culture cellulaire qui peut être nécessaire. Il convient de noter, cependant, que la préparation de l'échantillon peut être optimisée pour la détection de protéines à très faible abondance. La méthode de préparation de l'échantillon assistée par filtre (FASP), bien que robuste, peut entraîner une perte de peptide de plus de 50 % dans un échantillon. La préparation d'échantillons à phase solide (SP3) à un seul pot est une méthode efficace de production d'échantillons pour l'analyse de spectrométrie de masse qui minimise la perte d'échantillons26. La récupération accrue permise par la méthode SP3 de préparation de l'échantillon peut être une alternative utile dans ce flux de travail pour la quantification des protéines qui tombent près de la limite de détection.
Ce flux de travail protéomique a été appliqué à de nombreux appâts nucléaires, y compris les kinases, les ligases d'ubiquitine E3 et les membres d'échafaudages de complexes multisubunit. En supposant une validation appropriée des réactifs d'anticorps, l'exécution réussie de ce flux de travail se traduira par la détection de partenaires d'interaction nucléaire protéique à haute confiance pour une protéine d'intérêt.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Ce travail a été appuyé par une subvention du Grand Défi à W.M.O. du Linda Crnic Institute for Down Syndrome et par un accord de coopération DARPA 13-34-RTA-FP-007. Nous tenons à remercier Jesse Kurland et Kira Cozzolino pour leur contribution à la lecture et aux commentaires sur le manuscrit.
matériels
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin, 0.1% EDTA | Thermo Fisher Scientific | 25200056 | |
| 1.5 ml low-rention microcentrifuge tubes | Fisher Scientific | 02-681-320 | |
| 4-20% Mini PROTEAN TGX Precast Protein Gels | Bio-Rad | 4561096 | |
| acetone (HPLC) | Thermo Fisher Scientific | A949SK-4 | |
| Amicon Ultra 0.5 ml 30k filter column | Millipore Sigma | UFC503096 | |
| Benzamidine | Sigma-Aldrich | 12072 | |
| benzonase | Sigma-Aldrich | E1014 | |
| Chloroacetamide | Sigma-Aldrich | C0267 | |
| Dialysis tubing closure | Caroline Biological Supply Company | 684239 | |
| DTT | Sigma-Aldrich | 10197777001 | |
| EDTA | Sigma-Aldrich | EDS | |
| GAPDH antibody | Santa Cruz Biotechnology | Sc-47724 | |
| Glycerol | Fisher Scientific | 887845 | |
| Glycine | Sigma-Aldrich | G8898 | |
| HeLa QC tryptic digest | Pierce | 88329 | |
| HEPES | Fisher Scientific | AAJ1692630 | |
| insulin | Thermo Fisher Scientific | 12585014 | |
| iodoacetamide | Sigma-Aldrich | I1149 | |
| KONTES Dounce homogenizer 7 ml | VWR | KT885300-0007 | |
| Large Clearance pestle 7ml | VWR | KT885301-0007 | |
| Lysyl endopeptidase C | VWR | 125-05061 | |
| Magnesium Chloride | Sigma-Aldrich | 208337 | |
| Microcystin | enzo life sciences | ALX-350-012-C100 | |
| Nonidet P 40 Substitute solution | Sigma-Aldrich | 98379 | |
| p84 antibody | GeneTex | GTX70220 | |
| Phosphate Buffered Saline | |||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 | |
| Pierce BSA Protein Digest, MS grade | Thermo Fisher Scientific | 88341 | LCMS QC |
| Pierce C18 spin columns | Thermo Fisher Scientific | PI-89873 | |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | 90057 | For mass spectrometry sample prep |
| PMSF | Sigma-Aldrich | P7626 | |
| Potassium Chloride | Sigma-Aldrich | P9541 | |
| Protein A Sepharose CL-4B | GE Healthcare Bio-Sciences | 17-0780-01 | |
| Protein G Sepharose 4 Fast Flow | GE Healthcare Bio-Sciences | 17-0618-01 | |
| SDS | Sigma-Aldrich | L3771 | |
| Silica emitter tip | Pico TIP | FS360-20-10 | |
| Small Clearance pestle 7ml | VWR | KT885302-0007 | |
| Sodium Chloride | Sigma-Aldrich | S3014 | |
| Sodium Fluoride | Sigma-Aldrich | 201154 | |
| Sodium metabisulfite | Sigma-Aldrich | 31448 | |
| Sodium orthovanadate | Sigma-Aldrich | S6508 | |
| Spectra/ Por 8 kDa 24 mm dialysis tubing | Thomas Scientific | 3787K17 | |
| TC Dish 150, Standard | Sarstedt | 83.3903 | Tissue culture dish for adherent cells |
| TCA | Sigma-Aldrich | T9159 | |
| TCEP | Thermo Scientific | PG82080 | |
| TFA | Thermo Fisher Scientific | 28904 | |
| Thermo Scientific Orbitrap Fusion MS | Thermo Fisher Scientific | ||
| Trizma Base | Sigma-Aldrich | T6066 | |
| Urea | Thermo Fisher Scientific | 29700 | |
| Waters ACQUITY M-Class UPLC | Waters | ||
| Waters ACQUITY UPLC M-Class Column Reversed-Phase 1.7µm Spherical Hybrid (1.7 µm, 75 µm x 250 mm) | Waters | 186007484 | nanoflow C18 column |
Références
- Varjosalo, M., et al. The protein interaction landscape of the human CMGC kinase group. Cell Reports. 3, 1306-1320 (2013).
- Kimple, M. E., Brill, A. L., Pasker, R. L. Overview of Affinity Tags for Protein Purification. Current Protocols in Protein Science. 73, (2013).
- Mahmood, N., Xie, J. An endogenous 'nonspecific' protein detected by a His-tag antibody is human transcription regulator YY1. Data in Brief. 2, 52 (2015).
- Zordan, R. E., Beliveau, B. J., Trow, J. A., Craig, N. L., Cormack, B. P. Avoiding the ends: internal epitope tagging of proteins using transposon Tn7. Genetics. 200, 47-59 (2015).
- Gibson, T. J., Seiler, M., Veitia, R. A. The transience of transient overexpression. Nature Methods. 10, 715-721 (2013).
- Bronicki, L. M., et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. European Journal of Human Genetics. 23, 1482-1487 (2015).
- Antonarakis, S. E. Down syndrome and the complexity of genome dosage imbalance. Nature Reviews Genetics. , (2016).
- Dowjat, W. K., et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neuroscience Letters. 413, 77-81 (2007).
- Fotaki, V., et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Molecular and Cellular Biology. 22, 6636-6647 (2002).
- Hämmerle, B., Elizalde, C., Tejedor, F. J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. European Journal of Neuroscience. 27, 1061-1074 (2008).
- Funakoshi, E., et al. Overexpression of the human MNB/DYRK1A gene induces formation of multinucleate cells through overduplication of the centrosome. BMC Molecular and Cell Biology. 4, 12 (2003).
- Yu, D., Cattoglio, C., Xue, Y., Zhou, Q. A complex between DYRK1A and DCAF7 phosphorylates the C-terminal domain of RNA polymerase II to promote myogenesis. Nucleic Acids Research. , 1-14 (2019).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 76, 4350-4354 (1979).
- Wiśniewski, J. R., Zougman, A., Nagaraj, N., Mann, M. Universal sample preparation method for proteome analysis. Nature Methods. 6, 359-362 (2009).
- Tyanova, S., Temu, T., Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols. 11, 2301-2319 (2016).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13, 731-740 (2016).
- Mellacheruvu, D., et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nature Methods. 10, 730-736 (2013).
- Choi, H., et al. SAINT: Probabilistic scoring of affinity purificationg-mass spectrometry data. Nature Methods. 8, 70-73 (2011).
- Shannon, P., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 13, 2498-2504 (2003).
- Szklarczyk, D., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Research. 45, 362-368 (2017).
- Bindea, G., et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 25, 1091-1093 (2009).
- Guard, S. E., et al. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Scientific Reports. 9, 6539 (2019).
- Razick, S., Magklaras, G., Donaldson, I. M. iRefIndex: A consolidated protein interaction database with provenance. BMC Bioinformatics. 9, 405 (2008).
- Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 11, 319-324 (2014).
- Liu, Y., Beyer, A., Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 165, 535-550 (2016).
- Hughes, C. S., et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Molecular Systems Biology. 10, 757 (2014).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.