
Method Article
Assessing the Age-Specific Phagocytic Ability of Adult Drosophila melanogaster Hemocytes using an In Vivo Phagocytosis Assay
In This Article
Summary
This protocol describes an in vivo assay of phagocytosis used to assess and quantify the ability of young and aged Drosophila melanogaster hemocytes to phagocytose bacteria.
Abstract
Phagocytosis is an essential function of the innate immune response. This process is carried out by phagocytic hemocytes whose primary function is to recognize a wide range of particles and destroy microbial pathogens. As organisms age, this process begins to decline, yet little is known about the underlying mechanisms or the genetic basis of immunosenescence. Here, an injection based in vivo phagocytosis assay is used to assess age related changes in different aspects of phagocytosis, such as binding, engulfment, and degradation of internalized particles, by quantifying phagocytic events in hemocytes in adult Drosophila. Drosophila melanogaster has become an ideal model to investigate age related changes in innate immune function for many reasons. For one, many genetic components and functions of the innate immune response, including phagocytosis, are evolutionarily conserved between Drosophila and mammals. Because of that, results obtained from using this protocol are likely to be widely relevant to understanding the age related changes in immune function in a variety of organisms. Additionally, we note that this method provides quantitative estimates of hemocyte phagocytic ability, which could be useful for a variety of research topics, and need not be limited to studies of aging.
Introduction
The innate immune system, which consists of physical and chemical barriers to infection as well as cellular components, is evolutionary conserved across multicellular organisms1,2. As the first line of defense, the innate immune system plays a critical role in combating invading pathogens in all animals1,2,3. The components of the innate immune response include a wide range of cell types which are classified on the basis that they lack specificity and immunological memory2,3,4. In humans, these cell types include phagocytic monocytes and macrophages, neutrophils, and cytotoxic natural killer cells4,5. While having a functional immune system is imperative for host survival, it is clear that the function of immune cells declines with age, a phenomenon known as immunosenescence5,6. Being able to assess age related changes in the immune response, including different aspects of the process of phagocytosis, could aid in our understanding of immunosenescence. The procedure we describe here provides an effective and repeatable approach to evaluate and quantify phagocytic events by hemocytes in Drosophila melanogaster.
Drosophila is an ideal model for studying the immune response for many reasons. For one, there is an extensive set of genetic tools available that make it possible to easily manipulate gene expression in a tissue-dependent manner7. These tools include a collection of mutants, RNA interference stocks, GAL4/UAS stocks, and the Drosophila Genetic Reference Panel that contains 205 different inbred lines for which the entire genome sequences are catalogued8. The short life cycle of Drosophila and the large number of individuals produced allow researchers to test multiple individuals in a controlled environment, in a short period of time. This greatly improves the ability to identify subtle differences in immune responses to infection among genotypes, between sexes or across ages. Importantly, many genetic components and functions of the innate immune response, including phagocytosis, are evolutionarily conserved between Drosophila and mammals1,2.
In Drosophila, the process of phagocytosis that follows infection is carried out by phagocytic hemocytes called plasmatocytes, which are equivalent to mammalian macrophages9. Hemocytes are essential for recognizing a wide range of particles and clearing microbial pathogens9,10,11,12,13. These cells express a variety of receptors that must differentiate self from non-self, and initiate signaling events needed to carry out the phagocytic process10,11,12,13,14,15. Once a particle is bound, it starts to be internalized by reorganization of the actin cytoskeleton and remodeling of the plasma membrane to expand around the particle, forming a phagocytic cup11,12,13,14. During this process, another set of signals tells the cell to internalize the particle further by closing the phagocytic cup, forming a membrane-bound phagosome11,12,13,14,15. The phagosome then undergoes a maturation process, associating with different proteins and fusing with lysosomes, forming an acidic phagolysosome11,12,13,14,15. At this point, particles can efficiently be degraded and eliminated11,12,13,14,15. Drosophila studies have revealed that older flies (4 weeks of age) have a reduced ability to clear an infection compared to younger flies (1-week old), likely due, at least in part, to a decline in some aspects of phagocytosis16,17.
The method described here utilizes two separate fluorescently-labelled heat-killed E. coli particles, one bearing a standard fluorophore and one that is pH sensitive, to assess two different aspects of phagocytosis: the initial engulfment of particles, and the degradation of particles in the phagolysosome. In this assay, fluoro-particle fluorescence is observable when the particles are bound and engulfed by hemocytes, while pH sensitive particles fluoresce only in the low pH conditions of the phagosome. Fluorescent events can then be observed in hemocytes that localize along the dorsal vessel. We focus on hemocytes localized to the dorsal vessel, which provide an anatomical landmark to locate hemocytes that are known to contribute to bacterial clearance, and to consistently isolate them. However, hemocytes in other parts of the body and the hemolymph are also important for clearance. Although we have not studied this cell population, our general procedure could be applicable for phagocytic assays of these cells as well. One advantage of our approach is that we can quantify phagocytic events within individual hemocytes, allowing us to detect subtle variation in phagocytic processes. Other studies that visualize fluorescent events through the cuticle18,19 do not account for differences in the numbers of hemocytes present, which is especially important to consider in our case as total hemocyte counts are expected to change with age17.
Protocol
1. Collect and age Drosophila
- To generate same aged F1 flies for testing phagocytosis, add 5-10 virgin females and 5 males of the appropriate genotypes to a vial containing fresh fly food. We use a cornmeal molasses agar based food20, but the method should work regardless of the type of diet the flies are reared on. For this experiment we used Hemese (He)-GAL4; UAS-GFP flies to label hemocytes genetically.
NOTE: More flies can be used, but some lines of D. melanogaster may not mate or reproduce well when overcrowded, and overcrowding may have adverse effects on larval development21 and phagocytosis. - Maintain flies under the desired experimental conditions. For this experiment, maintain He-GAL4; UAS-GFP flies at 24 °C. Allow adult flies to mate for a week, and then remove adults. F1 flies will then be collected from these vials after eclosion for experimental use.
- Collect virgin flies throughout the week, or until the desired number of flies are collected. Virgins are not required; however, mating may impact immune response. If F1 flies are to be tested as virgins, separate males and females within 8 hours of eclosion and maintain in separate vials to prevent mating. Collect enough flies to allow assessment of phagocytosis in at least 10 flies per genotype/treatment/sex per age.
- If aging the flies to one week, collect at least 50 flies total, or 20 flies per treatment condition for each particle, either fluoro-particles or pH sensitive particles, to be injected. This will ensure a minimum of 10 flies to assay at the time of the experiment.
- If aging flies to more than 3 weeks, collect at least 100-150 flies total, or 50-75 flies per treatment condition to ensure there are enough flies available to measure phagocytosis. When aging flies in the laboratory, we typically use insect cages maintained at 24 °C in 12:12 L:D conditions, and change the food every other day. If vials are being used instead of cages, tip flies into new vials every 3-5 days, depending on the condition of the food in the vial. The number of flies needed will depend on how late in age phagocytosis will be analyzed, and the age-specific survival rates of that genotype in a particular environmental condition.
- If young and aged flies will be assessed, plan accordingly so that 1-week old and aged flies will be injected on the same day. This will minimize variations in particle concentration between experiments and will ensure that the effect of age on the phagocytic measurement is not confounded with the effect of the day the assay was performed.
- House the collected flies at 24 °C until they are 5-7 days old, or maintain the flies to the desired age.
2. Prepare fluorescently labeled particles
- Reconstitute heat-killed E. coli fluoro-particles or pH sensitive E. coli particles to a stock concentration of 20 mg/mL or 1 mg/mL, respectively. Other bacteria are available for use that may be more suitable for certain experiments, but refer to the manufacturer’s instructions for appropriate stock concentrations.
- For fluoro-particles, add 990 µL of 1x PBS (pH 7.4) or preferred buffer and 10 µL of 2 mM (20%) sodium azide. Vortex to mix.
NOTE: Sodium azide is a preservative that may be omitted; however, particles prepared without sodium azide do not last as long. Fluoro-particles must be used within 24 hours and pH sensitive particles must be used within 7 days. - For pH sensitive particles, add 1,980 µL of 1x PBS (pH 7.4) or preferred buffer and 20 µL of 2 mM (20%) sodium azide. Vortex to mix.
- Make multiple single-use 20 µL aliquots in 1.5 mL microcentrifuge tubes. Store fluoro-particles at -20 °C for up to a year, and pH sensitive particles at 4 °C for up to 6 months, protected from light.
- For fluoro-particles, add 990 µL of 1x PBS (pH 7.4) or preferred buffer and 10 µL of 2 mM (20%) sodium azide. Vortex to mix.
- On the day of injections, remove sodium azide from the particles before use. To do this, centrifuge the particles for 5 min at 15,000 x g at room temperature.
- Remove the supernatant and wash particles twice by resuspending in 50 µL of 1x PBS or preferred buffer, and centrifuge for 5 min at 15,000 x g.
- After the second wash, remove the supernatant and resuspend particles in 100 µL of 1x PBS or preferred buffer. Keep the solution in the tube and minimize exposure to light throughout the experiment. Once the sodium azide is removed, use fluoro-particles within 24 h, and use pH sensitive particles used within 5-7 days.
NOTE: In our previous experiments, we found that this concentration of particles provided countable numbers of phagocytic events and that the number of particles available for phagocytosis by hemocytes was not limiting17. However, users of this protocol may want to compare results using other concentrations to ensure that there is an adequate number of particles available for phagocytosis by hemocytes in the conditions of their experiments.- If sodium azide was omitted, dilute the particles 1:5 in the same buffer the particles were prepared with, for injections.
- Add a drop (~10 µL) of green food coloring to the particles. This makes it easier to ensure the flies have been injected.
3. Inject the flies
- Prepare glass needles for injections.
- Pull glass needles using a pipette puller. Set the pipette puller heater to 55 °C and the solenoid to 45. Use only the needles provided with the injector, as it is not guaranteed that other needles will work with the same precision.
- Fill a 1 mL sterile syringe with mineral oil, and attach the 30 gauge hypodermic (G) needle, provided with the nano-injector.
- Backfill the pulled capillary needle by inserting the 30 G needle into the blunt end of the pulled glass needle and fill with mineral oil. Slowly remove the 30 G needle, ensuring there are no air bubbles throughout the needle, as this can cause inaccurate injection volumes. The injector will not work properly without backfilling the needle.
- Using forceps, break off the tip of the needle to create an opening to allow ejection of solution.
- Assemble the nano-injector.
NOTE: Other injectors can be used. The method described below apply to the nano-injector. For other injectors, consult the user manual for instructions.- Set the injector to the desired volume (between 46 nL and 69 nL).
- Remove the collet and place the sealing O-ring, the white spacer with the indentation facing up to receive the back end of the needle, and the larger O-ring onto the metal plunger, in that order. Reattach the collet without tightening it.
- Insert the metal plunger into the blunt end of the oil-filled glass needle. Gently push the needle down, inserting it into the larger O-ring. Tighten collet until secure.
NOTE: If the metal plunger does not extend past the collet, press and hold ‘EMPTY’ until the plunger is visible. This makes it easier to ensure the plunger is inserted into the needle. - Press and hold ‘EMPTY’ until the injector beeps. This ejects most of the mineral oil from the needle, leaving a small volume of oil to act as a barrier between the two liquids, as well as removes air bubbles.
- Fill the needle with either fluoro-particles or pH sensitive particles by inserting the tip of the glass needle into the microcentrifuge tube containing the prepared particles.
- Press and hold ‘FILL’ until the injector beeps.
- Injections
- Transfer the flies that are to be injected into an empty vial. Immobilize the flies by placing the vial in ice. CO2 can also be used to immobilize the flies. However, note that, when using pH sensitive particles, elevated CO2 levels can artificially acidify any buffers being used and can elevate background fluorescence.
- Inject the flies in the sternopleural plate of the thorax (Figure 1A). The injection is successful if green dye is seen going into the fly (Figure 1B). If the fly does not turn green, make sure the needle is not clogged.
NOTE: Alternatively, flies can be injected in the abdomen, but keep the injection site consistent across all experiments. - Place injected flies into a new food vial, noting the time that the first and last fly was injected. To minimize experimental error due to the amount of time it takes to complete injections, complete injections in a timely manner. With practice, it should take no longer than 10 min to inject one set of flies. Lay the vial on its side until all of the flies have recovered, to prevent the flies from becoming stuck in the food.
- Allow flies to recover for 60-90 min, depending on experimental conditions. Here, a 60-min recovery time was used. Note that this recovery time range was optimal for counting phagocytic events in the experimental conditions in the Horn et al. study17. However, under some conditions, this may be too long to detect subtle differences in phagocytosis between treatment groups. It may be useful to carry out time course experiments as was done previously17 to determine the recovery time that will reveal maximal differences between control and experimental results. Whatever recovery time is chosen, keep this consistent across all experimental treatments.
- If injecting with fluoro-particles and pH sensitive particles on the same day, use a new needle for each solution. Do not inject individual flies with both particles if they both fluoresce red, since the result would not differentiate between the two aspects of phagocytosis, particle binding/engulfment vs. particle inclusion in the phagosome.
4. Dissecting the dorsal vessel
- After flies have recovered for 60-90 min, transfer all living flies to an empty vial and immobilize on ice.
- Transfer one fly at a time onto a silicone elastomer dissection plate.
NOTE: Prepare dissection plates at least 1-week prior to use, if curing at room temperature. To do this, prepare the elastomer and pour it into a 33 mm x 10 mm Petri dish, filling the dish about halfway. Gently tap the dish on a flat surface to minimize air bubbles. Let the plates sit at room temperature, undisturbed, for at least a week.- Under a dissecting stereo microscope, orient the fly ventral side up.
- Using insect pins, pin the fly onto the plate by inserting one pin through the thorax and another pin through the most posterior end of the abdomen, near the genitalia (Figure 2A). It may be useful to cut the pins in half before pinning the specimen so as not to obstruct the dissection. Repeat with up to 10 flies per dissection plate.
NOTE: Optional: Remove the wings and legs before pinning fly to plate. This will help prevent a bubble forming around the fly when media is added. - Once all flies have been pinned to the plate, add enough dissection media to cover the flies (~1 mL) using a transfer pipet.
- Using forceps or cuticle scissors, remove the head.
- Using cuticle scissors, make two horizontal incisions: one directly above the posterior pin in the abdomen, and another at the most anterior end of the abdomen, where the thorax and abdomen meet (Figure 2B, C). In Figure 2, the head was left intact to clarify orientation.
- Make a vertical incision, connecting the two horizontal incisions (the three cuts resemble the letter I). This will open up the abdominal cavity (Figure 2D).
- Using forceps, remove the internal organs and tissue, avoiding the dorsal vessel. If undisturbed, the transparent dorsal vessel can often be seen pulsating near the anterior end of the abdomen. Additional pins can be used to pin open newly cut ends of the cuticle (Figure 2E, F).
- Using cuticle scissors, remove the thorax. Alternatively, the thorax can be removed before mounting the dissected cuticles and dorsal vessel on a microscope slide.
- Repeat with remaining flies. dissect flies in a timely manner, to minimize possible variations in phagocytic rate. Once all flies have been dissected, leave the cuticles with the attached dorsal vessel pinned to the plate. All of step 5 will be carried out in the dissection plate. This will prevent damaging or accidentally discarding cuticles between steps.
5. Fixation and staining
- Fix dissected cuticles with attached dorsal vessel in 4% paraformaldehyde (PFA).
- Using a new disposable transfer pipet for each step, discard the dissection media, and replace with 1 mL of 4% PFA. Throughout the fixation and staining, keep the dissections protected from light as much as possible.
- Incubate at room temperature for 15 min with rocking at 20 rpm. Do not allow dissections to sit in fixative for more than 20 min, as this could start to damage the tissue.
- Wash cuticles 2x in 1x PBS + 0.1% Tween (PBST).
- Remove fixative and replace with 1 mL of 1x PBST.
- Wash at room temperature for 15 min with rocking.
- Repeat 1x.
NOTE: Dissected, fixed tissue can be stored at 4 °C, for up to 3 days, protected from light, after the first wash by replacing the wash with fresh PBST. Do not store fixed tissue if antibodies will be used. Antibodies should be used with fresh tissue for best results.
- Optional: Antibody staining. Antibodies can be used to clearly visualize the dorsal vessel clearly (Figure 3), or to detect hemocyte specific markers. This can ensure only cells along the dorsal vessel are counted or to mark the membrane of the hemocytes.
- Remove the second wash, add primary antibody at the appropriate dilution. Incubate overnight at 4 °C, with rocking.
- Remove primary antibody, and wash twice with PBST for 15 min with rocking.
- Add fluorescent secondary antibody. We recommend a green-fluorescent antibody so it does not obscure the fluorescent particles. Incubate at room temperature for 2 h, with rocking.
- Remove secondary antibody, wash twice for 15 min each with PBST.
- Stain with DAPI.
- Remove final wash and replace with 1 mL of DAPI diluted in PBST (1:1000).
- Stain for 20 min with rocking at room temperature.
- Remove DAPI, and wash 2x (repeat step 5.2).
- Replace final wash with fresh 1x PBST.
NOTE: Flies can be stored at 4 °C, for up to 3 days, protected from light, after the first wash by replacing the wash with fresh PBST.
6. Mounting cuticles onto microscope slides and imaging
- Prepare dissected cuticles.
- Under the dissecting stereomicroscope, cut off excess cuticle that could interfere with imaging.
- Using forceps, transfer the cuticles with attached dorsal vessel into a 1.5 mL microcentrifuge tube containing 70% glycerol. Placing the cuticles in glycerol will help remove PBST and allows for a clearer image during imaging.
- Mount cuticles onto the microscope slide.
- Add a few drops of 70% glycerol to a microscope slide.
- Using forceps, remove cuticles from the glycerol tube and place in glycerol on the slide.
- Under the dissecting stereo microscope, use forceps to orient cuticles ventral side up, ensuring the dorsal vessel is visible. The darker pigment of the cuticle will be face down.
- Add an additional drop of glycerol, if needed. This helps prevent air bubbles and allows for a clearer image.
- Gently place a coverslip over cuticles and seal edges with fingernail polish. Allow to dry for 10-15 min before proceeding. Image the flies immediately or store in a lightproof box at 4 °C.
- Image the dorsal vessel.
- Using a fluorescent microscope, use the structural interference system to generate optical sections of the dorsal vessel. A confocal microscope can alternatively be used which may provide additional accuracy. Obtain Z-stack images of the dorsal vessel using a 20x objective and preferred imaging software. Add a 10 mm scale bar, and properly label and save images as tiff files (Figure 4) or the desired format.
NOTE: The number of Z-stack images obtained can vary between dissections and depends on how well the dorsal vessel was dissected, and on the desired step size between images. Here, the step size was set to 0.49 mm. That number can range anywhere from 3 to 40 images per stack in our experience.
- Using a fluorescent microscope, use the structural interference system to generate optical sections of the dorsal vessel. A confocal microscope can alternatively be used which may provide additional accuracy. Obtain Z-stack images of the dorsal vessel using a 20x objective and preferred imaging software. Add a 10 mm scale bar, and properly label and save images as tiff files (Figure 4) or the desired format.
7. Analyze images
- Quantify fluorescent events using ImageJ.
- Open images and stack them: Image à Stacks à Images to Stack.
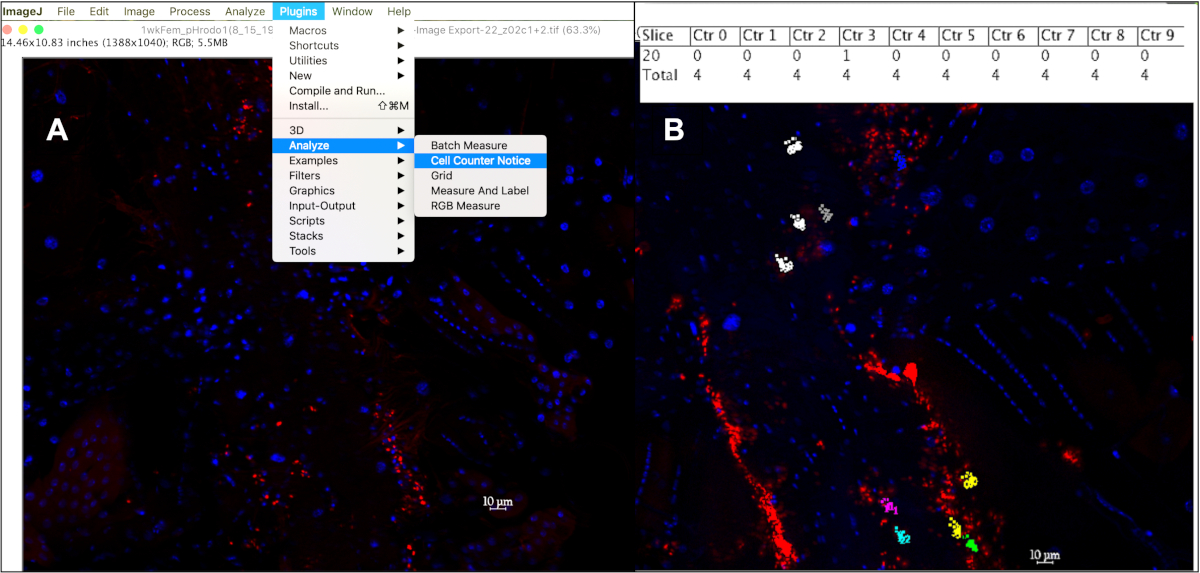
- Count only the ~1 mm sized fluorescent signals within a 10 mm diameter centered on a DAPI-positive nucleus by clicking on each event from at least 10 cells per fly, from at least 10 flies per age per particle type injected: Plugins à Analyze à Cell Counter Notice (Figure 5A). The cell counter notice tool assigns a different color to every cell tracked, with a colored dot that corresponds to every fluorescent event clicked on within that cell.
- To list the counter and stack position associated with each point, press ‘m’ or select Measure under the Analyze tab, or to display the number of events counted in each cell in a results table, press ‘alt +y’ (Figure 5B).
- Transfer the cell counts to a spreadsheet to be used for statistical analysis.
- Perform statistical analysis.
- Analyze differences in phagocytic events using either fixed-effects ANOVA or mixed-model nested ANOVA. Mixed models can be used to test the main fixed effects of age and genotype and the random effect of individuals nested within each genotype17.
NOTE: Investigators should be rigorous in testing the assumptions of any statistical procedure used to analyze the data.
- Analyze differences in phagocytic events using either fixed-effects ANOVA or mixed-model nested ANOVA. Mixed models can be used to test the main fixed effects of age and genotype and the random effect of individuals nested within each genotype17.
Results
To illustrate the described injection methods, Figure 1A shows the injection site on Drosophila melanogaster, as well as how food dye allows for a visual confirmation that the fly was injected (Figure 1B). The addition of food dye also aids in the recognition of a clogged needle. Injections can be performed in the abdomen, but keep the injection site consistent across experiments. This will help minimize possible variations between each experiment.
To visualize the fluorescently labeled particles within resident hemocytes along the dorsal vessel, we dissected the dorsal vessel and attached abdominal cuticle. Figure 2A-F outlines the dissection methods.
To assess the age-specific ability of young and aged flies to carry out phagocytosis, hemocytes along the dorsal vessel are visualized using a fluorescent microscope. To ensure that only cells along the dorsal vessel are counted, antibodies or GFP-tagged genes for certain blood cell markers or heart specific collagen, such as Hemese and Hemolectin, or Pericardin (Figure 3), respectively, can be used22,23,24. Fluorescently labeled E. coli particles are 1 mm in length, while hemocytes are 10 mm in diameter17. Only those fluorescent events located within a 10 mm diameter centered on a DAPI-positive nucleus are counted (Figure 4). To quantify fluorescent events, ImageJ software is used (Figure 5).

Figure 1: Injection site and visual verification. (A) Lateral side of thorax is pierced with a pulled-capillary needle. (B) Injections are visually verified by adding green food dye to particle solution. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Dorsal vessel dissection. (A) Pins are placed in the thorax and posterior abdomen (black arrows). (B-C) Two horizontal incisions (green arrows) are made at the posterior end of the abdomen (B), and anterior end (C). (D) A vertical incision (green arrow) is made down the middle of the abdomen, connecting the two horizontal cuts. (E) Optional pins (*) are used to filet open the abdominal cavity, exposing internal tissue. (F) Internal tissue (crop, gut, uterus, ovaries, fat bodies) is removed, exposing the dorsal vessel. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Ventral view of a dissected dorsal vessel from a 5-week old female injected with pH sensitive particles, stained with antibody directed against Pericardin (A). Dotted white line outlines the lateral side of the dorsal vessel, with arrow pointing towards the anterior region. (B) Enlarged image of (A): clusters of hemocytes (blue arrow) that were actively degrading bacteria, within the first aortic chamber of the dorsal vessel. (C) Enlarged image of (A) outlining the extracellular matrix (ECM) collagen-like protein, Pericardin (green arrow), that holds the dorsal vessel in place24. Please click here to view a larger version of this figure.
{kind=link}

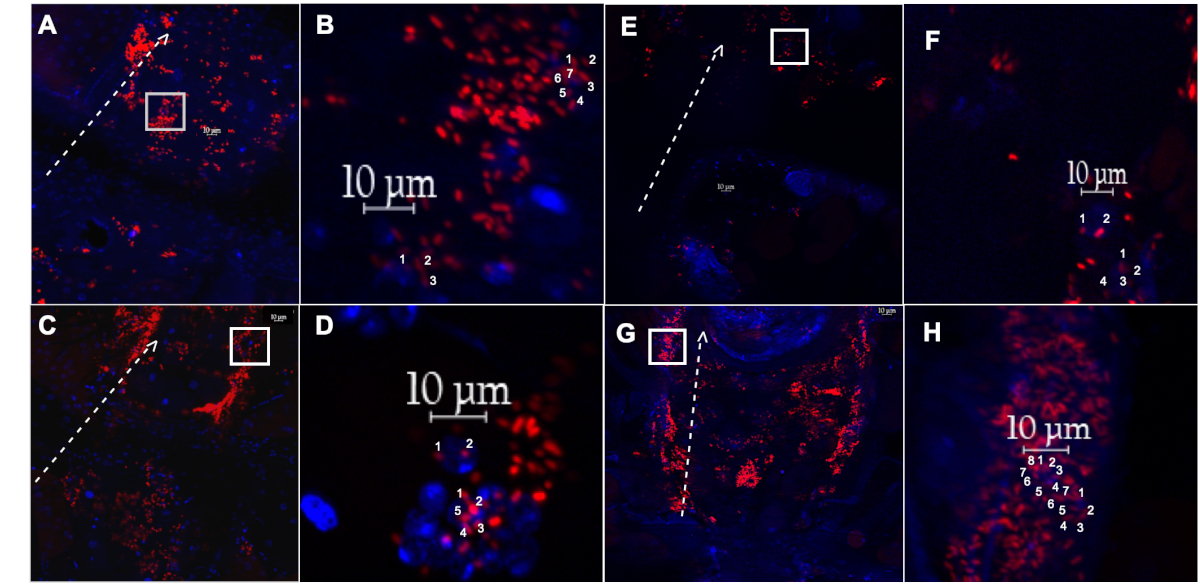
Figure 4: Dissected dorsal vessel and associated hemocytes from a female fly injected with pH sensitive particles, or fluoro-particles. (A) The dorsal vessel and associated hemocytes with engulfed pH sensitive-labelled E. coli particles (red), or (E) fluoro-labelled E. coli particles (red), isolated from a 1-week old fly, after recovering for 60 min. (B,F) Magnified inset of (A) and (E) (white box), respectively, showing two individual hemocytes with countable events. (C) The dorsal vessel and associated hemocytes with engulfed pH sensitive-labelled E. coli particles(red), or ( G) fluoro-labelled E. coli particles (red), isolated from a 5-week old fly, after recovering for 60 min. (D,H) Magnified inset of (C) and (G) (white box), respectively, showing two individual hemocytes with countable events. Dotted white line outlines the lateral side of the dorsal vessel, with arrow pointing towards the anterior region. Nuclei stained with DAPI (blue). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Quantifying phagocytic events within a 10 mm hemocytes using the cell counter in ImageJ. (A) After opening image(s) in ImageJ, the Cell Counter Notice tool can be used to keep track of phagocytic events per cell. (B) This tool will assign a different color to each cell selected to be counted, with each dot corresponding to a fluorescent event within that cell. Pressing ‘alt+y’ will display a table showing the number of events counted per cell. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The protocol described here is a reliable way to quantify different aspects of phagocytosis, under controlled experimental conditions. We note that we have only tested this procedure with gram negative bacterial particles and results may differ if gram positive bacterial particles are used. Indeed, it would be interesting to compare the phagocytic responses to both gram negative and gram positive bacteria in different experimental conditions. The use of a nano-injector allows for precise control over injection volumes, ensuring each fly is injected with the same volume of particles. One limitation to the protocol comes from inconsistencies in particle preparations. Particles will aggregate once frozen, so small variations in dilution volumes, or lack of vortexing, can affect particle concentration between experiments. To minimize possible variations in particle concentrations between ages, it is beneficial to inject 1- and 5-week old flies on the same day, using the same needle and particle solution. Another potential drawback is that during dissections, the dorsal vessel and/or cuticle can easily be damaged if pins are not handled properly. To avoid disrupting the dorsal vessel, minimize the number of pins used per dissection. The advantage to this dissection method is that all fixation, washing and staining steps can be performed in the dissection plate. Because the cuticles are pinned down, this prevents the cuticles from being lost between steps.
Compared to existing methods18,19,25,26,27,28,29, the described protocol has its advantages and limitations. By dissecting the dorsal vessel, we are able to visualize and quantify individual hemocytes at this location. This makes it possible to detect subtle variations in phagocytic activity between experimental groups. Other methods visualize fluorescently labeled particles by collecting hemocytes using a Bleed/Scrape assay19,25,26,27, or through an intact ventral cuticle18,19,28,29; however, individual hemocytes cannot be assessed when visualized through the dorsal cuticle. The advantage of this protocol, when compared to the Bleed/Scrape method is that our method allows us to assess only those hemocytes associated with the dorsal vessel, and does account for circulating cells or those along the body wall, which may be functionally different. Dissecting the dorsal vessel also removes the need to include a second round of injections with a fluorescence quencher, like Trypan blue19,26. This is because any particles not bound to or engulfed by a cell will be washed away during wash steps. Conversely, alternative methods may be easier to perform because they do not require dissections. While dissecting the dorsal vessel is easy to learn, this step adds a level of complexity that may not be feasible in some experimental designs.
Although the described use of this in vivo phagocytosis assay is to assess and quantify phagocytic events between different ages, this protocol is highly adaptable and can be used to analyze different aspects of phagocytosis between genotype, sex, or tissue type. With phagocytosis being of central importance for most multicellular animals, understanding how this process declines with age could lead to better therapeutic treatments for the aging population. This approach offers long-term potential for elucidating aspects of age-related changes in the immune response, with special focus on phagocytosis.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by grants from the National Institutes of Health R03 AG061484-02 and the UMBC College of Natural and Mathematical Sciences Becton Dickinson Faculty Research Fund.
Materials
| Name | Company | Catalog Number | Comments |
| 0.10 mm Insect pins | Fine Science Tools | 26002-10 | Here: pins are cut in half, and the sharp end is used |
| 1 mL sterile syringes | Becton Dickinson | 309602 | Filled with mineral oil to load needle |
| 15% Fetal Bovine Serum (FBS) | Gibco | 16000-044 | for dissection media |
| 16 % Paraformaldehyde | Electron Microscopy Sciences | 15710 | EM-grade, 4% working, diluted in 1X PBS |
| 1x Phosphate buffered saline (PBS) | Sigma | P3813 | |
| 3 mL Trasnfer Pipet | Falcon | 357524 | |
| 3.5" Glass Capillaries | Drummond | 3-000-203-G/X | 1.14mm O.D X 3.5" length X 0.53" I.D |
| 35x10 mm Petri dishes | Becton Dickinson | 351008 | Used as dissection plate, filled half way with Sylgard |
| 6x penicillin/streptomycin | Life Technologies | 15140-122 | for dissection media |
| 70% Glycerol | Sigma | G9012 | |
| Analog Vortex mixer | VWR | 58816-121 | |
| Biological point forceps, Dumont No. 5 | Fine Science Tools | 11295-10 | |
| DAPI (4',6-diamidino-2-phenylindole) | Life Technologies | D1306 | Diluted 1:1000 in 1x PBST |
| Drosophila strain | w[*]; P{w[+mC]=He-GAL4.Z}85, P{w[+mC]=UAS-GFP.nls}8 | ||
| E. coli (K-12 strain) BioParticles™, Alexa Fluor™ 594 conjugate | Life Technologies | E23370 | |
| Glass slides | Premiere | D17026102 | |
| Live cell imaging solution | Life Technologies | A14291DJ | preferred buffer for particle preparation and dilutions |
| Mineral oil | Mpbio | 194836 | |
| Nanoject II automatic nanoliter injector | Drummond | 3-000-204 | |
| Narrow Polystyrene Super Bulk Drosophila Vials | Genesee | 32-116SB | Size: 25 X 95 mm |
| Nutating Mixer | Fisher Scientific | 88-861-043 | Speed used: 20 rpm |
| pHrodo™ Red E. coli BioParticles™ Conjugate for Phagocytosis | Life Technologies | P35361 | |
| Schneider's Drosophila cell culture media (1x) | Gibco | 21720-024 | Dissection media, combine: Schneiders, FBS, and pen/strep; filter sterilize |
| Sodium azide | Sigma-Aldrich | S2002 | 2mM (or 20%) working |
| Spring scissors | Fine Science Tools | 15000-00 | |
| Sylgard 184 Silicone elastomer | Electron Microscopy Sciences | 24236-10 | Prepare according to provided protocol |
| Tween 20 | Sigma | P1379 | For PBS + 0.1% tween |
| Vertical Pipette Puller Model 700C | David Kopf Instruments | 812368 | Heater: 55? Solenoid: 45 |
| Zeiss AxioImager.Z1 fluorescent microscope | Zeiss | Here: Apotome structural interference system with Zeiss Zen imaging software |
References
- Abhyankar, V., Kaduskar, B., Kamat, S. S., Deobagkar, D., Ratnaparkhi, G. S. Drosophila DNA/RNA methyltransferase contributes to robust host defense in ageing animals by regulating sphingolipid metabolism. The Journal of Experimental Biology. 221, (2018).
- DeVeale, B., Brummel, T., Seroude, L. Immunity and aging: the enemy within. Aging Cell. 3 (4), 195-208 (2004).
- Gomez, C. R., Nomellini, V., Faunce, D. E., Kovacs, E. J. Innate immunity and aging. Experimental Gerontology. 43 (8), 718-728 (2008).
- Panda, A., et al. Human innate immunosenescence: causes and consequences for immunity in old age. Trends in Immunology. 30 (7), 325-333 (2009).
- Aw, D., Silva, A. B., Palmer, D. B. Immunosenescence: emerging challenges for an ageing population. Immunology. 120 (4), 435-446 (2007).
- Min, K. -. J., Tatar, M. Unraveling the molecular mechanism of immunosenescence in Drosophila. International Journal of Molecular Sciences. 19 (9), 2472 (2018).
- Brand, A. H., Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118, 401-415 (1993).
- Hales, K. G., Korey, C. A., Larracuente, A. M., Roberts, D. M. Genetics on the fly: a primer on the Drosophila model system. Genetics. 201 (3), 815-842 (2015).
- Banerjee, U., Girard, J. R., Goins, L. M., Spratford, C. M. Drosophila as a genetic model for hematopoiesis. Genetics. 211 (2), 367-417 (2019).
- Rämet, M., Manfruelli, P., Pearson, A., Mathey-Prevot, B., Ezekowitz, R. A. B. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature. 416 (6881), 644-648 (2002).
- Garschall, K., Flatt, T. The interplay between immunity and aging in Drosophila. F1000Research. 7, (2018).
- Brennan, C. A., Anderson, K. V. Drosophila: The genetics of innate immune recognition and response. Annual Review of Immunology. 22 (1), 457-483 (2004).
- Rosales, C., Uribe-Querol, E. Phagocytosis: a fundamental process in immunity. BioMed Research International. 9042851, (2017).
- Kinchen, J. M., Ravichandran, K. S. Phagosome maturation: going through the acid test. Nature Reviews. Molecular Cell Biology. 9 (10), 781-795 (2008).
- Stuart, L., et al. A systems biology analysis of the Drosophila phagosome. Nature. 445 (7123), 95-101 (2007).
- Mackenzie, D. K., Bussière, L. F., Tinsley, M. C. Senescence of the cellular immune response in Drosophila melanogaster. Experimental Gerontology. 46 (11), 853-859 (2011).
- Horn, L., Leips, J., Starz-Gaiano, M. Phagocytic ability declines with age in adult Drosophila hemocytes. Aging Cell. 13 (4), 719-728 (2014).
- Elrod-Erickson, M., Mishra, S., Schneider, D. Interactions between the cellular and humoral immune responses in Drosophila. Current Biology. 10 (13), 781-784 (2000).
- Garg, A., Wu, L. P. Drosophila Rab14 mediates phagocytosis in the immune response to Staphylococcus aureus. Cellular Microbiology. 16 (2), 296-310 (2014).
- . Bloomington Drosophila Stock Center: Indiana University Bloomington Available from: https://bdsc.indiana.edu/innformation/recipes/molassesfood.html (2019)
- Fellous, S., Lazzaro, B. P. Larval food quality affects adult (but not larval) immune gene expression independent of effects on general condition. Molecular Ecology. 19 (7), 1462-1468 (2010).
- Kurucz, E., et al. Hemese, a hemocyte-specific transmembrane protein, affects the cellular immune response in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 100 (5), 2622-2627 (2003).
- Goto, A., et al. A Drosophila haemocyte-specific protein, hemolectin, similar to human von Willebrand factor. Biochemical Journal. 359, 99-108 (2001).
- Cevik, D., Acker, M., Michalski, C., Jacobs, J. R. Pericardin, a Drosophila collagen, facilitates accumulation of hemocytes at the heart. Developmental Biology. 454 (1), 52-65 (2019).
- Ghosh, S., Mandal, S., Mandal, L. Detecting proliferation of adult hemocytes in Drosophila by BrdU incorporation and PH3 expression in response to bacterial infection. Wellcome Open Research. 3, (2018).
- Hao, Y., Yu, S., Luo, F., Jin, L. H. Jumu is required for circulating hemocyte differentiation and phagocytosis in Drosophila. Cell Communication and Signaling. 16 (1), 95 (2018).
- Petraki, S., Alexander, B., Brückner, K. Assaying blood cell populations of the Drosophila melanogaster larva. Journal of Visualized Experiments. (105), (2015).
- Gyoergy, A., et al. Tools allowing independent visualization and genetic manipulation of Drosophila melanogaster macrophages and surrounding Tissues. G3: Genes, Genomes, Genetics. 8 (3), 845-857 (2018).
- Bosch, P. S., et al. Blood cells of adult Drosophila do not expand, but control survival after bacterial infection by induction of Drosocin around their reservoir at the respiratory epithelia. BioRxiv. 578864, (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved