
Method Article
Generation of Murine Primary Colon Epithelial Monolayers from Intestinal Crypts
In This Article
Summary
In this protocol, we describe how to generate murine primary epithelial colon monolayers directly from intestinal crypts. We provide experimental approaches to generate confluent monolayers on permeable filters, confluent monolayers for scratch wound healing and biochemical studies, and sparse and confluent monolayers for immunofluorescence analysis.
Abstract
The intestinal epithelium is comprised of a single layer of cells that act as a barrier between the gut lumen and the interior of the body. Disruption in the continuity of this barrier can result in inflammatory disorders such as inflammatory bowel disease. One of the limitations in the study of intestinal epithelial biology has been the lack of primary cell culture models, which has obliged researchers to use model cell lines derived from carcinomas. The advent of three dimensional (3D) enteroids has given epithelial biologists a powerful tool to generate primary cell cultures, nevertheless, these structures are embedded in extracellular matrix and lack the maturity characteristic of differentiated intestinal epithelial cells. Several techniques to generate intestinal epithelial monolayers have been published, but most are derived from established 3D enteroids making the process laborious and expensive. Here we describe a protocol to generate primary epithelial colon monolayers directly from murine intestinal crypts. We also detail experimental approaches that can be used with this model such as the generation of confluent cultures on permeable filters, confluent monolayer for scratch wound healing studies and sparse and confluent monolayers for immunofluorescence analysis.
Introduction
Intestinal epithelial cells (IEC) line the intestines forming a selectively permeable barrier that allows nutrient and water absorption while prevent microorganisms and toxins to enter the body 1. The intestinal mucosa is composed of luminal projections called villi (only present in small intestine) and invaginations named crypts. Villi and the surface of colon crypts are covered by differentiated epithelial cells while the base of the crypts is comprised of stem cells that make the rapid renewal of the intestinal epithelia, which has a turnover from 3 to 7 days. Intestinal stem cells (ISC) are not only important for maintaining gut homeostasis but for adequate repair of damaged epithelia2.
Study of the intestinal epithelia biology was limited by the lack of primary cell cultures with transformed cell lines being the only tool available. Intestinal epithelial model cell lines are not capable to accurately replicate the physiology of the normal intestinal epithelium. The development of 3D cultures derived from ISC provided intestinal mucosal biologists with in vitro models that resemble in vivo gut mucosal conditions3. Crypts can be easily isolated from murine samples, embedded in a basement membrane matrix medium (e.g., Matrigel) and grown in conditioned media containing Wnt3a, R-spondin, and Noggin, generating 3D structures known as enteroids (small intestine) or colonoids (large intestine)4. Enteroids and colonoids are polarized spheroidal structures where the apical domain is facing an internal lumen and the basolateral region is in direct contact with the extracellular matrix. Enteroids and colonoids contain all major differentiated intestinal epithelial sub-types such as Enterocytes/Colonocytes, Paneth, Enteroendocrine and Goblet cells and they appear in relatively the same proportions as they do in the section of the gut where they were isolated from5. Even though 3D enteroids and colonoids represent a major advance in the study of intestinal development and physiology, these models present certain drawbacks such as limited access to the apical surface of the epithelial cells (lumen) and the ability to scale cultures up or down to achieve high-throughput screening of molecules of interest. To overcome these limitations, protocols to obtain primary 2D cultures of IEC derived from 3D enteroids/colonoids were generated. 2D enteroids/colonoids grow as sheet of cells just as model cell lines do and are ideal to study intestinal wound repair, host-pathogen interactions, and regenerative medicine amongst others. Several published papers describe how to generate 2D monolayers from 3D structures or directly from intestinal crypts, (see6,7,8,9,10,11) but these methods tend to be labor intensive and hard to reproduce. A fast, simple, and reproducible method to obtain monolayers directly from freshly isolated mouse intestinal crypts is outlined in this protocol.
Here we explain in detail the process for crypt extraction with minimal generation of debris, extracellular matrix choice and different surfaces and applications for this technique. This experimental approach was optimized for colon crypts, but similar results are obtained when applied for small intestine.
Protocol
All procedures described below have been approved and conducted in accordance with the guidelines set by University of Michigan Institutional Animal Care and Use Committee.
1. Preparation of reagents for crypt isolation and culture (prepare in tissue culture hood)
- 50 mM ethylenediamine tetraacetic acid (EDTA): Add 50 mL 0.5 M stock to 450 mL of phosphate buffered saline, without calcium (Ca2+) and magnesium (Mg2+) to prepare 500 mL. In this protocol PBS will refer to PBS without calcium and magnesium unless otherwise stated.
- Shake buffer: Dissolve 7.4 g sucrose (43.3 mM) and 5 g sorbitol (54.9 mM) in PBS to prepare 500 mL.
- L-WRN (L-Wnt-3A, R-spondin and Noggin) media: Supplement Advanced Dulbecco's Modified Eagle Medium/Ham's F-12 (DMEM/F12) (780 mL) with 20% Fetal Bovine Serum (FBS) (200 mL), 1x commercially available glutamine supplement (10 mL), 100 U/mL penicillin and, 100 g/mL streptomycin (10 mL), and filter sterilize with 0.22 µm filter.
- Get L-WRN cells through ATCC, grow in T175 flasks, and select using Geneticin and Hygromycin. Media is changed and collected for 12 days.
NOTE: Every batch of media is tested for Wnt activity using a TOPflash Wnt Reporter assay. In this case the Michigan Medicine Translational Tissue Modeling Laboratory protocols (https://www.umichttml.org/protocols) were followed. The TOPflash HEK 293 cell line is grown to confluency in a T75 flask, trypsinized and plated onto a 96-well plate. The following day different dilutions of the collected media is added to cells and incubated in a 5% CO2 incubator at 37 °C overnight. Next day, the cells are lysed, and Firefly Luciferase assay is performed according to manufacturer's instructions. The assay is normalized using recombinant Wnt-3A. The media is divided into 25 mL aliquots in 50 mL conical tubes and store at -80 °C.
- Get L-WRN cells through ATCC, grow in T175 flasks, and select using Geneticin and Hygromycin. Media is changed and collected for 12 days.
- Base media: For 500 mL, supplement Advanced DMEM/F12 (448 mL) with 2x commercially available glutamine supplement (10 mL), 20 mM HEPES (10 mL), 100 U/mL penicillin and, 100 g/mL streptomycin (10 mL), 2 mM N-acetyl-L-cysteine (2 mL), N2 supplement (10 mL) and B27 supplement (20 mL), filter sterilize with 0.22 µm filter. Divide the media into 25 mL aliquots in 50 mL conical tube and store at -80 °C.
- LWRN complete media: Combine 25 mL LWRN media with 25 mL of base media and supplement with 200 ng/mL Epidermal growth factor (EGF) (20µL) and 2x antibiotic-antimycotic solution (1 mL). Store the complete media at 4 °C.
- Collagen and Laminin: Dissolve 5 mg powder in 5 mL of filter sterilized 100 mM acetic acid (add 60 µL of acetic acid stock to 9.94 mL of molecular grade water) to produce a stock concentration of 1 mg/mL. Rotate at 4 °C for 4 h and make 100 µL aliquots in 0.2 mL tubes. Freeze at -20 °C. Laminin is purchased at a stock concentration of 100 µg/mL.
- Complete media without growth factors (CMGF-): Supplement Advanced DMEM/F12 (500 mL) with 1x commercially available glutamine supplement (5 mL), 10 mM HEPES (5 mL), 100 U/mL penicillin and, 100 g/mL streptomycin (5 mL).
- Differentiation media: To 9.2 mL of CMGF- media, add 200 µL of B27 supplement, 100 µL of N2 supplement, 20 µL of N-acetyl-L-cysteine, 500 µL of Noggin media12 (made from Noggin-producing cells) and 2 µL of EGF to make 10 mL of differentiation media.
2. Preparation of plates, chamber slides and cell culture membrane inserts
- Coating 48-well plate and chamber slides for plating 2D monolayer: Use the coating solution which constitutes laminin (1:40 dilution, see Table of Materials) and collagen (1:30 dilution) in cold Dulbecco's Phosphate buffered saline, with Ca2+ and Mg2+ (DPBS). Add 200 µL of coating solution to each well and pre-incubate the plate/chamber slide in a 5% CO2 incubator at 37 °C for at least 2 h.
- Coating 0.4 µm cell culture membrane inserts: Make 1:30 dilution of collagen in molecular grade water and add 200 µL to each insert. Keep the plate containing the membrane insert on ice at 4 °C for 30 min. Following 30 min incubation, keep the plate in a 5% CO2 incubator at 37 °C for 1.5-2 h. Polyester and polycarbonate membrane inserts yield comparable results.
NOTE: Any tissue culture plate can be seeded (up and downscaling can be performed) using this protocol by adjusting the collagen/laminin volume to obtain full coverage of the plating surface.
3. Crypt isolation
NOTE: Before starting the dissection, prepare collagen and/or laminin coated plate/membrane inserts/chamber slide and leave them in a 5% CO2 incubator at 37 °C. Prepare a clean work bench and sterile surgical instruments appropriate for surgery, and a biological safety cabinet for culturing 2D monolayers. Confirm all other standard equipment for 2D monolayer culturing such as humidified CO2 incubator, tabletop centrifuges (maintained at 4 °C), microscope and pipettes (including serological pipettes) are ready.
- Use C57Bl/6 mice, 8-12 weeks old. Euthanize mice using an approved method of euthanasia.
- Spray the mice carcasses with 70% ethanol (EtOH) solution to clean the dissection area and remove excess EtOH using paper tissue.
NOTE: Ensure that the crypt isolation reagents (such as PBS, 50 mM EDTA, shake buffer) crypts are kept cold. These reagents can be prepared one-day prior and are good to use for at least 3 months when stored at 4 °C. Also, make sure that the LWRN complete media is kept in a bead/water bath maintained at 37 °C until use. - Using clean dissection scissors and forceps, dissect out the colon from the rectum to the cecum. Hold the end of the colon using forceps and very gently flush off the feces using ice cold PBS in a 10 mL syringe, fitted with a 20 G feeding tube (Figure 1A). Make sure not to rupture the colon.
- Remove the proximal colon. This will be the portion of the colon closest to the ileocecal junction.
- With forceps, slide the distal colon gently onto the 20 G feeding tube, tying off the colon at the end of the tube by the tip with 4-0 silk suture thread (Figure 1B).
- Invert the colon inside out using one's fingers over the end tied off and tie the other end with 4-0 silk suture thread. Using surgical scissors, cut colon below the tip of the feeding tube (Figure 1C,D,E).
- Using the plunger of a 1.25 mL repeat syringe, gently open the untied end of the inverted colon onto the tip of a 1.25 mL repeat syringe. Slide the inverted colon onto the end of the syringe and tie tightly with 4-0 silk suture thread (Figure 1F,G).
- Insert the plunger into the syringe and inflate the colon to form a sausage. Inflate until the colon sausage looks turgent with no visible wrinkles (Figure 1H).
- Place syringe/colon in 15 mL tube with 5 mL of Cell Recovery Solution on ice for 20 min, inflate and deflate the colon once every 5 min (Figure 1I). The sausage must remain inflated during the incubation.
- Tie off using 4-0 silk suture thread below the tip of the repeat syringe with the colon inflated. Cut the sausage off the repeat syringe and place in 15 mL tube containing 10 mL of 50 mM (2mM for small intestine) EDTA for 40 min and rotate at 4 °C (Figure 1J,K).
- Decant off the EDTA solution and replace with 5 mL of shake buffer. Shake the sausage manually in vertical position (vigorously) for 2 min.
- Decant the shaking solution into a new 15 mL tube and repeat shaking step for a total of 10 mL of crypts in shaking buffer.
- Take 20 µL of the crypt suspension in a Petri dish and count the number of crypts under a microscope. Calculate the crypt concentration in crypts/µL. Depending on the concentration, dilute the samples to obtain 5 crypts/ µL at the moment of plating (1000 crypts/cm2).
- Spin the tube with isolated crypts using tabletop centrifuge at 400 x g for 10 min at 4 °C.
- In the meantime, remove 48-well plate/insert/chamber slide from the incubator and place it in biosafety cabinet. Aspirate the coating solution using P200 and leave the plate with the lid slightly offset until cells are ready to be plated.
4. Culturing 2D monolayer
NOTE: For a detailed protocol on how to generate intestinal epithelial monolayers from 3D colonoids check protocols by Estes and Kovbasnjuk labs (7,11).
- Remove the shaking buffer using 10 mL serological pipette. Make sure that the pellet is intact and may using P1000 to remove any liquid left. Re-suspend the pellet in 3 mL of LWRN complete media and pipette up and down with P1000. Add 200 µL of crypts to each well of a pre-coated 48-well plate/chamber slide and incubate in a 5% CO2 incubator at 37 °C.
- The following day aspirate the media using P200 and add fresh media. The cells become confluent in 24-48 h.
- For cell culture membrane inserts, add 200 µL crypts (5 crypts/µL) to top of the inserts and 600 µL of complete L-WRN media to the bottom part. The following day aspirate the media using P200 and add fresh media only to the top chamber. Incubate the plate in a 5% CO2 incubator at 37 °C. Transepithelial Electrical Resistance (TEER) is measured every day using Epithelial Volt/Ohm meter (EVOM).
NOTE: If the culture has a TEER reading greater than 300 Ω.cm2, they are meant to be confluent. Confluency is achieved in 3-4 days. Change media every 2 days.
Results
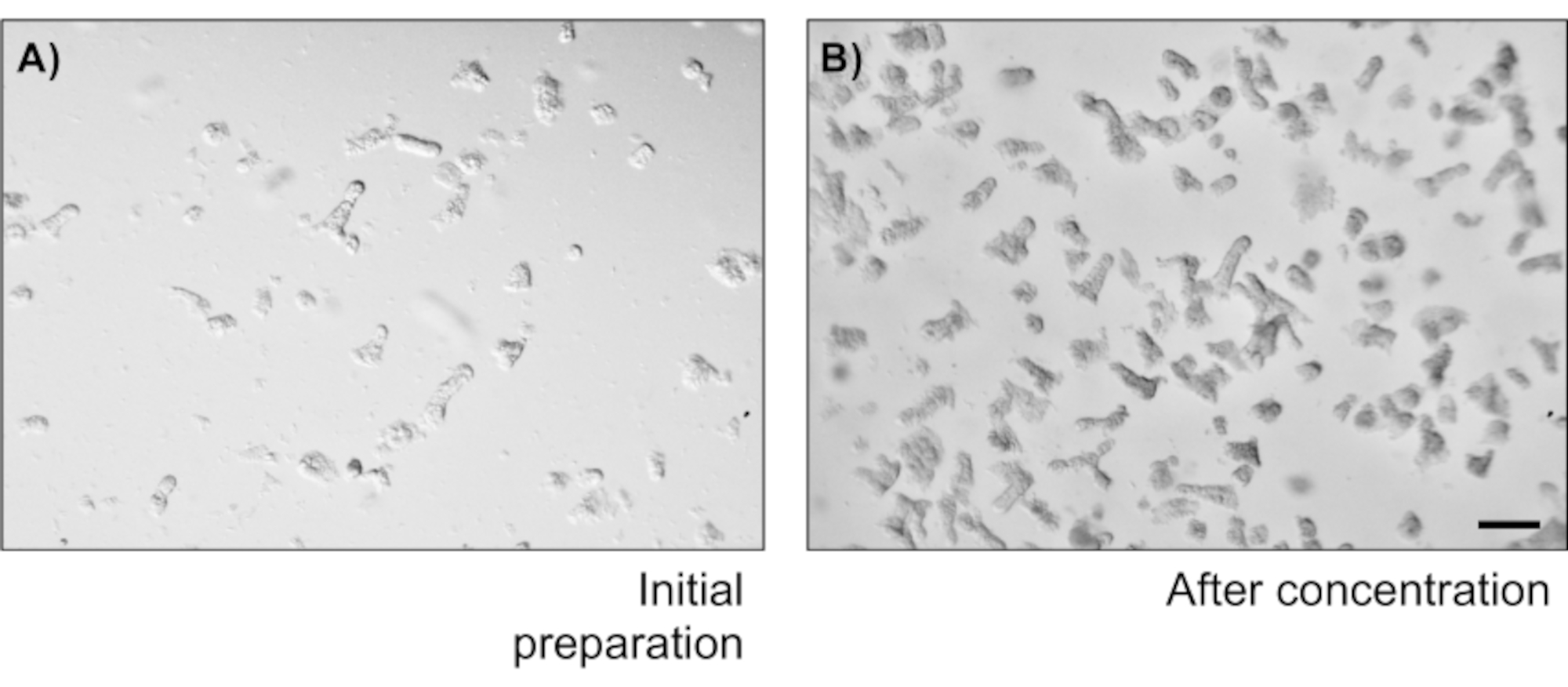
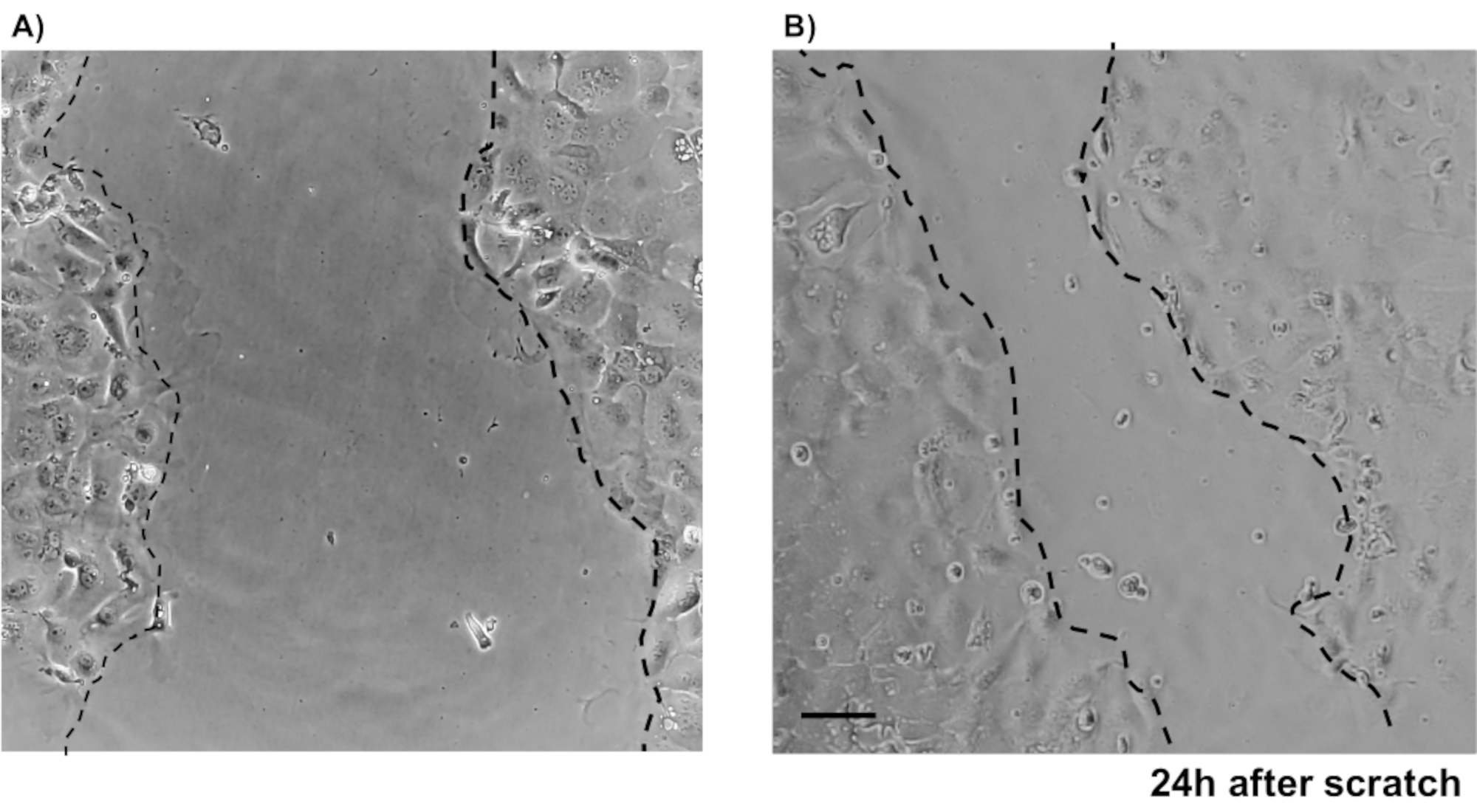
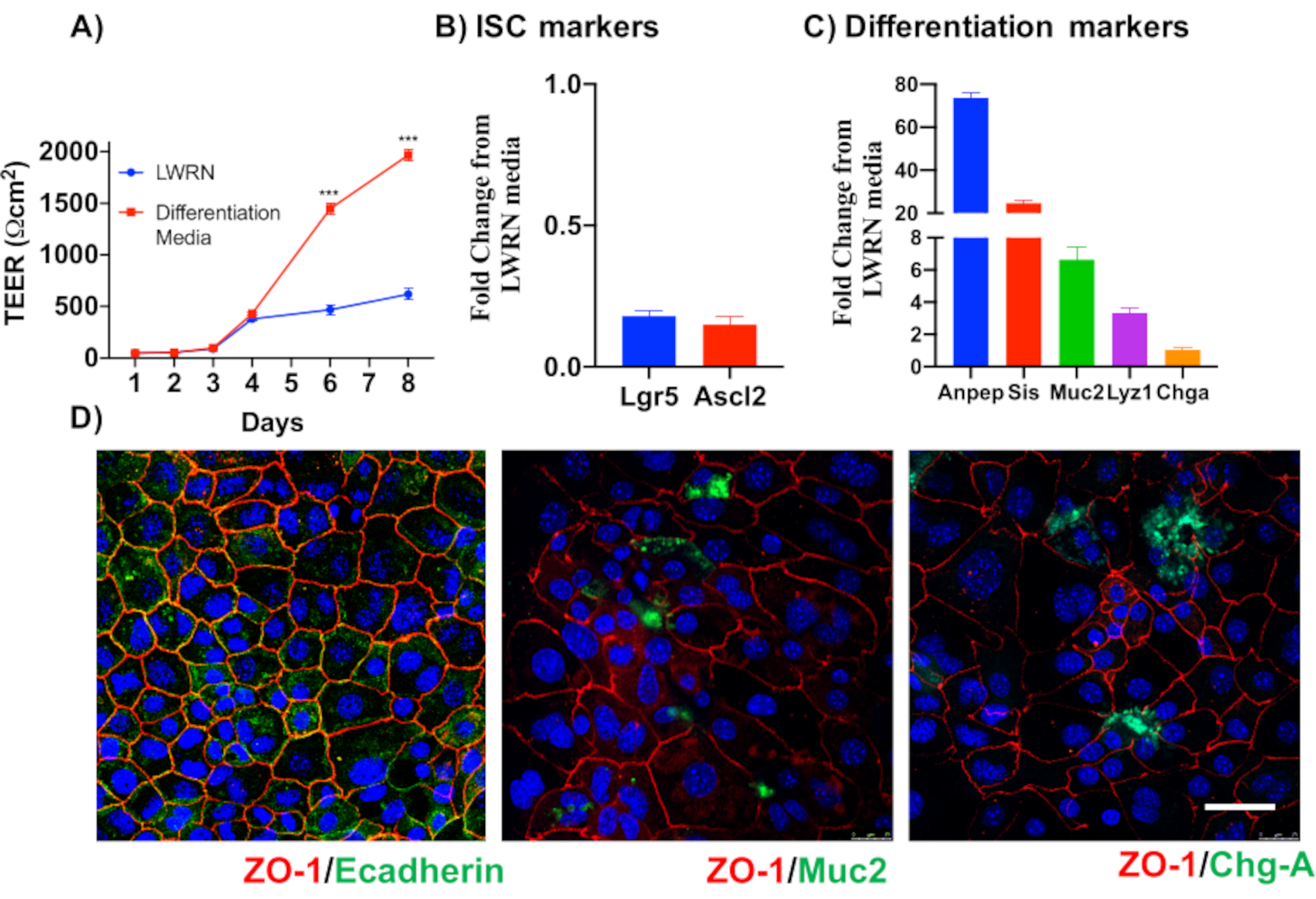
To illustrate the reliability of the primary epithelial colon monolayer cultures, a summary of the crypt isolation and representative images derived from the protocol is shown. The user must have in mind that the isolated crypts are cultured in sterile conditions, so a correct dissection and cleaning of the colon is a priority. Figure 1 features key steps during crypt isolation. Isolated crypts (Figure 2A) are now counted and concentrated (Figure 2B) to obtain a concentration of 5 crypts/µL. After a concentrated preparation of crypts, the cells will be plated in the desired format (culture dish, membrane inserts or chamber slides) and incubated with the appropriate media depending on experimental needs. Figure 3 shows culture progression after 24 and 48 h of culture in 48-well plates. The cells are incubated until the desired confluency is reached. To exemplify possible applications of this method, we allowed 48-well plate wells to reach confluency and proceeded to do a scratch-wound assay. Figure 4 depicts a freshly created scratch (Figure 4A) in a 2D colonoid monolayer and the same wound 24 h after (Figure 4B). It is clear that the culture is still healthy and viable and there is wound repair. To generate differentiated monolayers, media is changed from LWRN to differentiation media. Differentiation is achieved by showing high TEER values (Figure 5A), decrease of ISC markers ((Leucine-rich repeat-containing G-protein coupled receptor 5 (Lgr5) and Achaete-scute like 2 (Ascl2)) and increase of differentiation markers ((Alanyl aminopeptidase (Anpep), Mucin 2 (Muc2), lysozyme 1 (Lyz1), Sucrose iso-maltase (Sis) and Chrmogranin A (Chga)) (Figure 5B,C) by PCR. Other markers as CDX2 and KRT20 can also be included in this panel. In addition to mRNA expression levels, the appearance of sub-types of differentiated epithelial cells in 2D colonoids grown in differentiation conditions is also shown by immunofluorescence (Muc2 and Chga; Figure 5D).

Figure 1: Sample preparation for healthy monolayers generation. Sample preparation is crucial for generation of healthy monolayers. Key steps of the isolation process are depicted in this figure to make it easier to the reader. The colon is excised from the mouse, making sure there is no fur leftovers. Carefully remove the feces making sure you do not perforate the colon; this is of vital importance as the colon needs to be able to hold air and be inflated and deflated. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Crypt isolation count and concentration. (A) Colonic crypts after shaking the colon sausages. The image depicts a field of a 20 µL drop. (B) Crypt concentration in order to obtain 5 crypts/µL. Scale bar: 1 mm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Primary IEC Monolayer growth. (A) 2D IEC monolayers after 24 h of plating and removal of cell debris. (B) Confluent 2D IEC monolayers 48 h after plating. Scale bar: 10 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Scratch wound assays using murine primary IEC. Images showing wound healing after a scratch was made in epithelial colon monolayer. Scale bar: 5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Differentiation of epithelial colon monolayers. (A) Differentiation media creates tight epithelial barriers as demonstrated by TEER. Differentiation media causes a drop in mRNA expression of (B) stem cells markers and upregulation of (C) differentiation markers. (D) Markers of specialized differentiated epithelial cells such as Muc2 and Chromogranin-A can also be detected via immunofluorescence of direct 2D colonoids. Scale bar: 5 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Our protocol provides a fast, reproducible, and reliable method to generate direct primary 2D IEC monolayers. One of the main differences in our protocol compared to previously published protocols to generate colon epithelial monolayers is that we do not cut the colon in small pieces to liberate the crypts. Instead, we adapted a protocol to separate intestinal epithelium from mesenchyme13 by a combination of chemical and mechanical forces to release crypts in an extremely clean preparation, (Figure 2), providing the researcher with ideal material to generate primary cultures. Our isolation method can be also used to generate 3D enteroids and colonoids. It is important to do a crypt count every time an experiment is performed to normalize the number of crypts that are being plated. Variables such as colon sausage turgor, speed of isolation, user expertise can affect the number of crypts isolated. The suggested plating crypt concentration mentioned in the protocol is a starting point, but every user to account for user driven variables must optimize it. We have used this technique with male and female WT mice ranging from 8 to 20 weeks and we have not seen major differences in cell survival, in theory crypts isolated from younger mice have better chances to survive. Crypts are plated in excess as only a small percentage of them attach to the surface and survive. A balance where there are enough crypts to have a 50% confluency one day after plating but not too many crypts where the dying crypts will have a cytotoxic effect is the goal. LWRN media must be carefully removed 24 hours after plating to eliminate dead crypts and debris, this must be done carefully to avoid detaching cells that are already growing as a monolayer.
After initial removal of LWRN media, the user must decide if the experimental conditions require primary IEC monolayers that remain closer to stem cells and add fresh LWRN media or if differentiation of the monolayer is desired, replace with differentiation media. The single most important factor in this protocol is to assure the integrity of the colon during the isolation process. If a rupture occurs, the sausage can be shortened to eliminate the damaged area. Before putting the sausage in EDTA be sure that the knots are as tight as possible. If the sausage is deflated after the EDTA incubation the protocol can be continued with little to no effect in the overall crypt yield. If the repeat syringe is not available, a regular micropipette tip attached to a regular syringe can also be used for the process of inflation and deflation. Also, if no cell recovery solution is available, the inflation and deflation steps can be done in EDTA (2mM small intestine, 50 mM colon), but this substitution is not recommended. If confluency is not required, crypts can grow in plates coated only with collagen and even in uncoated plates. Only use uncoated plates cases there is no other option, but the cells will not grow healthy as they would in collagen-coated plates. When it comes to culture health and stability, monolayers plated in plastic are healthy for 4 to 5 days while monolayers plated in transwells can be carried for up to 8 days.
One of the main limitations of this method is the cell media required to grow the epithelial colon monolayers. LWRN cells are available at ATCC, but LWRN conditioned media generation is labor intensive and requires access to a fluorescent spectrometer to determine Wnt activity. Differentiation media requires a number of reagents that are added fresh before use, which makes it a tedious process. Finally, most of these reagents are costly and is easy to burn the reagents in a fast pace. If a laboratory desires to establish this technique without previous intestinal primary cell culture, it is highly recommended to find a collaborator/college with experience and train one of their members.
Maintenance of 3D culture could be expensive due to cost of basement membrane matrix medium and high volumes of conditioned media needed for organoid cultures, but it has the advantage of using a reduced number of mice and the generated structures can be passaged many times. Enteroids (derived from small intestine) are relatively easy to isolate and maintain while colonoids are more delicate, grow in a slower pace and have a more limited passage capacity. Monolayer generation from 3D colonoids require a disproportionate amount of 3D structures, that make these kinds of experiments time consuming and costly. On the contrary, direct epithelial colon monolayer prep is fast and is a quick way to obtain the results. One colon prep can generate a confluent area of 75 cm2 (10 to 15 mL of conditioned media, replaces once) 2 to 3 days after plating (this area would require 144 wells of 3D colonoids, which means almost 6mL of Matrigel and more than 250 mL of conditioned media). The lower consumption of media, low-cost maintenance of the cell culture and ability to perform functional tests and fast downstream processing are big advantages of epithelial colon monolayers.
This protocol is a valuable tool in the study of intestinal epithelial cell biology in areas such as cell-adhesion, polarity, and differentiation. It gives the advantage to generate primary cell cultures from genetically modified mice (knock-out, overexpressing, reporters). The primary intestinal epithelial monolayers allow easy access to the apical and basolateral surfaces (when plated on transwells) allowing the study of permeability, barrier and transepithelial migration of different cells types. Finally, this model can be useful in different fields like host-pathogens interactions, epithelial damage and repair and drug discovery.
Disclosures
The authors do not have any conflicts of interest.
Acknowledgements
This work was supported by a Crohn's and Colitis Foundation Career Development Award (544599, to MQ) and the NIH grants (DK055679, DK089763, DK059888, to AN). We would like to thank the Michigan Medicine Translational Tissue Modeling Laboratory for their continuous help and access to their reagents and protocols.
Materials
| Name | Company | Catalog Number | Comments |
| Advanced DMEM/F12 | Gibco | 12634-010 | |
| Antibiotic Antimycotic solution | Corning | 30-004CI | |
| B27 supplement (50X) | Gibco | 12587-010 | |
| Cell Recovery Solution | Corning | 354253 | |
| Collagen from human placenta (type IV) | Sigma-Aldrich | C5533 | |
| D-Sorbitol | Sigma | 85529-250G | |
| D-Sucrose | Fisher Scientific | BP220-1 | |
| Dulbecco’s phosphate buffered saline, with Ca2+ and Mg2+ (DPBS) | Corning | 21-030-CV | |
| Epithelial Volt/Ohm meter | World Precision Instruments | 0-10KΩ with STX2 (EVOM2) | |
| Ethylenediamine tetraacetic acid (EDTA) | Lonza | 51201 | |
| Fetal Bovine Serum (FBS) | Corning | 35-016-CV | |
| Firefly Luciferase assay | Biotium | 30085-2 | |
| Geneticin | Gibco | 10131-035 | |
| GlutaMAX (100X) | Gibco | 35050-061 | |
| HEPES (1M) | Corning | 25060CI | |
| Human recombinant EGF | R&D systems | 236-EG | Stock Concentration: 500µg/mL |
| Human recombinant Wnt-3A | R&D systems | W3a-H-005 | |
| Hygromycin B | Invitrogen | 10687010 | |
| LWRN cells | ATCC | CRL-3276 | |
| Molecular grade water | Corning | 46-000-CV | |
| N2 supplement (100X) | Gibco | 17502-048 | |
| N-acetyl-L-cysteine | Sigma-Aldrich | A9165-5G | Stock Concentration: 500mM |
| Noggin | Conditioned media | - | |
| Nunc Lab-Tek Chamber slide system | Sigma-Aldrich | C7182-1PAK | |
| Pencillin-Streptomycin (10,000U/mL) | Corning | 30002CI | |
| Phospahte buffered saline, Ca2+ and Mg2+ free (PBS) | Corning | 21-040-CV | |
| Plastic 20G feeding tube | Fisher Scientific | 50-810-46 | |
| rh-laminin-521 | Gibco | A29248 | Stock concentration: 100µg/mL |
| Roboz Surgical 4-0 Silk Black Braided 100YD | Fisher Scientific | NC9452680 | |
| TOPflash HEK293 cells | ATCC | CRL-3249 | |
| Transwell Permeable supports (0.4µm) | Corning | 3470 |
References
- Quiros, M., Nusrat, A. Contribution of wound-associated cells and mediators in orchestrating gastrointestinal mucosal wound repair. Annual Reviews in Physiology. 81, 189-209 (2019).
- Blutt, S. E., et al. Use of organoids to study regenerative responses to intestinal damage. American Journal of Physiology-Gastrointestinal and Liver Physiology. 317 (6), 845-852 (2019).
- Zhang, M., Liu, Y., Chen, Y. G. Generation of 3D human gastrointestinal organoids: principle and applications. Cell Regeneration. 9 (1), 6(2020).
- Sato, T., et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 459 (7244), 262-265 (2009).
- Zachos, N. C., et al. Human enteroids/colonoids and intestinal organoids functionally recapitulate normal intestinal physiology and pathophysiology. Journal of Biological Chemistry. 291 (8), 3759-3766 (2016).
- Kozuka, K., et al. Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Reports. 9 (6), 1976-1990 (2017).
- In, J. G., Foulke-Abel, J., Clarke, E., Kovbasnjuk, O. Human colonoid monolayers to study interactions between pathogens, commensals, and host intestinal epithelium. Journal of Visualized Experiments. (146), e59357(2019).
- Cardenas, D., et al. Two- and three-dimensional bioengineered human intestinal tissue models for cryptosporidium. Methods in Molecular Biology. 2052, 373-402 (2020).
- Moon, C., VanDussen, K. L., Miyoshi, H., Stappenbeck, T. S. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunology. 7 (4), 818-828 (2014).
- VanDussen, K. L., et al. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut. 64 (6), 911-920 (2015).
- Zou, W. Y., et al. Human intestinal enteroids: New models to study gastrointestinal virus infections. Methods in Molecular Biology. 1576, 229-247 (2019).
- Heijmans, J., et al. ER stress causes rapid loss of intestinal epithelial stemness through activation of the unfolded protein response. Cell Reports. 3 (4), 1128-1139 (2013).
- Nik, A. M., Carlsson, P. Separation of intact intestinal epithelium from mesenchyme. Biotechniques. 55 (1), 42-44 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved