
Method Article
Flexible Organic Electronic Devices for Pulsed Electric Field Therapy of Glioblastoma
In This Article
Summary
This work describes the development of flexible interdigitated electrodes for implementation in 3D brain tumor models, namely, in vitro culture, in ovo model, and in vivo murine model. The proposed method can be used to evaluate the effects of pulsed electric fields on tumors at different levels of complexity.
Abstract
Glioblastoma is difficult to eradicate with standard oncology therapies due to its high degree of invasiveness. Bioelectric treatments based on pulsed electric fields (PEFs) are promising for the improvement of treatment efficiency. However, they rely on rigid electrodes that cause acute and chronic damage, especially in soft tissues such as the brain. In this work, flexible electronics were used to deliver PEFs to tumors and the biological response was evaluated with fluorescent microscopy. Interdigitated gold electrodes on a thin, transparent parylene-C substrate were coated with the conducting polymer PEDOT:PSS, resulting in a conformable and biocompatible device. The effects of PEFs on tumors and their microenvironment were examined using various biological models. First, monolayers of glioblastoma cells were cultured on top of the electrodes to investigate phenomena in vitro. As an intermediate step, an in ovo model was developed where engineered tumor spheroids were grafted in the embryonic membrane of a quail. Due to the absence of an immune system, this led to highly vascularized tumors. At this early stage of development, embryos have no immune system, and tumors are not recognized as foreign bodies. Thus, they can develop fast while developing their own vessels from the existing embryo vascular system, which represents a valuable 3D cancer model. Finally, flexible electrode delivery of PEFs was evaluated in a complete organism with a functional immune system, using a syngenic, orthograft (intracranial) mouse model. Tumor spheroids were grafted into the brain of transgenic multi-fluorescent mice prior to the implantation of flexible organic electrode devices. A sealed cranial window enabled multiphoton imaging of the tumor and its microenvironment during treatment with PEFs over a period of several weeks.
Introduction
Glioblastoma multiforme (GBM) is a highly invasive tumor and therefore difficult to eradicate with standard treatments such as resection, radiotherapy, and chemotherapy. Despite multimodal treatments, prognosis remains very poor and most of the patients experience disease progression within 1 year of diagnosis1,2. Recently, the development of bioelectric treatments has shown great potential to improve existing therapies. These therapies use the delivery of pulsed electric fields (PEF), typically in a single treatment session, to disrupt cellular membrane integrity and the microenvironment of tumors. This cell membrane disruption, also known as electroporation, can be reversible or irreversible depending on the electric field intensity and the number of pulses. Irreversible electroporation (IRE) is applied as a non-thermal tissue ablation technique in which electric pulses cause fatal damage to cellular membranes leading to cell death3. Reversible electroporation is applied in electrochemotherapy (ECT), an established technique that consists of the delivery of PEFs in combination with chemotherapy drugs to enhance drug uptake in cancer cells4. Moreover, recent studies demonstrated calcium electroporation as an alternative to ECT with high efficiency for cancer treatment, which is also inexpensive and induces fewer side effects5. Despite these promising advances, PEFs are generally applied using rigid, metallic electrodes which are known to cause damage to soft tissue6. The brain is particularly sensitive to such invasive devices where the mechanical mismatch induces inflammation and astroglial scarring7.
In this context, a flexible PEF delivery system in combination with 3D models of glioblastoma tumors is presented, from microfabrication to a murine model. Conformal electrodes are made with standard thin-film microfabrication processes, including the use of soft and biocompatible materials such as parylene-C, gold, and PEDOT:PSS8,9. An interdigitated electrode design is used to cover a large surface area while maintaining adequate transparency for imaging between the electrode fingers10. For the tumor model, 3D spheroids of glioblastoma cells expressing a genetically encoded fluorescence reporter are produced using a variation of the liquid-overlay 96-well plate method11. The spheroids are grafted into the chorioallantoic membrane of a quail embryo, resulting in an in ovo model that has been extensively used to study angiogenesis or drug toxicology12,13. Tumors can be grafted and vascularized by the embryo's vasculature in the absence of an immune system at this stage of embryonic development12. Flexible electrodes are then placed on top of the vascularized tumor to study the effect of PEF delivery on the spheroid and its vasculature. Finally, these effects are investigated on a complete living organism, including tumor microenvironment and immune system, by implanting engineered spheroids into the brain parenchyma of murine models14. Flexible electrodes are placed on top of the insertion site and the craniotomy is sealed with a glass window, allowing repeated two-photon imaging over several weeks.
These methods will be useful for people interested in various domains ranging from microelectronics engineering to oncology applications. The microfabrication protocol can be used and adapted for any application requiring thin-film metal electrodes coated with PEDOT:PSS. Further, the biological models developed for the evaluation of antitumor electrical treatments will be of general interest for the investigation of the differentiation of cellular, vascular, and immune response to implanted materials.
Protocol
All experimental procedures were performed in accordance with the French legislation and in compliance with the European Community Council Directive of November 24, 1986 (86/609/EEC) for the care and use of laboratory animals. The research on animals was authorized by the Direction Départementale des Services Vétérinaires des Bouches-du-Rhône and approved by the ethical committee of Provence Cote D'Azur (Apafis # 22689-2019100414103054).
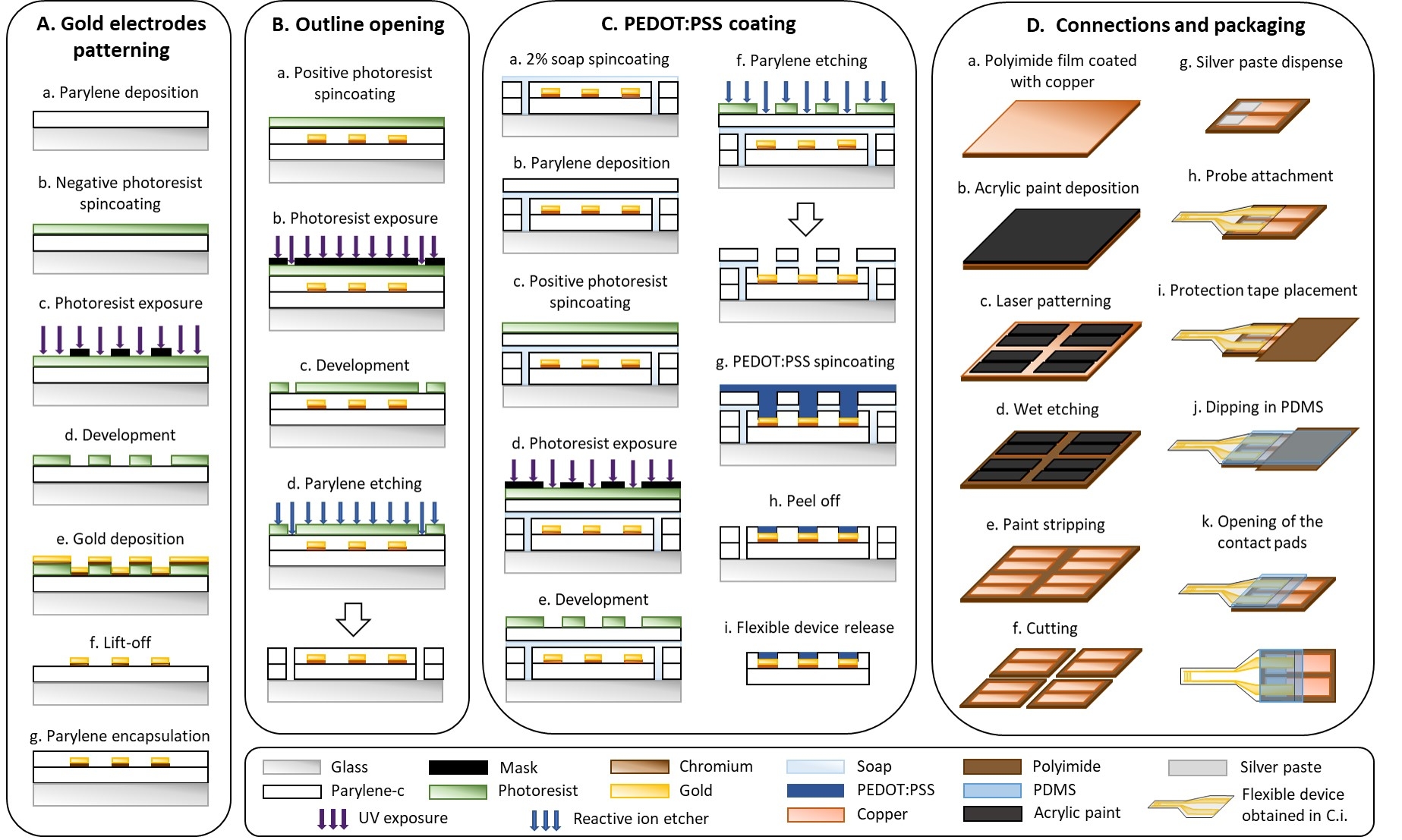
1. Flexible device microfabrication (Figure 1)
- Cleaning of the glass slides
- Sonicate glass slides in 2% soap solution for 15 min. Rinse them with water.
- Sonicate again in a mixture of 80% of pure acetone and 20% of pure isopropanol for 15 min.
CAUTION: These solvents are harmful and flammable. Wear personal protective equipment (PPE) and handle them under a fume hood. - Rinse the slides with isopropanol and dry with an air gun.
NOTE: Ensure that acetone doesn't dry on the substrates during the whole process.
- Gold electrode patterning (Figure 1A)
- Deposit a 3 µm layer of parylene-C (PaC) with a parylene deposition system (Figure 1Aa).
- Place the cleaned glass slides in the deposition chamber. Spray soap on the chiller, let it dry, and insert it into the designated cold trap of the deposition system. This anti-adhesive facilitates easy removal of PaC from the chiller after the deposition.
CAUTION: PaC is an irritant and poses a health danger. Wear gloves while handling it. - Weigh 6 g of PaC in an aluminum boat and place it in the furnace. Evacuate the machine (P = 10 mTorr) and start the deposition with the following parameters: TChiller = -100 °C, TFurnace = 690 °C, TVaporizer = 175 °C, and TChamber = 135 °C.
- When the deposition is finished and the temperature of the vaporizer is below 40 °C, turn off the chiller, vaporizer, and furnace. Vent the machine and collect the samples.
- Place the cleaned glass slides in the deposition chamber. Spray soap on the chiller, let it dry, and insert it into the designated cold trap of the deposition system. This anti-adhesive facilitates easy removal of PaC from the chiller after the deposition.
- Activate the surface of the samples by oxygen plasma treatment for 30 s (100 W, 50 sccm).
- Spin-coat the plasma-treated samples with a negative photoresist at 1,000 x g for 40 s. Place the samples on a hot plate at 110 °C for 2 min (Figure 1Ab).
CAUTION: The photoresist solution is flammable and causes irritation; wear PPE and handle it under a fume hood. - Place an i-line filter in the beamline of the UV broadband contact aligner and expose the photoresist through a mask that features the interdigitated electrode design (Figure 1Ac).
NOTE: Interdigitated electrodes with a gap of 50 or 250 µm were designed using a layout editor and photomasks were ordered from a company that produces polyester photomasks by laser photoplotting. - Bake the above samples at 110 °C on a hot plate for 3 min and let them cool down to room temperature for 5 min. Immerse samples in a metal-ion-free developer for 3 min to remove the non-exposed photoresist. Rinse samples with water and dry them with an air gun (Figure 1Ad).
CAUTION: The developer solution is an irritant; wear PPE and handle under a fume hood. - Activate the surface of the samples by oxygen plasma treatment for 60 s (100 W, 50 sccm).
- Deposit a 20 nm adhesion layer of chromium and a 300 nm layer of gold with a thermal evaporator as follows (Figure 1Ae).
- Vent the evaporator machine and clip the samples (facing down) on the upper round plate with metal screws. Fill the dedicated crucibles, respectively, with chromium and gold. Seal and evacuate the machine to reach a pressure below 5·10-6 Torr. Start the rotation of the sample holder.
- Select the crucible containing chromium and slowly increase the current going through it until a deposition rate of 0.2 Å·s-1 is reached. Open the shutter and wait until 20 nm of chromium is deposited. Close the shutter and slowly ramp down the current until 0 mA.
- Select the crucible containing gold and slowly increase the current going through it until a deposition rate of 0.2 Å·s-1 is reached. Open the shutter to evaporate gold, wait until 10 nm of gold is deposited, and then increase the deposition rate to 1.5 Å·s-1 until approximately 300 nm is deposited. Close the shutter and slowly ramp down the current to 0 mA.
- Let the samples cool down to room temperature for 15 min after deposition. Stop the rotation of the sample holder, vent the machine, and collect the samples.
- Immerse the samples in a beaker with acetone. Place the beaker on a shaking plate set at 110 rpm for 15 min to lift off the photoresist. Rinse the samples with isopropanol and dry them with an air gun (Figure 1Af).
- Activate the surface of the samples by oxygen plasma treatment for 30 s (100 W, 50 sccm).
- Deposit a 3 µm insulation layer of PaC with a parylene deposition system (see step 1.2.1) (Figure 1Ag).
- Deposit a 3 µm layer of parylene-C (PaC) with a parylene deposition system (Figure 1Aa).
- Outline opening (Figure 1B)
- Spin-coat the samples with a positive photoresist at 600 x g for 35 s. Place it on a hot plate at 110 °C for 2 min (Figure 1Ba).
CAUTION: The photoresist solution is flammable and causes irritation; wear PPE and handle under a fume hood. - Make sure there is no i-line filter in the beamline of the UV broadband contact aligner and expose the photoresist through a mask that features the outline of the device with a UV broadband contact aligner (Figure 1Bb).
- Immerse the samples in a metal-ion-free developer for 4 min to remove the exposed photoresist. Rinse the samples with water and dry them with an air gun (Figure 1Bc).
- Etch the outline through the two layers of PaC with a reactive ion etcher (160 W, 22 min, O2: 50 sccm, CF4: 10 sccm) (Figure 1Bd).
- Remove the remaining photoresist with acetone, rinse with isopropanol, and dry the samples with an air gun.
- Spin-coat the samples with a positive photoresist at 600 x g for 35 s. Place it on a hot plate at 110 °C for 2 min (Figure 1Ba).
- PEDOT:PSS coating (Figure 1C)
- Spin-coat a 2% soap solution at 70 x g for 35 s (Figure 1Ca).
- Deposit a 3 µm sacrificial layer of PaC with a parylene deposition system (see step 1.2.1) (Figure 1Cb).
- Spin-coat positive photoresist at 600 x g for 35 s. Place the samples on a hot plate at 110 °C for 2 min (Figure 1Cc).
- Make sure there is no i-line filter in the beamline of the UV broadband contact aligner and expose the photoresist through a mask that features the active surface of the electrodes (Figure 1Cd).
- Immerse the samples in a metal-ion-free developer for 4 min to remove the exposed photoresist. Rinse the samples with water and dry them with an air gun (Figure 1Ce).
- Etch the PaC with a reactive ion etcher to open the active surface of the electrodes (160 W, 24 min, O2: 50 sccm, CF4: 10 sccm). Check with a microscope that there is no residual PaC on the active surface (Figure 1Cf).
- Remove the remaining photoresist with acetone, rinse with isopropanol and dry the samples with an air gun.
- Activate the surface of the samples using oxygen plasma treatment for 90 s (100 W, 50 sccm).
- Mix a commercial dispersion of chemically polymerized PEDOT:PSS with 5 vol% of ethylene glycol (EG), and 0.1 vol% of dodecylbenzene sulfonic acid (DBSA). Sonicate for 15 min. Add 1 wt% of (3-glycydyloxypropyl) trimethylsiloxane (GOPS) and sonicate for 5 min. Filter the solution through a 1.2 µm filter.
CAUTION: EG is an irritant and poses a health danger. DBSA is an irritant and corrosive. GOPS is corrosive. Wear appropriate PPE and handle these chemicals under a fume hood.
NOTE: The total volume depends on the number of samples. For 10 standard glass slides, prepare at least 20 mL that would correspond to the following quantities: 18.78 mL of PEDOT:PSS, 1 mL of EG, 20 µL of DBSA, and 200 µL of GOPS. - Spin-coat four layers of PEDOT:PSS solution at 150 x g for 35 s. After deposition of each layer, bake the samples at 110 °C for 60 s on a hot plate and cool them down to room temperature for 5 min before spinning the next layer (Figure 1Cg).
- Remove the sacrificial PaC layer by immersing the samples in water (Figure 1Ch).
- Bake the samples at 140 °C for 1 h.
- Immerse the samples in deionized water for 30 min to remove the remaining soap and low-molecular-weight compounds in the PEDOT:PSS film and to detach samples from the glass substrate (Figure 1Ci).
- Connections and packaging (Figure 1D)
- Deposit a thin layer of acrylic paint on a polyimide film that is coated with copper (Figure 1Da). Use an aerosol to obtain a homogeneous layer of paint (Figure 1Db).
- Pattern the acrylic paint with a laser (75 kHz, 7 W, 1 laser pass, 400 mm·s-1) to obtain two rectangular contact pads (5 mm x 15 mm; 1.5 mm gap) (Figure 1Dc).
- Wet etch the copper with saturated 30% (w/v) ferric chloride (FeCl3) in water for 15 min at 40 °C (Figure 1Dd).
CAUTION: FeCl3 is an irritant and corrosive; handle it with gloves under a fume hood. - Strip the acrylic paint with acetone by slightly rubbing it with a cloth (Figure 1De).
- Cut the patterned polyimide film into rectangular shapes (15 mm x 30 mm) with a laser (15 kHz, 10 W, 30 laser passes, 130 mm·s-1) (Figure 1Df).
- Dispense silver paste with a three-axis dispensing machine at a pressure of three bar with a 330 µm diameter needle (5 m·min-1) (Figure 1Dg).
CAUTION: Silver paste is an irritant; handle with gloves. - Align and attach the PaC probe with the polyimide film under a binocular microscope using tweezers (Figure 1Dh).
NOTE: Alignment marks can be patterned in step 1.5.2 to facilitate the positioning of the probe on the contact pads. - Bake at 140 °C for 2 h in the oven.
- Place a 1 cm2 polyimide protection tape on the contact pads (Figure 1Di).
- Dip the interface where the PaC probe and polyimide film are connected in PDMS (Figure 1Dj).
- Bake it for 2 h at 50 °C.
- Remove the protection tape to open the contact pads (Figure 1Dk).
NOTE: Microfabrication of in vitro devices is similar but steps 1.2.1, 1.3, and 1.5must be skipped.
2. Generation of Glioblastoma GCaMP6f stable cell line
- Lentivirus production
- In a 75 cm² flask, culture a HEK 293T-derived cell line optimized for lentivirus production in 10 mL of Dulbecco's Modified Eagle's Medium (DMEM) containing 4.5 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate and supplemented with 10% tetracycline-free fetal bovine serum (FBS), 100 units·mL-1 of penicillin and 100 µg·mL-1 of streptomycin for least for 3 days until 80% confluence.
- Remove the medium from the flask. Gently rinse the cells with 10 mL of Phosphate-Buffered Saline (PBS).
- Add 1 mL of 0.25% Trypsin/EDTA solution and incubate the flask for 5 min at 37 °C.
CAUTION: Trypsin/EDTA solution poses a health danger; wear PPE and handle under a fume hood. - Add 8 mL of culture medium. Gently flush the cell suspension.
- Count the cells and plate 4 x 106 cells in a Petri dish in 8 mL of culture medium.
- The next day, dilute 25 µg of the plasmid containing the gene GCaMP6f and a selection marker conferring resistance to puromycin in a total volume of 600 µL of water. Add it to a tube of transfection reagent. Vortex for 10 s at 3,000 rpm and incubate the tube at room temperature for 10 min to allow the production of nanoparticles.
- Add the contents of the tube dropwise on the culture of HEK 293 T cells and rock gently by hand. Incubate cells at 37 °C for at least 4 h.
- Replace the media containing nanoparticle complexes with fresh media and return the cells at 37 °C.
- Three days later, collect the supernatant and centrifuge at 500 x g for 10 min to remove cellular debris. Collect the liquid phase containing viral particles.
NOTE: The virus production in the supernatant can be confirmed using a quantitative lentiviral titer test and can be stored at -80 °C for at least 2 years.
- Glioblastoma cells transduction
- In a 75 cm² flask, culture glioblastoma cells in 10 mL of DMEM containing 1 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate and supplemented with 10% tetracycline-free FBS, 100 units·mL-1 of penicillin, and 100 µg·mL-1 of streptomycin for at least 4 days.
- Discard the medium and add the supernatant obtained in step 2.1.9 on the target cells.
- Add 5 µg·mL-1 of Hexadimethrine Bromide in the medium to improve transduction. Incubate for 6 h at 37 °C. Replace the medium with 10 mL of fresh medium.
CAUTION: Hexadimethrine Bromide is an irritant. Handle it with gloves.
- Generation of a stable cell line
- Two to three days after transduction, add 10 mL of DMEM containing 1 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, 10% FBS, 100 units·mL-1 of penicillin, 100 µg·mL-1 of streptomycin and supplemented with puromycin to kill the non-transduced cells. Culture cells in this media for at least 3 days.
CAUTION: Puromycin is an irritant; handle it with gloves.
NOTE: Sensitivity of cells to puromycin must be tested before transduction by culturing cells in their recommended medium containing different concentrations of puromycin. One day later, check the cells with a microscope. Choose the adequate concentration at which the majority of cells are dead but few are still alive, to ensure that the antibiotic is not too toxic and might kill the transfected cells as well. - Remove the medium and rinse the cells with 10 mL of PBS.
- Add 1 mL of 0.25% of a Trypsin/EDTA solution and incubate the flask for 5 min at 37 °C.
- Add 8 mL of culture medium. Gently flush the cell suspension.
- Collect 100 µL of cell suspension and measure cell concentration with a cell counter. Aspirate 50 µL of cell suspension in a handheld automated cell counter with a 60 µm sensor.
- Seed 1 cell/well in a 96-well plate. For example, for a concentration of 1 x 103 cells per mL, add 1 / (1 x 103), i.e., 0.001 mL of cell suspension per well. Seed every well to increase the chances of success. Complete with culture medium to reach a total volume of 200 µL per well.
- One day later, find each well containing one cell and check its fluorescence (λexc = 490 nm and λem = 530 nm). Mark the wells containing only one transfected cell. Continue the growth for a few days until the well is almost confluent.
- Discard the medium and rinse the cells with 200 µL of PBS. Add 100 µL of 0.25% Trypsin/EDTA solution and incubate the 96-well plate for 5 min at 37 °C.
- Add 100 µL of medium and gently flush the cell suspension. Transfer the cell suspension in a Petri dish. Add 5 mL of medium and let the cells grow for a few days until the Petri dish is almost confluent.
- Discard the medium and rinse the cells with 5 mL of PBS. Add 1 mL of 0.25% Trypsin/EDTA solution and incubate the Petri dish for 5 min at 37 °C.
- Add 6 mL of medium and gently flush the cell suspension. Transfer the cell suspension to a T25 flask. Continue the growth for a few days until the flask is almost confluent.
- Discard the medium and rinse the cells with 5 mL of PBS. Add 1 mL of 0.25% Trypsin/EDTA solution and incubate the T25 flask for 5 min at 37 °C. Add 7 mL of DMEM containing 1 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, and supplemented with 10% FBS, 100 units·mL-1 of penicillin, and 100 µg·mL-1 of streptomycin. Gently flush the cell suspension.
- Split the cell suspension to four T25 flasks (2 mL per flask) and add 5 mL of medium to each flask. Let the cells grow for a few days until the flasks are almost confluent.
- Repeat step 2.3.12 for three flasks and keep the last flask for step 3.1.3. Transfer the cell suspension in a 15 mL conical tube and centrifuge at 150 x g for 5 min. Discard the supernatant and resuspend the cell pellet in 900 µL. Gently mix the cells to maintain a homogeneous cell suspension.
- Transfer the cell suspension into cryogenic storage vials. Add 100 µL of dimethyl sulfoxide. Place the cryovials at -80 °C overnight. Transfer frozen cells to liquid nitrogen for further experiments.
NOTE: The efficiency of the transfection can be assessed by adding 5 µM ionomycin calcium salt in the medium and checking the induced increase of fluorescence under a fluorescence microscope (λexc = 490 nm and λem = 530 nm).
- Two to three days after transduction, add 10 mL of DMEM containing 1 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, 10% FBS, 100 units·mL-1 of penicillin, 100 µg·mL-1 of streptomycin and supplemented with puromycin to kill the non-transduced cells. Culture cells in this media for at least 3 days.
3. 3D models
- Spheroid culture
- Prepare a solution of 1% (w/v) agarose in deionized (DI) water.
- Add 100 g of agarose powder in 100 mL of DI water and heat the solution in a microwave oven until all the powder is dissolved. Stir the solution regularly to avoid clumps. Autoclave the solution for 20 min at 120 °C.
- Once retrieved from the autoclave, add 75 µL of agarose solution per well in a 96-well plate carefully. Deposit it on the side of the well to form a meniscus, resulting in a non-adherent round bottom. Let it solidify for 15 min at room temperature.
- Detach the cells (step 2.3.12) from the flask obtained in step 2.3.14.
- Add 10,000 cells per well of glioblastoma cells and complete to reach a total volume of 150 µL per well with DMEM containing 1 g·L-1 of glucose, L-glutamine, sodium pyruvate, and sodium bicarbonate, and supplemented with 10% fetal bovine serum (FBS), 100 units·mL-1 of penicillin and 100 µg·mL-1 of streptomycin.
- Incubate cells at 37 °C without moving the plate for 3 days. Then, replace half of the media with fresh media every 2 days with a multi-channel pipette until further experiments. Keep the pipette tip in the upper part of the well to avoid damage to the agarose or the spheroid itself.
NOTE: The size of the spheroids depends on the number of cells seeded and the cell line, so it must be adapted depending on the experiments.
- Prepare a solution of 1% (w/v) agarose in deionized (DI) water.
- The in ovo model
- Place fertilized eggs of Japanese quail (C. japonica) in an incubator (37 °C and 57% humidity) on trays with an automatic rotator that turns eggs every 2 h. This day is considered Embryonic Day (ED) 0.
- Wash plastic weighing boats by placing them in 70% (w/v) ethanol. Take out the weighing boats and dry them under a fume hood.
NOTE: From this point, experiments are not performed in sterile conditions. However, clean conditions are required to avoid the development of mold on the embryos. - On ED3, gently open the eggs using a tweezer with thin tips pre-washed with 70% (w/v) ethanol. Pour the embryo into a plastic weighing boat, cover it with another weighing boat and place it in a standard humidified incubator at 37 °C for 3 days.
- On ED6, make a small incision in the chorioallantoic membrane (CAM) with a 23 G needle.
- Using a pipette, place a 7-day-spheroid on the incision, and return the embryo to the incubator for 3 days, until further experiments.
NOTE: A fluorescent dye can be injected into the eye of the embryo to visualize blood vessels. - On the day of the experiment, place the flexible probe on top of the vascularized tumor using a micromanipulator.
- The in vivo model
NOTE: This part of the protocol is adapted from the one previously published in reference14. Adult multicolor fluorescent AMU-Neuroinflam mice (B6.Cg-Tg(Thy1-CFP)23Jrs(Ly6a-EGFP)G5Dzk(Itgax-EYFP)1Mnz/FD) were used; these mice present labeling of a subpopulation of Thy1+ neurons by transgenic expression of ECFP, labeling of peripheral LyzM+ inflammatory cells by transgenic expression of EGFP and labeling of a subtype of microglia expressing EYFP under the control of Cd11c+. In brief, the animals are lightly sedated with 1.5% isoflurane for 2 mins before any treatment or injection. Before the surgery, the animals are anesthetized with Ketamine (120 mg/kg; IP) and Xylazine (12 mg/kg; IP). Then, 3% Lidocaine gel is applied locally to alleviate any pain in the ears associated with the fixation of stereotactic support. Then, 0.25% Bupivacaine solution is administered to the surgical site to alleviate any pain due to the craniotomy. Once the mouse was prepared for surgery, a craniotomy of 4 mm diameter was performed according to reference14. With a 26 G needle, a hole was made in the dura-mater in the middle of the craniotomy and the tumor spheroid was injected with the injection system described in reference14. Additionally, as described here, a flexible electrode was placed on the GCamp6 or DsRed expressing tumor spheroid before sealing the craniotomy with a glass window.- Place a drop of Dulbecco's Phosphate-Buffered Saline (DPBS) so that it covers the craniotomy. Place the flexible electrode onto the drop of DPBS, and gently place the back of the probe with contact pads onto the mouse's back (Figure 4B).
NOTE: Use sterile gloves and a “tip only” technique. Change the gloves if a non-sterile surface is contacted. Provide thermal support during this procedure. - Touch the DPBS drop with a small piece of paper to absorb DPBS until the probe can lay flat on the dura and follow the curvature of the brain. Ensure that a small layer of DPBS remains below the electrodes without escaping from the side of the electrode. This ensures a barrier against glue spillover during the next steps.
NOTE: Sterilize all equipment before use. - Place a small drop of silicone adhesive onto the probe and cover it with a 5 mm round cover glass. Push the cover glass down until the silicone is evenly distributed and the distance between the cover glass and the probe is minimal. Push the cover glass down for another 30 s so that silicone can solidify.
- To secure the cover glass, quickly apply superglue on its sides and push it down until the glue cures to solid.
- Using a toothpick, apply superglue at the probe's neck taking care that the superglue gets drawn under the neck to provide stable support for it.
- Cover the skull with dental cement to build a chronic cap. Take special care to cover the edges of the cover glass only.
- Lift the back of the probe and apply cement underneath the neck of the probe. Rest the probe onto the cement before it cures. Push down the neck of the probe gently with blunt forceps so that its surface is at the same level as that of the cover glass and not in the way of the microscope objective during the experiment.
- Cover the top of the probe neck with not more than 1.5 mm of dental cement layer to achieve a firm hold on the probe. Build a cement well presenting a 1.5 mm ridge at a distance of 1-2 mm around the cover glass to create a basin for the immersion fluid for the two-photon imaging (Figure 4C).
- After the cement has cured, apply buprenorphine post-surgical analgesics (0.05 mg/kg, 0.1 mL per 10 g of body weight subcutaneously) and maintain the animal in a warm atmosphere until it wakes up. This includes proximity to an infrared light bulb as well as wrapping the animal in a paper towel.
NOTE: Place a thermometer at the level of the mouse to monitor the temperature. - Characterize impedance in the 1-10 kHz range using a potentiostat.
- Let the animal recover from the surgery for at least 10 days. Administer anti-inflammatory drugs immediately after surgery and continue monitoring the animal's state to provide appropriate post-operative analgesia.
- Place a drop of Dulbecco's Phosphate-Buffered Saline (DPBS) so that it covers the craniotomy. Place the flexible electrode onto the drop of DPBS, and gently place the back of the probe with contact pads onto the mouse's back (Figure 4B).
4. Pulsed Electric Field (PEF) delivery and imaging
- Place the specimens under a fluorescence microscope. In the case of 3D models, tumors can only be observed from the top.
NOTE: For the in ovo model, experiments were performed under an epifluorescence microscope (but is also possible with a two-photon microscope), whereas experiments on the in vivo model were performed under a two-photon microscope (Figure 6). - Connect a pulse generator to the contact pads of the devices, using pogo pin connectors (in vitro) or crocodile clips (in ovo and in vivo) (Figure 4A). Set the desired parameters (number of pulses, voltage, pulse duration, frequency) and apply PEFs by running the generator (Figure 4A). Measure fluorescence simultaneously to observe the effects of the PEFs in real-time.
Results
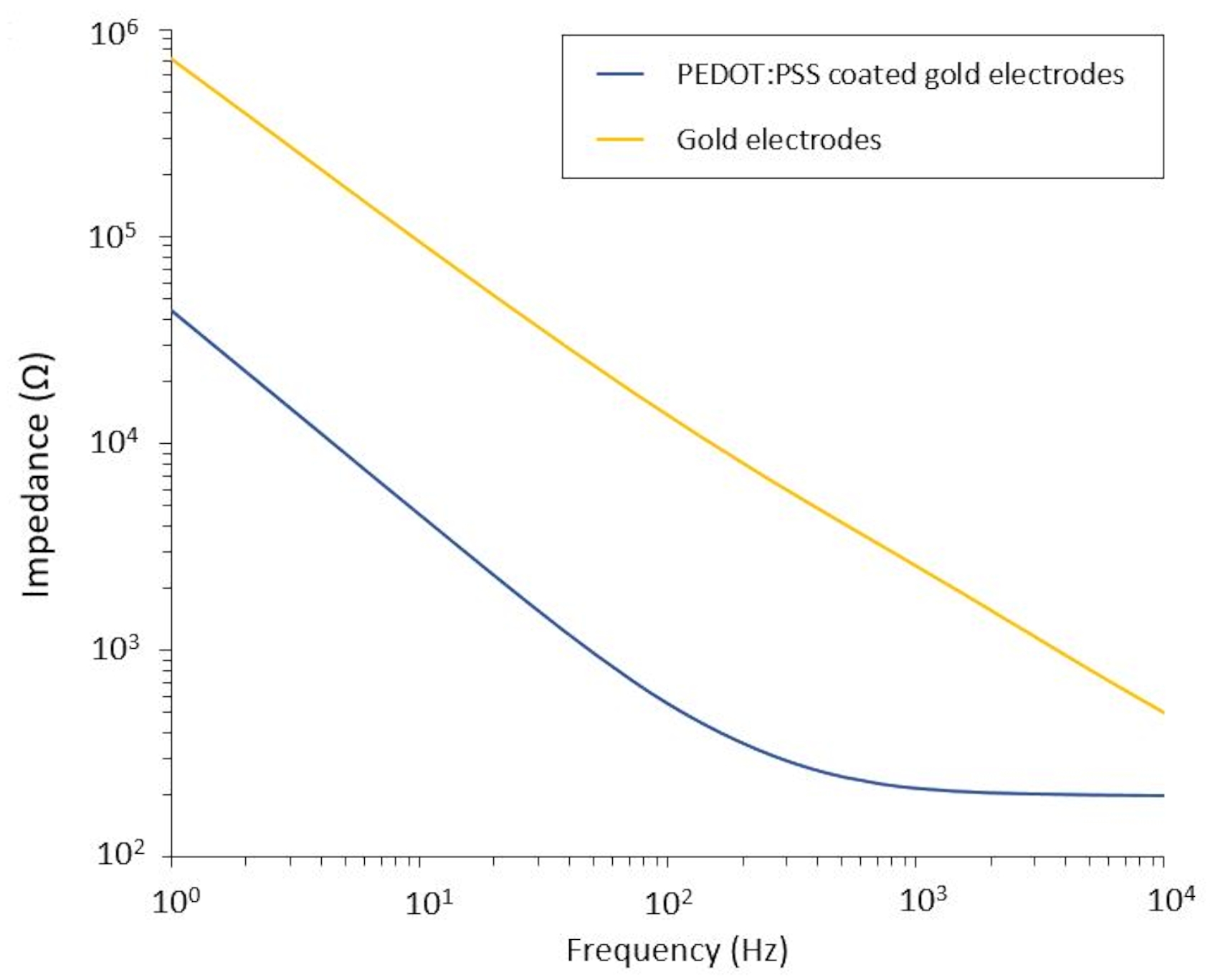
This protocol allows the application to two glioblastoma models in which a flexible PEF delivery system is integrated. Following microfabrication and packaging steps, flexible electrodes are characterized in saline solution by electrochemical impedance spectroscopy (EIS) to assess and validate their performance. The PEDOT:PSS-coated electrodes show the typical capacitive and resistive dominated regions separated by a cut-off frequency, whereas the uncoated electrodes display only capacitive behavior (Figure 2).
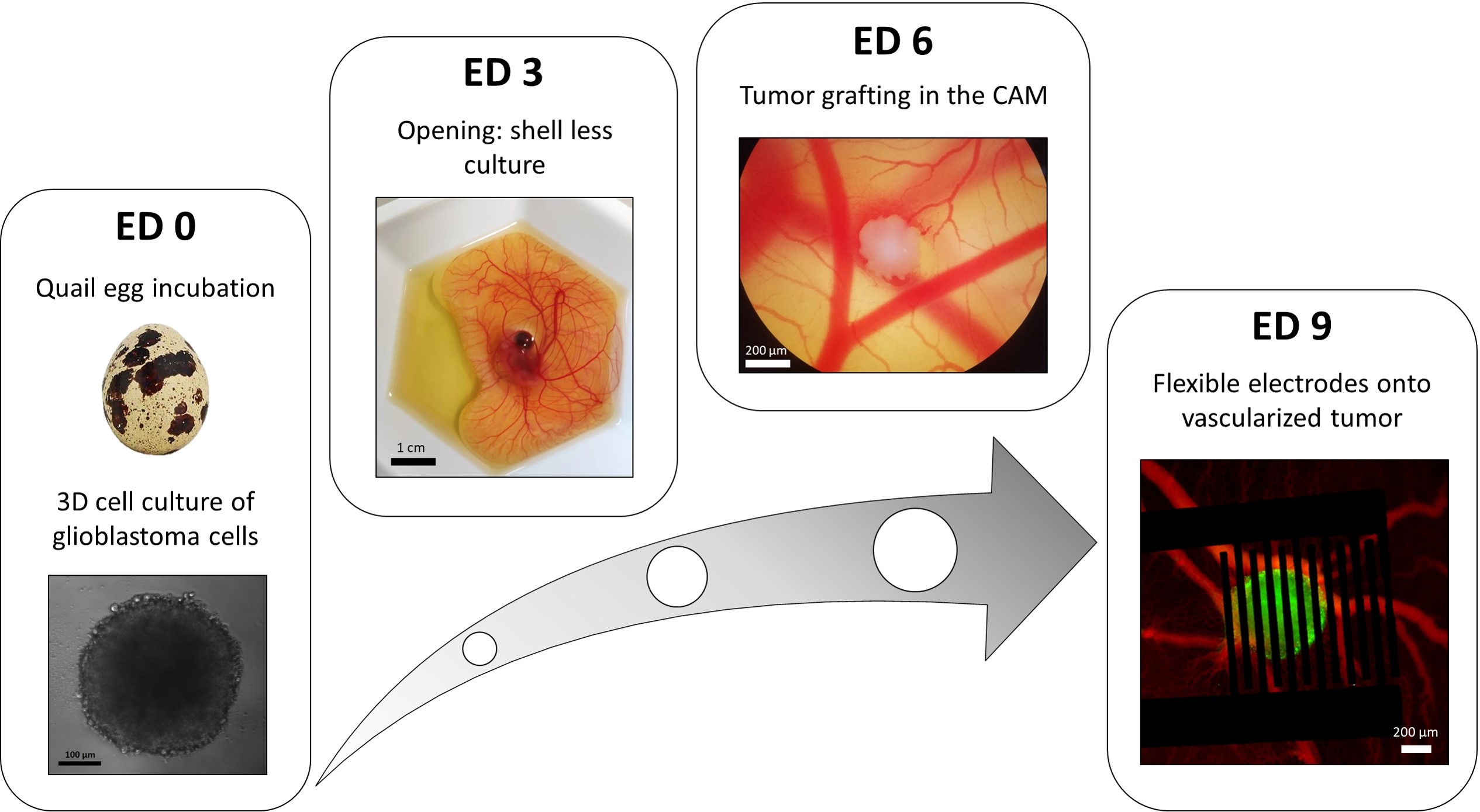
A variation of the liquid-overlay 96-well plate method is used to grow 3D tumors made of transfected glioblastoma cells stably expressing a fluorescent intracellular calcium reporter. The growth of the spheroids can be observed with a bright-field microscope (Figure 3; ED 0). At least 2 or 3 days are needed to obtain spherical and dense spheroids, depending on the cell line and the number of cells seeded.
In the in ovo model, spheroids are grafted in the chorioallantoic membrane of a quail embryo (Figure 3; ED 6). The success of the graft can be assessed by fluorescence microscopy a few days later, as living cells have intracellular calcium, and are thus fluorescent (Figure 3; ED 9). The vascularization of the tumor can be observed under a fluorescence microscope by injecting a fluorescent dye into the blood vessels (Figure 3; ED9). However, it might not always be possible to visualize the blood vessels inside the tumor as the spheroid is very dense. The flexible interdigitated electrodes are placed on top of the vascularized tumor (Figure 3; ED 9) and connected to a pulse generator. The probe must be gently placed to avoid bleeding of the embryo; otherwise, the fluorescent dye can spread, which obstructs any observation by imaging. Correct delivery of the pulse to the biological environment can be verified by measuring the current going through the circuit. Imaging of these in ovo models allows real-time monitoring of the effect of PEFs on the intracellular calcium in a 3D glioblastoma tumor, as well as the vasoconstriction induced on the tumor's vasculature, avoiding any influence of other cell types including the immune system15.
The study of the PEF effect on glioblastoma can also be performed in a more complete and predictive model. Indeed, the in vivo model described above14 consists of grafting a 3D glioblastoma tumor in the brain parenchyma of a mouse (Figure 4). The injection site of the tumor is plugged by a cross-linked dextran gel hemi-bead, to recapitulate the physiological biophysical constraints during the growth of the tumor. Although described in reference14, it is worth re-emphasizing that it is critically important that the dextran hemi-bead be precisely superglued to the dura mater; otherwise, the tumor can escape through the open dura and completely cover the brain, making the imaging impossible. For any chronic imaging, tissue ingrowth that takes place as the cranial window heals poses a serious barrier, as the new tissue is non-transparent and makes images foggy or non-usable. Therefore, after inserting and gluing the hemi-bead, the sidewalls of the opened cranial window need to be sealed with a thin layer of superglue meticulously placed all around the cavity wall, without letting the superglue slip or flow onto the dura. When the flexible probe is placed on top of the tumor injection site, no bubbles can stay under the probe, for two reasons. Firstly, imaging cannot proceed when bubbles are present. Secondly, bubbles serve as insulators, thus changing the electrical stimulation properties. After taking the precautions described above, the craniotomy is sealed with a glass window cemented to the skull to allow chronic imaging over weeks. As the tumor consists of GCaMP or DsRed expressing cells, the injection can be confirmed with a fluorescence microscope. The electrochemical impedance of the electrodes must be measured to validate the performance after implantation. Compared to the impedance in saline solution, an increase of the impedance is expected in vivo at frequencies above 100 Hz due to the presence of a biological environment (Figure 5). Vascularized neural parenchyma and tumor infiltration can be observed and characterized through the transparent substrate over weeks by two-photon microscopy (Figure 6). The use of transgenic animals expressing fluorescent proteins in cells of interest (immune cells and neurons) can, for example, allow demonstration of the minimal inflammatory process induced by electrode implantation alone (Figure 6A) or show the presence of microglia and monocytes 26 days after implantation of a PEF stimulated electrode implanted on top of a growing GBM tumor (Figure 6B1). In the latter case, both peripheral-monocyte-derived cells and brain-resident microglial cells were found around and inside the tumor (Figure 6B2). On the day of PEF delivery, contact pads of the flexible electrodes can be connected to the pulse generator, directly under the two-photon microscope. Overall, this model can be used to investigate the effect of bioelectric treatments over time using various types of cells involved in brain tumor development, up to a depth of around 500 µm.

Figure 1: Microfabrication of flexible electrodes. (A) Gold electrode patterning and Parylene C substrate. (B) Outline opening. (C) PEDOT:PSS coating. (D) Connections and packaging. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Electrochemical impedance spectroscopy of flexible gold electrodes and PEDOT:PSS coated cold electrodes in a saline solution. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: The in ovo model of glioblastoma. ED 0: Spheroid observed with a bright-field microscope. ED 3: Shell less culture of a quail embryo 3 days after opening. ED 6: Tumor implanted in the CAM observed with a bright-field microscope. ED 9: Flexible device placed on the vascularized tumor (tumor in green and blood vessels in red). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: The in vivo application. (A) Scheme for in vivo experiments. (B) Probe placement before the application of cover glass and acrylic resin. (C) Completed probe implantation. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Electrochemical impedance spectroscopy of flexible gold electrodes in a saline solution compared to an implanted probe. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Intravital multispectral two-photon imaging through electrodes. (A) Tiled image of the healthy brain surface in a control multifluorescent AMU-Neuroinflam mouse 3 days after electrode implantation. Cyan shows dendritic arborization of layer 5 pyramidal neurons, green shows recruited granulocytes and monocytes, and yellow shows activated microglia and dendritic cells. Pink shows infrared diffusion due to heat accumulation. (B1) Similar image as in A but 26 days after tumor spheroid implantation 200 µm deep inside the cortex immediately followed by electrode implantation. Note the accumulation of green and yellow immune cells. (B2) Similar image as in B1 but 100 µm below the surface of the electrodes. Note the presence of blue neuronal dendritic arborization in the periphery of the red tumor mass itself infiltrated by yellow microglia and dendritic cells. Deep blue shows a second harmonic signal from the peritumoral collagen. (B3) Zoomed view of B2 showing the presence of interneuron somas (indicated by arrows) in the vicinity of the tumor. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The approach described in this work enables brain tumor models with an integrated PEF delivery system to study the effect of PEFs at different levels of biological organization. The microfabrication protocol consists of standard thin-film processes, which provide a large degree of freedom in electrode design that can be adapted to the specific application. Sometimes, an additional thermal annealing step can be useful at the end of the fabrication, to reduce bending of the electrodes that occurred during manufacturing.
The use of a stable glioblastoma cell line expressing a fluorescent calcium indicator avoids all complications linked with dye delivery and retention, especially in 3D tumors that are very dense16. Indeed, a high expression level is observed over a long period compared to standard chemical fluorescent calcium indicators17. This protocol can be applied to various cell lines, as it is commonly used for imaging neural activity11. Here, human and murine cell lines were used (U87 and Gl261 for implantation in immunodeficient or immunocompetent mice, respectively). Indeed, recent studies showed that the U87 cell line is different from that of the original cells as many mutations were acquired over years of cell culture, affecting experimental reproducibiliy18. The method used for the preparation of 3D tumors is high-throughput, reproducible, and allows the generation of spheroids of a specific size depending on the cell line, the number of cells at seeding, and the time of growth19. However, these spheroids are dense, which presents a disadvantage when imaging at the core of the tumor.
The in ovo model is useful as a first approach to study the effect of PEF on 3D tumors and their vasculature, without interactions with other cell types that are present in the brain. This model is inexpensive, fast, high-throughput, and raises fewer ethical issues than animal models. It is important to maintain the integrity of the embryo throughout the entire experiment, as it could affect its survival and the quality of the imaging. Special care must be taken while opening the quail egg, to avoid damages to the embryonic membrane. The graft and the placement of the flexible electrodes must also be performed carefully, to avoid bleeding that could kill the embryo. Injection of fluorescent dye in the blood vessels allows simultaneous visualization of the tumor cells and vascularization with fluorescence microscopy. The intraocular injection must be performed carefully to avoid dye leaking into the embryonic liquid, which could cause a residual fluorescence in the background that degrades the quality of the imaging. This model can also be used for following drug uptake, as it allows access to the circulatory system. However, experiments are limited by the 12-day survival time of the embryo, thus allowing 7 days of observation, which is significantly shorter than the in vivo model21.
The in vivo brain tumor model can be monitored for 4 to 5 weeks before animals reach an ethical experimental endpoint determined by a sudden 20% weight loss. It is well tolerated and remains in place if the connecting tail of the electrode is not too long. Otherwise, animals tend to scratch the flipping connector, which might ultimately be torn, hence preventing subsequent connection to the stimulator. This 4-week period is nevertheless valuable to cover the different stages of glioblastoma development. When comparing the tumor cell densities in the same volume of interest at different time intervals, the evolution of the tumor growth kinetics can be observed. In particular, enhanced tumor growth was observed at the time of the immune switch22. A similar study in the presence of a stimulating electrode would inform on the effect of PEF on tumor proliferation rate and tumor sensitivity to immune elimination. In comparison to in ovo model, the in vivo model can be seen as a valuable preclinical model to study the impact of immune cells on tumor progression and their contribution to the therapeutic effect of PEF. This protocol is adapted from a previous article with the addition of a flexible electrode device on the tumor before placing a cranial window14. Both the acute and chronic bioelectric treatments of tumors can be characterized by direct and subsequent observations with two-photon microscopy given that initial stimulation is expected to induce cell death and to trigger lasting dysregulation of the immune response.
The connections of the flexible probe are easily accessible under the two-photon microscope. Electrical stimulation parameters can thus be adjusted in real-time based on the observed effect on the neural tissue and/or the targeted cells, similar to how a medical doctor would perform interventional procedures while observing MRI or CT images of his patient. A final consideration is the importance of careful sealing of the electrode on the brain with superglue and silicone glue to prevent tissue regrowth.
In conclusion, the protocol described here represents an innovative model to study the effect of PEF therapy with flexible organic polymer electrodes for glioblastoma tumor models. The two models exhibit different levels of complexity such that cellular, vascular, or immune effects can be separated for a better understanding of the mechanisms of action. Conformal, superficial electrodes reduce the iatrogenic damage while enabling disruption of the tumor microenvironment, triggering vasoconstriction or dysregulation of intracellular calcium15.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
The work reported here was supported by the French National Research Agency (ANR-18-CE19-0029). The authors warmly thank S.M. Bardet for her contribution to the generation of a stable GCaMP6f cell line and D. O'Connor for her help with the in ovo model.
Materials
| Name | Company | Catalog Number | Comments |
| (3-Glycidyloxypropyl)trimethoxysilane | Sigma | 440167 | GOPS |
| 0.25% Trypsin-EDTA (1X) | Gibco | 25200-056 | |
| 4-Dodecylbenzenesulfonic acid | Sigma | 44198 | DBSA |
| 96-well plate | Falcon | 353075 | |
| Acetone | Technic | 530 | |
| Acrylic resin | Fischer scientific | NC1455685 | |
| agarose | Sigma | A9539 | |
| autoclave | Tuttnauer | 3150 EL | |
| AZ 10XT | Microchemicals | Positive photoresist | |
| AZ 826 MIF Developer | Merck | 10056124960 | Metal-ion-free developer for the negative photoresist |
| AZ Developer | Merck | 10054224960 | Metal-ion-free developer for the positive photoresist |
| AZ nLof 2070 | Microchemicals | Negative photoresist | |
| Buprenorphine | Axience | ||
| Carprofen | Rimadyl | ||
| Centrifuge Sorvall Legend X1R | Thermo Scientific | 75004260 | |
| CMOS camera Prime 95B | Photometrics | ||
| CO2 incubator HERAcell 150i | Thermo scientific | ||
| DAC board | National Instruments | USB 6259 | |
| Déco spray Pébéo | Cultura | 3167860937307 | Black acrylic paint |
| Dextran Texas Red 70.000 | Thermofisher | D1830 | |
| Die bonding paste "Epinal" | Hitachi | EN-4900GC | Silver paste |
| Dimethyl sulfoxide | Sigma | D2438 | |
| Dispensing machine | Tianhao | TH-2004C | |
| Dulbecco’s Modified Eagle’s Medium + GlutaMAX™-I | Gibco | 10567-014 | |
| Dulbecco's Modified Eagle's Medium | Sigma | D6429 | |
| Egg incubator COUVAD'OR 160 | lafermedemanon.com | ||
| Ethylene glycol | Carl Roth | 6881.1 | |
| Fertilized eggs of Japanese quail | Japocaille | ||
| Fetal Bovine Serum | VWR | S181BH | |
| Flask | Greiner | 658170 | |
| Fluorescence macroscope | Leica MZFLIII | ||
| Gl261 | DSMZ | ACC 802 | |
| Gold pellets - Dia 3 mm x 6 mm th | Neyco | ||
| Handheld automated cell counter | Millipore | PHCC00000 | |
| Heating and drying oven | Memmert | UF110 | |
| Hexadimethrine Bromide Sequa-brene | Sigma | S2667 | |
| hot plate Delta 6 HP 350 | Süss Microtec | ||
| Illumination system pE-4000 | CoolLed | ||
| Infrared tunable femtosecond laser (Maï-Taï) | Spectra Physics (USA) | ||
| Ionomycin calcium salt | Sigma | I3909 | |
| Kapton tape SCOTCH 92 33x19 | 3M | Polyimide protection tape | |
| Lab made pulse generator | |||
| Labcoter 2 Parylene Deposition system PDS 2010 | SCS | ||
| Lenti-X 293 T cell line | Takara Bio | 63218 | HEK 293T-derived cell line optimized for lentivirus production |
| Lenti-X GoStix Plus | Takara Bio | 631280 | Quantitative lentiviral titer test |
| Mask aligner MJB4 | Süss Microtec | ||
| Micro-90 Concentrated cleaning solution | International Products | M9050-12 | |
| Microscope slides 76 x 52 x 1 mm | Marienfeld | 1100420 | |
| Needles 30G | BD Microlance 3 | 304000 | |
| PalmSens4 potentiostat | PalmSens | ||
| parylene-c : dichloro-p-cyclophane | SCS | 300073 | |
| PCB Processing Tanks | Mega Electronics | PA104 | |
| PEDOT:PSS Clevios PH 1000 | Heraeus | ||
| penicillin / streptomycin | Gibco | 15140-122 | |
| Petri dish | Falcon | 351029 | |
| pGP-CMV-GCaMP6f | Addgene | 40755 | plasmid |
| Phosphate Buffer Saline solution | Thermofisher | D8537 | |
| Plasma treatment system PE-100 | Plasma Etch | ||
| PlasmaLab 80 Reactive Ion Etcher | Oxford Instruments | ||
| Plastic mask | Selba | ||
| Plastic weigh boat 64 x 51 x 19 mm | VWR | 10770-454 | |
| Poly-dimethylsiloxane: SYLGARD 184 Silicone Elastomer Kit | Dow chemicals | 1673921 | |
| Polyimide copper film 60 µm (Kapton) | Goodfellow | IM301522 | |
| Propan-2-ol | Technic | 574 | |
| Protolaser S | LPKF | ||
| puromycin | Gibco | A11103 | |
| Round cover glass 5 mm diameter | Fischer scientific | 50-949-439 | |
| Scepter Sensors - 60 µm | Millipore | PHCC60050 | |
| Silicone adhesive Kwik-Sil | World Precision Instruments | ||
| spin coater | Süss Microtec | ||
| Spin Coater | Laurell | WS-650 | |
| Super glue | Office depot | ||
| tetracycline-free fœtal bovine Serum | Takara Bio | 631105 | |
| Thermal evaporator Auto 500 | Boc Edwards | ||
| Two-photon microscope | Zeiss LSM 7MP | ||
| U87-MG | ATCC | HTB-14 | Human glioblastoma cells |
| Ultrasonic cleaner | VWR | ||
| Vortex VTX-3000L | LMS | VTX100323410 | |
| Xfect single shots reagent | Takara Bio | 631447 | Transfection reagent |
References
- Koshy, M., et al. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. Journal of Neuro-Oncology. 107 (1), 207-212 (2012).
- Davis, M. E. Glioblastoma: Overview of disease and treatment. Clinical Journal of Oncology Nursing. 20, 2-8 (2016).
- Edd, J. F., Horowitz, L., Davalos, R. V., Mir, L. M., Rubinsky, B. In vivo results of a new focal tissue ablation technique: irreversible electroporation. IEEE transactions on Bio-Medical Engineering. 53 (7), 1409-1415 (2006).
- Breton, M., Mir, L. M. Microsecond and nanosecond electric pulses in cancer treatments. Bioelectromagnetics. 33 (2), 106-123 (2012).
- Frandsen, S. K., et al. Direct therapeutic applications of calcium electroporation to effectively induce tumor necrosis. Cancer Research. 72 (6), 1336-1341 (2012).
- Lee, J. H., Kim, H., Kim, J. H., Lee, S. -. H. Soft implantable microelectrodes for future medicine: prosthetics, neural signal recording and neuromodulation. Lab on a Chip. 16 (6), 959-976 (2016).
- Lee, H., Bellamkonda, R. V., Sun, W., Levenston, M. E. Biomechanical analysis of silicon microelectrode-induced strain in the brain. Journal of Neural Engineering. 2 (4), 81-89 (2005).
- Fattahi, P., Yang, G., Kim, G., Abidian, M. R. A review of organic and inorganic biomaterials for neural interfaces. Advanced Materials. 26 (12), 1846-1885 (2014).
- Lecomte, A., Degache, A., Descamps, E., Dahan, L., Bergaud, C. In vitro and in vivo biostability assessment of chronically-implanted Parylene C neural sensors. Sensors and Actuators B: Chemical. 251, 1001-1008 (2017).
- Dijk, G., Ruigrok, H. J., O'Connor, R. P. PEDOT:PSS-coated stimulation electrodes attenuate irreversible electrochemical events and reduce cell electropermeabilization. Advanced Materials Interfaces. 8 (19), 2100214 (2021).
- Chen, T. -. W., et al. Ultra-sensitive fluorescent proteins for imaging neuronal activity. Nature. 499 (7458), 295-300 (2013).
- Ribatti, D. Chapter 5 Chick embryo chorioallantoic membrane as a useful tool to study angiogenesis. International Review of Cell and Molecular Biology. 270, 181-224 (2008).
- Valdes, T. I., Kreutzer, D., Moussy, F. The chick chorioallantoic membrane as a novel in vivo model for the testing of biomaterials. Journal of Biomedical Materials Research. 62 (2), 273-282 (2002).
- Ricard, C., Stanchi, F., Rougon, G., Debarbieux, F. An orthotopic glioblastoma mouse model maintaining brain parenchymal physical constraints and suitable for intravital two-photon microscopy. Journal of Visualized Experiments: JoVE. (86), e51108 (2014).
- Lefevre, M. C. Integrating flexible electronics for pulsed electric field delivery in a vascularized 3D glioblastoma model. npj Flexible Electronics. 5, 19 (2021).
- Perry, J. L., Ramachandran, N. K., Utama, B., Hyser, J. M. Use of genetically-encoded calcium indicators for live cell calcium imaging and localization in virus-infected cells. Methods. 90, 28-38 (2015).
- Blömer, U., et al. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. Journal of Virology. 71 (9), 6641-6649 (1997).
- Lenting, K., Verhaak, R., ter Laan, M., Wesseling, P., Leenders, W. Glioma: experimental models and reality. Acta Neuropathologica. 133 (2), 263-282 (2017).
- Hickman, J. A., et al. Three-dimensional models of cancer for pharmacology and cancer cell biology: Capturing tumor complexity in vitro/ex vivo. Biotechnology Journal. 9 (9), 1115-1128 (2014).
- Tay, S. L. M., Heng, P. W. S., Chan, L. W. The CAM-LDPI method: a novel platform for the assessment of drug absorption. Journal of Pharmacy and Pharmacology. 64 (4), 517-529 (2012).
- Kundeková, B., Máčajová, M., Meta, M., Čavarga, I., Bilčík, B. Chorioallantoic Membrane Models of Various Avian Species: Differences and Applications. Biology. 10 (4), 301 (2021).
- Ricard, C., et al. Phenotypic dynamics of microglial and monocyte-derived cells in glioblastoma-bearing mice. Scientific Reports. 6 (1), 26381 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved