Method Article
RNA Fluorescence in situ Hybridization (FISH) to Visualize Microbial Colonization and Infection in Caenorhabditis elegans Intestines
In This Article
Summary
Intestinal microbes, including extracellular bacteria and intracellular pathogens like the Orsay virus and microsporidia (fungi), are often associated with wild Caenorhabditis nematodes. This article presents a protocol for detecting and quantifying microbes that colonize and/or infect C. elegans nematodes, and for measuring pathogen load after controlled infections in the lab.
Abstract
The intestines of wild Caenorhabditis nematodes are inhabited by a variety of microorganisms, including gut microbiome bacteria and pathogens, such as microsporidia and viruses. Because of the similarities between Caenorhabditis elegans and mammalian intestinal cells, as well as the power of the C. elegans system, this host has emerged as a model system to study host intestine-microbe interactions in vivo. While it is possible to observe some aspects of these interactions with bright-field microscopy, it is difficult to accurately classify microbes and characterize the extent of colonization or infection without more precise tools. RNA fluorescence in situ hybridization (FISH) can be used as a tool to identify and visualize microbes in nematodes from the wild or to experimentally characterize and quantify infection in nematodes infected with microbes in the lab. FISH probes, labeling the highly abundant small subunit ribosomal RNA, produce a bright signal for bacteria and microsporidian cells. Probes designed to target conserved regions of ribosomal RNA common to many species can detect a broad range of microbes, whereas targeting divergent regions of the ribosomal RNA is useful for narrower detection. Similarly, probes can be designed to label viral RNA. A protocol for RNA FISH staining with either paraformaldehyde (PFA) or acetone fixation is presented. PFA fixation is ideal for nematodes associated with bacteria, microsporidia, and viruses, whereas acetone fixation is necessary for the visualization of microsporida spores. Animals were first washed and fixed in paraformaldehyde or acetone. After fixation, FISH probes were incubated with samples to allow for the hybridization of probes to the desired target. The animals were again washed and then examined on microscope slides or using automated approaches. Overall, this FISH protocol enables detection, identification, and quantification of the microbes that inhabit the C. elegans intestine, including microbes for which there are no genetic tools available.
Introduction
Caenorhabditis elegans has emerged as a powerful model system to study innate immunity and host-microbe interactions in the intestinal epithelial cells1,2. Due to having a transparent body and only 20 intestinal cells, C. elegans represents a convenient system for monitoring the processes of microbial intestinal colonization and infection in the context of an intact organism. Nematode intestinal cells share multiple morphological and functional similarities with mammalian intestinal epithelial cells, making them a tractable in vivo model for dissection of processes that govern microbiome colonization and pathogen infection3,4,5,6.
Wild C. elegans feed on a variety of microbes that colonize and infect the intestine, and sampling of these nematodes has resulted in the discovery of viruses, eukaryotes (fungi, oomycetes), and bacteria that naturally associate with this host7,8,9,10. The Orsay virus was found infecting the intestine and is currently the only known natural virus of C. elegans9. Microsporidia are fungal-related obligate intracellular pathogens that are the most commonly found infection in wild-caught Caenorhabditis, with several species having been discovered infecting C. elegans and related nematodes8,11. Many bacteria are commonly found inhabiting the intestinal lumen of wild-caught C. elegans and several species have been established as a natural model for the C. elegans microbiome (CeMbio)6,12,13,14. Discovering and characterizing microbes that naturally colonize and/or infect C. elegans is essential to understanding the genetic mechanisms that govern these host-microbe interactions, as well as visualizing novel microbial processes that only occur in the context of an intact host animal.
After sampling, wild nematodes are screened via differential interference contrast (DIC) microscopy to look for phenotypes that are indicative of infection or colonization. For example, changes in the stereotypical granulated appearance of the intestinal cells can be associated with the presence of an intracellular parasite infection8. Specifically, the loss of the gut granules and decreased cytosolic viscosity are signs of viral infection, whereas the reorganization of gut granules into 'grooves' may indicate infection with microsporidia in the genus Nematocida8,9. Because there is a wide variety of microbes present in wild C. elegans samples, it can be difficult to distinguish among microbes through DIC microscopy. Information regarding the spatial distribution of microbes within the host may also be difficult to detect due to the small size of many microbes15. Additionally, culturing any particular microbes of interest in vitro is not always possible, leading to difficulties in detection and/or quantification.
RNA fluorescence in situ hybridization (FISH) provides a method to fluorescently label microbes by utilizing fluorescent probes that bind to the RNA of the small ribosomal subunit (SSU) in fixed cells. If analysis of morphological characteristics suggests a particular class of microbe, FISH probes that target specific or broad classes of such microbes can be used. For example, EUB338 is considered a universal probe for the bacterial SSU and is commonly used to detect a wide range of bacteria16. The protocol described here uses single-stranded DNA probes that are end-labeled with a fluorophore and specifically designed to be complementary to the target SSU of the microbe of interest, although there are previously designed probes available16. The main advantage of targeting the SSU of microbes is the relatively large abundance of this RNA, which typically comprises 80%-90% of all RNA in the cell, leading to staining with a very high signal-to-noise ratio17. Probes can also be designed to target RNA to detect viruses, like the Orsay virus9,18, which are often present in very high copies in infected cells if the virus is actively replicating.
Depending on the results with known probes, it may be necessary to obtain further sequence information to design more specific probes for species confirmation in situ. A common approach is to use universal primers against conserved regions of the SSU (16S for bacteria and 18S for eukaryotes) to amplify (via PCR) regions that are more divergent8. Using this sequence information, probes with more species-specificity can be designed. These FISH probes can then enable the identification of microbes in a culture-independent manner8. Additionally, RNA FISH can give insight into unique morphological colonization and infection characteristics, including filamentation or tissue localization patterns19,20. Different colored FISH probes can be used simultaneously, which allows for visual distinction between microbes in wild nematode samples, as well as observation of microbe-microbe dynamics inside a host15,20. Furthermore, RNA FISH staining can be applied to host-pathogen interaction studies where infection and colonization of a known species can be easily quantified manually or through automated approaches to provide insights on pathogen load, for example, in comparing C. elegans mutants that have either increased or decreased resistance to infection21.
Protocol
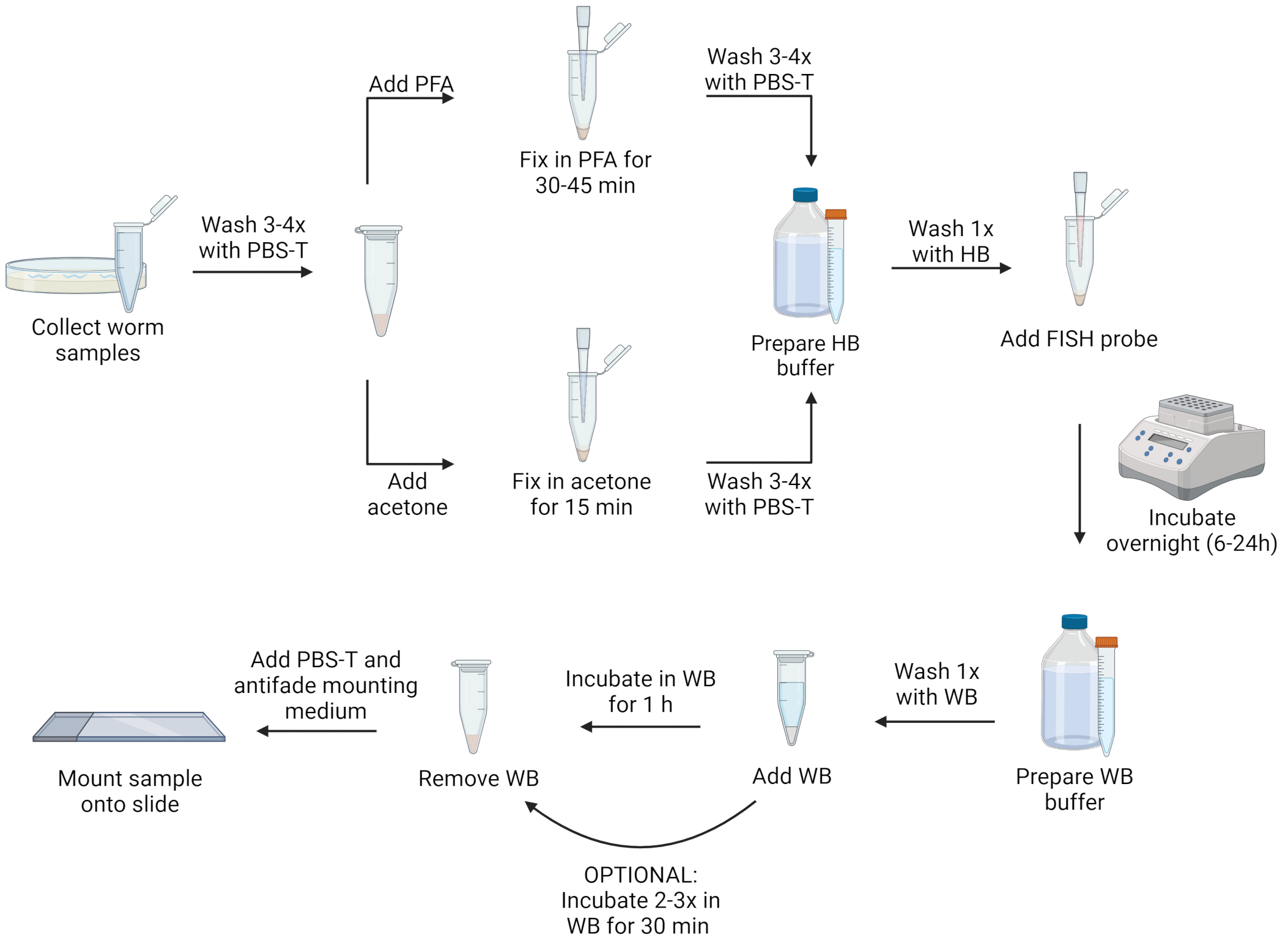
NOTE: Nematodes may be fixed with either paraformaldehyde solution (PFA) or acetone. PFA allows for better visualization of morphology than acetone and can preserve signals from transgenic green fluorescent protein (GFP), which is destroyed by acetone. However, acetone fixation is necessary to permeabilize microsporidian spores to enable labeling this life stage. Furthermore, acetone can be more convenient than PFA because it is less toxic, and samples can be stored for several days in acetone in a -20 °C freezer without the need for removing the fixative. Below are two separate protocols, using either PFA solution or acetone as a fixative. For a visualization of the protocol steps, see Figure 1.
1. FISH staining with PFA fixation
- Prepare nematodes with associated microbes
- Grow nematodes with the desired microbe of interest on standard Nematode Growth Media (NGM) plates seeded with the appropriate food source. Incubate the nematodes at 20 °C until the desired life stage is reached.
- Add 2 mL of M9 minimal salts media (42 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 19 mM NH4Cl) + 0.1% Tween 20 to NGM plates containing the Caenorhabditis strain infected or colonized with the desired microbe to be visualized.
NOTE: The addition of detergent is to prevent nematodes from sticking to pipettes and microfuge tubes, and to help pellet L1-L2 stage nematodes. 0.1% Triton-X can be used in place of Tween 20. - Pipette up the nematodes from the plates using a glass Pasteur pipette and bulb, and transfer them to marked 1.5 mL microfuge tubes.
NOTE: Glass pipettes are preferred because nematodes can stick to plastic pipettes, but the addition of detergent (Tween 20 or Triton-X) can minimize this issue. - Using a microcentrifuge, spin down the samples containing the colonized or infected nematodes at 2,000 x g for 60 s for L1 animals, or 500 x g for 60 s for L4 or adult animals. All subsequent centrifugation steps will be performed at the selected speed.
- Remove the supernatant from the microfuge tubes using a pipette. Avoid disturbing the nematode pellet by carefully removing the supernatant down to 100 µL above the pellet. This can be estimated using marked 1.5 mL microfuge tubes.
- Wash nematodes to eliminate external contamination
- Add 1 mL of 1x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) + 0.1% Tween 20 (PBS-T) to microfuge tubes.
- Spin down the samples in a microcentrifuge at the appropriate speed (see step 1.1.4). Using a pipette, remove all but 100 µL of the supernatant. This can be estimated using marked 1.5 mL microfuge tubes.
- Repeat the above two steps 2-3x.
NOTE: Three total washes are typically sufficient; however, additional washes can be performed to remove any excess external contamination.
- Fix nematodes with PFA
- In the fume hood, add 33 µL of 16% PFA to the microfuge tube containing 100 µL of supernatant above the nematode pellet obtained from step 1.2.3 for a final concentration of 4% PFA.
CAUTION: PFA is a carcinogen. Contact with PFA may result in skin sensitization and irritation and eye damage. PFA releases toxic fumes that may lead to respiratory irritation or sensitization. When using PFA, work in a fume hood with proper personal protective equipment and refer to appropriate safety data sheets before use. - Incubate the samples containing the nematodes colonized or infected with the microbe of interest for 30-45 min at room temperature. Following incubation, store the samples in 70% ethanol at 4 °C until the protocol is ready to be continued.
NOTE: A shorter incubation period is better for maintaining the GFP signal in transgenic strains due to the degradation of GFP by PFA over time. Longer incubation times allow the fixative to better permeabilize into the samples. It is best to determine the incubation time empirically depending on the sample.
- In the fume hood, add 33 µL of 16% PFA to the microfuge tube containing 100 µL of supernatant above the nematode pellet obtained from step 1.2.3 for a final concentration of 4% PFA.
- Remove the PFA solution
- Spin down the samples in a microcentrifuge at the appropriate speed (see step 1.1.4). With a pipette, remove as much of the supernatant as possible without disturbing the pellet.
CAUTION: The supernatant contains PFA, which is toxic. Discard the supernatant and at least the first two washes as toxic waste in a fume hood. - Add 0.5 mL of PBS-T to the samples in the microfuge tubes.
- Follow and repeat steps 1.4.1 and 1.4.2 2-3x with PBS-T.
NOTE: Performing more washes will help reduce the background signal. At least four washes in total are recommended. - Following the last wash, spin down the samples and remove the supernatant, leaving the pellet undisturbed.
- Spin down the samples in a microcentrifuge at the appropriate speed (see step 1.1.4). With a pipette, remove as much of the supernatant as possible without disturbing the pellet.
- Prepare hybridization buffer (HB) and wash the nematodes
- Prepare 1 mL of HB (900 mM NaCl, 20 mM Tris pH 7.5, 0.01% SDS) per sample.
NOTE: HB should be prepared fresh before each use to avoid precipitation. However, a general buffer (900 mM NaCl, 20 mM Tris pH 7.5) can be made in advance and stored at room temperature until HB is needed. Prior to use, prepare 1 mL per sample of the general buffer and add SDS to a final concentration of 0.01%. - Add 800 µL of HB to the microfuge tubes containing the nematode pellet. Pellet the samples in the microcentrifuge (see step 1.1.4). Remove the supernatant without disturbing the pellet.
- Prepare 1 mL of HB (900 mM NaCl, 20 mM Tris pH 7.5, 0.01% SDS) per sample.
- Hybridize the FISH probe to the desired target sequence
- Mix 100 µL per sample of prepared HB with the desired FISH probe to a final concentration of 5-10 ng/µL probe.
NOTE: FISH probes are 15-23 -mer oligos, antisense to the SSU of the microbe of interest, and labeled with a colored fluorophore attached to the 5' or 3' end (see Table 1 for probes used here). Generally, stock FISH probes are stored at 1 mg/mL. - Add 100 µL of HB containing FISH probe to each sample. Mix by gently flicking or inverting the tubes.
NOTE: Different colored FISH probes may be added simultaneously to visualize multiple fluorescent signals in the same sample (see Figure 1B). - Incubate the samples overnight (6-24 h) in a dry bath at 46-54 °C or a thermal mixer at 46-54 °C at 1,200 rpm.
NOTE: Incubation at 46-48 °C is typically used for hybridization. However, this temperature may need to be adjusted depending on the melting temperature of the FISH probe. In general, the hybridization temperature is 4 °C below the melting temperature.
- Mix 100 µL per sample of prepared HB with the desired FISH probe to a final concentration of 5-10 ng/µL probe.
- Remove the FISH probe and wash nematodes
- Prepare 3 mL of wash buffer (WB) (900 mM NaCl, 20 mM Tris pH 7.5, 5 mM EDTA, 0.01% SDS) per sample.
NOTE: WB should be prepared fresh before each use to avoid precipitation. However, a general buffer (900 mM NaCl, 20 mM Tris pH 7.5) can be made in advance and stored at room temperature until the wash buffer is needed. Prior to use, prepare 3 mL per sample of the general buffer and add EDTA to a final concentration of 5 mM and SDS to a final concentration of 0.01%. - Centrifuge the samples at the appropriate speed (see step 1.1.4). Remove the HB using a pipette, while being careful to leave the nematode pellet undisturbed.
- Add 1 mL of prepared WB to each sample.
- Centrifuge the samples at the appropriate speed (see step 1.1.4). Remove the WB using a pipette, while being careful to leave the pellet undisturbed.
- Add 1 mL of prepared WB to each sample.
- Incubate the samples for 1 h at 48-56 °C in a dry bath (or thermal mixer at 48-56 °C at 1200 rpm). If incubating in a dry bath, gently invert the tubes every 15-20 min.
NOTE: In general, 48 °C is used as the standard wash temperature; however, this temperature may need to be adjusted if there is high background signal. The wash temperature is often 2 °C higher than the hybridization temperature. Incubation time in WB can be shortened to 30 min to further reduce background, after which, steps 1.7.4 and 1.7.5 should be repeated. This is to be followed by one (for bacteria) or two (for microsporidia sporoplasms) 30 min incubation periods at 48 °C. - Centrifuge the samples at the appropriate speed (see step 1.1.4). Using a pipette, remove the wash buffer, while being careful to leave the pellet undisturbed.
- Add 100-500 µL of PBS-T to each of the samples. At this point, the samples can be stored in PBS-T at 4 °C for up to a week until the protocol is ready to be continued.
- Prepare 3 mL of wash buffer (WB) (900 mM NaCl, 20 mM Tris pH 7.5, 5 mM EDTA, 0.01% SDS) per sample.
- Mount the nematodes
- Centrifuge the samples at the appropriate speed (see step 1.1.4). Remove as much PBS-T as possible without disturbing the nematode pellet.
- (Optional) Add 20 µL of antifade mounting medium with DAPI (Table of Materials) to the samples.
- Load a 20 µL pipettor with a 200 µL pipette tip and use scissors to cut the tip of the pipette off to allow larger nematodes to be pipetted.
- With the cut pipette tip, transfer 5-10 µL of the pellet onto a microscope slide. Cover with a 22 x 22 coverslip. To store the slides, seal the edges of the coverslip with nail polish and keep them in a dark box at 4 °C until ready for further use.
2. FISH staining with acetone fixation
- Prepare nematodes with associated microbes as described in step 1.1.
- Wash nematodes to eliminate external contamination as described in step 1.2.
- Fix nematodes with acetone
- Remove supernatant without disturbing the pellet and add 1 mL of lab-grade acetone to the sample.
CAUTION: Acetone is a highly flammable liquid and vapor. Although it can be purchased over the counter as nail polish remover, it is important to remember that acetone causes serious eye irritation, and may cause drowsiness or dizziness. - Incubate the samples containing nematodes colonized or infected with the microbe of interest for 15 min at room temperature. Following incubation, the samples can be stored in acetone at -20 °C for up to 2 weeks until the protocol is ready to be continued.
NOTE: Do not use this protocol with transgenic C. elegans strains expressing GFP (or its allelic mutated forms) if it is desired to maintain the fluorescence, as acetone destroys this signal.
- Remove supernatant without disturbing the pellet and add 1 mL of lab-grade acetone to the sample.
- Remove the acetone
- Spin down the samples in a microcentrifuge at the appropriate speed (see step 1.1.4). With a pipette, remove the supernatant without disturbing the pellet.
CAUTION: The supernatant contains acetone. Discard the supernatant and at least the first two washes as toxic waste in a fume hood. - Add 0.5 mL of PBS-T to the samples in the microfuge tubes.
- Follow and repeat steps 2.4.1 and 2.4.2 2-4x with PBS-T.
NOTE: Performing more washes will help reduce the background signal. It is recommended to perform four washes in total. - After the last wash, spin down the samples and remove the supernatant, ensuring that the pellet is undisturbed.
- Spin down the samples in a microcentrifuge at the appropriate speed (see step 1.1.4). With a pipette, remove the supernatant without disturbing the pellet.
- Prepare hybridization buffer (HB) and wash the nematodes as described in step 1.5.
- Hybridize the FISH probe to the desired target sequence as described in step 1.6.
- Remove FISH probe and wash nematodes
- Prepare 1.1 mL of wash buffer (WB) (900 mM NaCl, 20 mM Tris pH 7.5, 5 mM EDTA, 0.01% SDS) per sample.
NOTE: WB should be prepared fresh before each use to avoid precipitation. However, a general buffer (900 mM NaCl, 20 mM Tris pH 7.5) can be made in advance and stored at room temperature until a wash buffer is needed. Prior to use, prepare 1.1 mL per sample of the general buffer and add EDTA to a final concentration of 5 mM and SDS to a final concentration of 0.01%. - Centrifuge the samples at the appropriate speed (see 1.1.4). Remove the HB using a pipette, while being careful to leave the nematode pellet undisturbed.
- Add 100 µL of prepared WB to each sample.
- Centrifuge the samples at the appropriate speed (see 1.1.4). Remove the WB using a pipette, while being careful to leave the pellet undisturbed.
- Add 1 mL of prepared WB to each sample.
- Incubate the samples for 1 h at 48-56 °C in a dry bath (or thermal mixer at 48-56 °C at 1,200 rpm). If incubating in a dry bath, gently invert the tubes every 15-20 min.
NOTE: In general, 48 °C is used as the standard wash temperature; however, this temperature may need to be adjusted if there is high background signal. The wash temperature is often 2 °C higher than the hybridization temperature. - Centrifuge the samples at the appropriate speed (see step 1.1.4). Using a pipette, remove the wash buffer, while being careful to leave the pellet undisturbed.
- Add 100-500 µL of PBS-T to each of the samples. At this point, the samples can be stored in PBS-T at 4 °C for up to a week until the protocol is ready to be continued.
- Prepare 1.1 mL of wash buffer (WB) (900 mM NaCl, 20 mM Tris pH 7.5, 5 mM EDTA, 0.01% SDS) per sample.
- Mount the nematodes as described in step 1.8.
Results
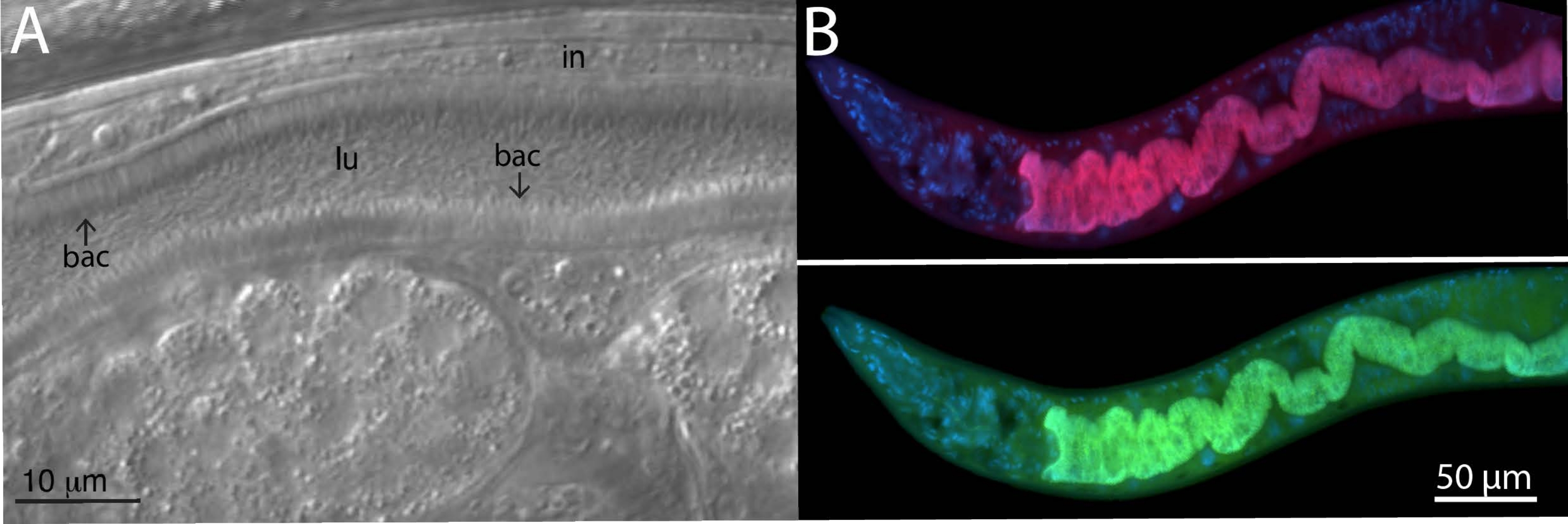
For analyzing microbiome bacteria, specific and universal FISH probes to bacterial 16S were utilized on wild-isolated animals. Wild Caenorhabditis tropicalis strain (JU1848) was sampled from the Nouragues forest near a small river in the French Guiana from rotting palm tree fruits22. Under the differential interference contrast (DIC) microscope, this nematode strain was found to be colonized with a bacterium that appears to directionally adhere to the intestinal epithelium (Figure 2A). JU1848 was then selectively cleaned to eliminate other microbial contaminants and enrich for the desired adhering bacterium23. Using the universal PCR method, the bacterium was identified as a new species in the Alphaproteobacteria class. A FISH probe labeled with Cal Fluor Red 610 was then designed specifically to the 16S rRNA sequence of this bacterium to allow fluorescent visualization of colonization within C. tropicalis (Figure 2B). A universal 16S rRNA FISH probe capable of binding many species of bacteria (EUB338) was labeled with 6-carboxyfluorescin (FAM) and was also added to this sample. The green and red fluorescent signals overlap completely, suggesting that most bacteria colonizing the intestines are the adhering Alphaproteobacteria bacterium. These animals were fixed in PFA before staining.
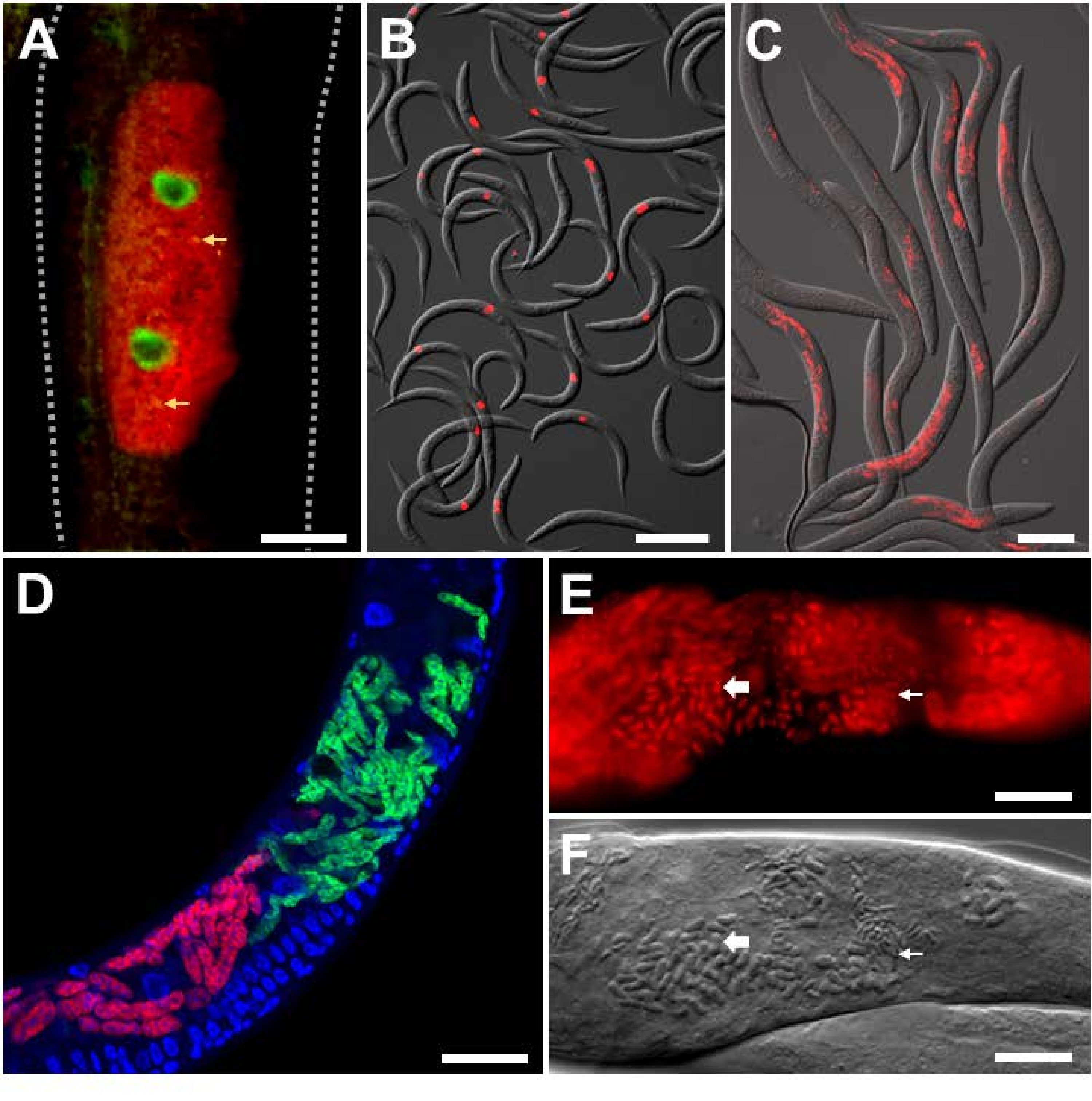
For analyzing experimental infection in the lab with intracellular pathogens of known identity, Orsay virus and microsporidian-specific FISH probes were utilized on C. elegans with a wild-type background. The Orsay virus is a positive-strand RNA virus from the Nodaviridae family, and the only natural viral pathogen found in C. elegans. The bipartite RNA genome of the Orsay virus consists of RNA1 and RNA2 segments, and FISH probes targeting both of these segments have been developed (Figure 3A,B)9,18. In the intestine, viral RNA is sensed by RIG-I homolog DRH-124, which is required for activation of the transcriptional defense program named the Intracellular Pathogen Response (IPR)25,26,27. The transcription of antiviral IPR genes is at least partially controlled by the ZIP-1 transcription factor21. Here, the expression of ZIP-1::GFP is seen localized in the intestinal nuclei of cells that show positive Orsay virus FISH staining in the cytoplasm (Figure 3A)21. Multiple animals stained with Orsay-specific FISH are shown to indicate the strength of this signal for easy quantification (Figure 3B). Animals shown in Figure 3A,B were fixed in PFA.
The microsporidian parasite named Nematocida parisii, meaning nematode-killer from Paris, is an obligate intracellular pathogen of the intestine. Several FISH probes that label 18S rRNA of N. parisii have been used, including fluorescently tagged MicroA and MicroB probes. Multiple animals stained with MicroB FISH are shown to indicate the strength of this signal for easy quantification (Figure 3C). Additionally, C. elegans is infected by other closely related microsporidia. Co-infection of N2 with N. parisii and the related N. ausubeli can be distinguished using this FISH protocol by designing species-specific FISH probes that compete against each other for binding to a divergent region on the 18S rRNA (Figure 3D)28. In this example, the N. parisii FISH probe has perfect base-pairing to the N. parisii 18S rRNA, but a 7 bp mismatch to N. ausubeli 18S rRNA. The converse is true for the N. ausubeli probe. As such, each species-specific FISH probe will outcompete for binding to the cognate species 18S over the non-cognate species. Additionally, the use of DAPI to stain nuclei allows for better localization of the infection in the context of the whole animal, especially for the intestine which has large, easily identifiable nuclei. Figure 3C,D contains animals that were fixed in PFA. Later infections with N. parisii result in the development of meronts into spores. To visualize N. parisii spores, the animals must be fixed in acetone as it penetrates the spore wall better than PFA (Figure 3E,F)8. The resulting FISH staining demonstrates the small and large rod-shaped structures, that likely correspond with N. parisii spores, which are stained with N. parisii-specific probes in red.

Figure 1: Visual representation of FISH protocol. Created with Biorender.com. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: FISH staining of wild C. tropicalis JU1848 strain colonized with adhering bacteria in the intestines. (A) Nomarski image depicting thousands of thin bacilli bacteria (bac) directionally binding to the intestine (in) of JU1848, creating a hair-like phenotype within the lumen (lu). This figure panel is adapted from Morgan, E. et al. (2021)23. (B) FISH staining of JU1848, fixed in PFA, using a red labeled probe (b002_16S_A-CF610) designed to target the 16S rRNA sequence of the adhering bacterium (top) and a green-labeled universal FISH probe (EUB338-FAM) designed to target the 16S of bacteria (bottom). DAPI staining of host nuclei is shown in blue. See Table 1 for probe sequences. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: FISH staining of C. elegans infected with intracellular pathogens. (A,B) FISH staining of C. elegans expressing ZIP-1::GFP and infected with the Orsay virus, which were fixed with PFA before staining to preserve the GFP signal. Orsay 1 Red and Orsay 2 Red probes were used for pathogen staining. (A) The composite image consists of merged red and green fluorescent channels. Nuclear ZIP-1::GFP expression is induced upon Orsay virus infection and is shown in green. Autofluorescence from the gut granules is shown in yellow and indicated with yellow arrows. Dotted lines outline the nematode body. Scale bar = 25 µm. (B) The composite image consists of merged red fluorescent and DIC channels. Scale bar = 200 µm. (C,D) FISH staining of wild-type C. elegans infected with microsporidia that were fixed in PFA. (C) FISH staining of wild-type C. elegans infected with N. parisii and fixed in PFA. MicroB-CF610 probe was used for pathogen staining. The composite image consists of merged red fluorescent and DIC channels. Scale bar = 100 µm. (D) FISH staining of wild-type C. elegans co-infected with N. parisii and N. ausubeli in the intestine. The two pathogens were co-stained using a pair of specific FISH probes that compete for binding to the same region of the 18S rRNA. N. parisii was stained using MicroF-CF610 (red) and N. ausubeli was stained using MicroSp1A-FAM (green). DAPI staining of host nuclei is seen in blue. Scale bar = 25 µm. (E) FISH staining with acetone-fixed wild-type C. elegans infected with N. parisii spores. MicroA-CF610 (red) was used for staining (red). Scale bar = 15 µm. (F) Nomarski image depicting N. parisii spores seen in (E). Scale bar = 15 µm. In (E) and (F), the small and large rod-shaped structures are labeled with small and large arrows, respectively, that correspond to N. parisii spores. See Table 1 for probe sequences. The image shown in (A) is adapted from Lažetić, V. et al. (2022)21. Images shown in (B) and (C) are adapted from Reddy, K. C. et al. (2019)26. Images shown in (E) and (F) are adapted from Troemel, E. R. et al. (2008)8. Please click here to view a larger version of this figure.
{kind=link}
| Probe name | Probe specificity | Probe fluorophore | Probe sequence |
| EUB338-FAM | Bacterial 16S (universal) | 5' 6-fluorescein (FAM) | GCTGCCTCCCGTAGGAGT |
| b002_16S_A-CF610 | Alphaproteobacteria 16S | Cal Fluor Red 610 (CF610) | TGTACCGACCCTTAACGTTC |
| Orsay1 Red | Orsay virus RNA1 | Cal Fluor Red 610 (CF610) | GACATATGTGATGCCGAGAC |
| Orsay2 Red | Orsay virus RNA2 | Cal Fluor Red 610 (CF610) | GTAGTGTCATTGTAGGCAGC |
| MicroA-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | CTCTGTCCATCCTCGGCAA |
| MicroB-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | CTCTCGGCACTCCTTCCTG |
| MicroF-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | AGACAAATCAGTCCACGAATT |
| MicroSp1A-FAM | Nematocida ausubeli 18S | 5' 6-fluorescein (FAM) | CAGGTCACCCCACGTGCT |
Table 1: List of FISH probe sequences. All FISH probes were commercially purchased with the fluorophore attached to the 5' end (via custom oligonucleotide synthesis; see Table of Materials) and the oligonucleotides were purified by reverse-phase HPLC.
Discussion
Wild C. elegans are naturally associated with a variety of microbes. Researchers can use RNA FISH to detect and identify these microbes as well as gain insight into their localization in the context of a whole animal. Microbes with desirable or interesting phenotypes can be identified through this method and then isolated for further characterization and sequencing. The abundance of numerous bacterial isolates from wild C. elegans may also be quantified via RNA FISH29. By using the protocol described here, it is also possible to observe known microorganisms inside their hosts and learn more about their interactions. Importantly, Orsay virus and microsporidia are obligate parasites and cannot be cultured independently of the host, so FISH is the standard visualization tool. Colonization or infection can also be quantified through RNA FISH using nematodes grown on plates seeded with a desired culturable bacteria of interest. In addition to staining microorganisms in C. elegans intestine, this protocol can be used for other nematode strains like C. tropicalis or Oscheius tipulae19,23.
The main advantage of the FISH protocol is that it offers a simple, quick, and robust method to stain microbes associated with C. elegans. Images produced from FISH staining have a high signal-to-noise ratio, which is achieved by utilizing FISH probes that target the abundant RNA of the SSU within the sample. Because there are typically 30x or higher levels of rRNA than rDNA, most of the signal from FISH staining with probes that target rRNA is due to rRNA rather than rDNA30. Furthermore, RNA FISH makes it possible to see infection or colonization within the context of the whole animal. This visualization is facilitated through co-staining host nuclei with DAPI and/or using fluorescent-marked strains of C. elegans to better highlight the localization of infection or colonization within the sample. For example, microsporidian-specific FISH was used to determine the tissue tropism of Nematocida displodere by using a panel of C. elegans strains with GFP expression in different tissues20. Additionally, this protocol is amenable to changes that allow for researchers to determine the ideal conditions suitable for their specific needs (e.g., adjusting the fixation period, increasing the hybridization temperature).
One critical step in the FISH protocol is fixing the samples. The incubation period following the addition of the fixative is necessary to allow time for the agent to permeabilize the sample. Longer incubation times are not ideal for samples containing transgenic fluorescent proteins due to protein degradation by PFA over time. For samples containing GFP, it is imperative to determine the optimal fixation time to allow for permeabilization, while still maintaining the GFP signal.
FISH can be used to stain for bacteria, viruses, or microsporidia in C. elegans. However, the best type of fixative agent used for FISH depends on the sample and downstream requirements. This protocol presents a PFA solution as the primary fixative agent to stain bacteria and viruses. However, PFA is not sufficient for the visualization of microsporidian spores as it cannot penetrate the spore wall. For visualization of spores, acetone should be used instead. Although, PFA fixation is efficient for FISH labeling of other life stages of microsporidia, including sporoplasms, meronts, and sporonts. Other major differences are seen between acetone fixation and PFA fixation; acetone is more convenient because samples can be quickly stored in the freezer after adding, without the need for washing. However, acetone quickly kills any existing GFP in a transgenic host. PFA is the preferred fixative if it is important to preserve some physiological structures in the host, as acetone-fixed animals appear to be more degraded, making identification of some tissues more difficult. Because the samples are fixed, this FISH protocol does not allow for live imaging of host-microbe interactions in vivo. However, a pulse-chase infection time course followed by FISH staining of samples at various time points can allow one to see some dynamics of microbial infection19,20,31.
Another critical step throughout the protocol is the thorough washing of the samples before and after hybridization. Before hybridization, when collecting the worms into the microfuge tubes, excess bacteria, or other microbes from the NGM plates can be carried with the worm sample. Three washes with PBS-T are standard; however, more washes may be necessary to sufficiently eliminate external microorganisms, especially when using heavily contaminated, wild-isolated C. elegans. When viewing the mounted samples after FISH, there may be some residual FISH probe that produces large amounts of signal in the background of the sample. The wash temperature and the number of washes are important to remove the excess and non-specifically bound probe. To reduce the background fluorescence, it is possible to perform two or three washes with 1 mL of WB every 30 min, instead of one wash with 1 mL of WB for an hour. Different FISH probes may require different wash temperatures. Typically, the wash temperature is 2 °C above the hybridization temperature, but this can be increased if there is too much background fluorescence (high noise).
The FISH protocol utilizes fluorescent probes designed to target species-specific microbial RNA, but FISH probes can be designed for other high-copy transcripts. Other FISH probes may have different melting temperatures, so incubation steps may need to be performed at a higher or lower temperature than described. FISH staining can identify the spatial distribution of microbial colonization or infection within the host, allowing for the characterization of host-microbe and microbe-microbe interactions. One limitation is that only a few conventional fluorophores can be used simultaneously, which reduces the number of different microorganisms that can be detected via FISH at the same time. This limits its use for complex microbiome studies in C. elegans. However, multicolor rRNA-targeted FISH utilizes probes labeled with non-canonical fluorophores that can increase the number of distinct microbial group labels15. Another limitation is that it is difficult to distinguish between closely related species, especially bacteria, that have SSU sequences that are highly similar. However, the extreme sequence divergence between microsporidia species helps to facilitate their differentiation with this protocol (Figure 3)32,33.
Overall, this FISH protocol describes a technique to detect microorganisms within C. elegans. It allows researchers to use a transparent and genetically tractable model system to detect and quantify colonization and infection within the context of an intact animal, as well as identify unique microbial behavior or morphology within the host. A preprint version of this manuscript was posted during review34.
Disclosures
The authors have no conflicts of interest.
Acknowledgements
Thank you to Dr. Marie-Anne Félix for providing us with wild nematode strains. This work was supported by NSF under CAREER Grant 2143718 and California State University under a CSUPERB New Investigator Award to RJL, NIH under R01 AG052622 and R01 GM114139 to ERT, and by an American Heart Association Fellowship to VL.
Materials
| Name | Company | Catalog Number | Comments |
| 10% SDS | Invitrogen | AM9822 | |
| Acetone | Fisher Scientific | A-11-1 | |
| Antifade mounting serum with DAPI (Vectashield) | Vectalab | NC9524612 | |
| EDTA | Fisher Scientific | S311-500 | |
| FISH probes (see Table 1) | LGC Biosearch Technologies | FISH probes were commercially purchased via custom oligonucleotide synthesis | |
| KCl | Fisher Scientific | P217 | |
| KH2PO4 | Fisher Scientific | P-286 | |
| Na2HPO4 | Fisher Scientific | S375-500 | |
| NaCl | Fisher Scientific | S-671 | |
| NH4Cl | Fisher Scientific | A-661 | |
| Paraformaldehyde | Electron Microscopy Science | 50-980-487 | CAUTION: PFA is a carcinogen. Handle appropriately |
| Thermal mixer | Eppendorf | 5384000020 | |
| Tris base | Fisher Scientific | BP152 | |
| Triton X-100 | Fisher Scientific | BP-151 | |
| Tween-20 | Fisher Scientific | BP337-500 |
References
- Pukkila-Worley, R., Ausubel, F. M. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Current Opinion in Immunology. 24 (1), 3-9 (2012).
- Balla, K. M., Troemel, E. R. Caenorhabditis elegans as a model for intracellular pathogen infection. Cellular Microbiology. 15 (8), 1313-1322 (2013).
- Dimov, I., Maduro, M. F. The C. elegans intestine: organogenesis, digestion, and physiology. Cell and Tissue Research. 377 (3), 383-396 (2019).
- Bossinger, O., Fukushige, T., Claeys, M., Borgonie, G., McGhee, J. D. The apical disposition of the Caenorhabditis elegans intestinal terminal web is maintained by LET-413. Developmental Biology. 268 (2), 448-456 (2004).
- Szumowski, S. C., Botts, M. R., Popovich, J. J., Smelkinson, M. G., Troemel, E. R. The small GTPase RAB-11 directs polarized exocytosis of the intracellular pathogen N. parisii for fecal-oral transmission from C. elegans. Proceedings of the National Academy of Sciences. 111 (22), 8215-8220 (2014).
- Samuel, B. S., Rowedder, H., Braendle, C., Félix, M. A., Ruvkun, G. Caenorhabditis elegans responses to bacteria from its natural habitats. Proceedings of the National Academy of Sciences. 113 (27), 3941-3949 (2016).
- Zhang, F., et al. Caenorhabditis elegans as a model for microbiome research. Frontiers in Microbiology. 8, 485 (2017).
- Troemel, E. R., Félix, M. -. A., Whiteman, N. K., Barrière, A., Ausubel, F. M. Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biology. 6 (12), 2736-2752 (2008).
- Felix, M. A., et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biology. 9 (1), 1000586 (2011).
- Osman, G. A., et al. Natural infection of C. elegans by an oomycete reveals a new pathogen-specific immune response. Current Biology. 28 (4), 640-648 (2018).
- Zhang, G. A large collection of novel nematode-infecting microsporidia and their diverse interactions with Caenorhabditis elegans and other related nematodes. PLOS Pathogens. 12 (12), 1006093 (2016).
- Clark, L. C., Hodgkin, J. Commensals, probiotics and pathogens in the Caenorhabditis elegans model. Cell Microbiology. 16 (1), 27-38 (2014).
- Dirksen, P., et al. CeMbio - The Caenorhabditis elegans microbiome resource. G3. Genes, Genomes, Genetics. 10 (9), 3025-3039 (2020).
- Berg, M., et al. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. The ISME Journal. 10 (8), 1998-2009 (2016).
- Michael, L., Markus, S., Petra, P., Holger, D. A multicolor fluorescence in situ hybridization approach using an extended set of fluorophores to visualize microorganisms. Frontiers in Microbiology. 10, 1383 (2019).
- Fuchs, B. M., et al. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Applied and Environmental Microbiology. 64 (12), 4973-4982 (1998).
- O'Neil, D., Glowatz, H., Schlumpberger, M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Current Protocols in Molecular Biology. 103 (1), 4-19 (2013).
- Franz, C. J., et al. Santeuil and Le Blanc viruses primarily infect intestinal cells in Caenorhabditis nematodes. Virology. 448, 255-264 (2014).
- Tran, T. D., Ali, M. A., Lee, D., Luallen, R. J. Bacterial filamentation as a mechanism for cell-to-cell spread within an animal host. Nature Communications. 13 (1), 1-11 (2022).
- Luallen, R. J., et al. Discovery of a natural microsporidian pathogen with broad tissue tropism in Caenorhabditis elegans. PLOS Pathogens. 12 (6), 1005724 (2016).
- Lažetić, V., et al. et al The transcription factor ZIP-1 promotes resistance to intracellular infection in Caenorhabditis elegans. Nature Communications. 13 (1), 1-16 (2022).
- Félix, M. -. A., et al. Species richness, distribution and genetic diversity of Caenorhabditis nematodes in a remote tropical rainforest. BMC Evolutionary Biology. 13 (1), 10 (2013).
- Morgan, E., Longares, J. F., Félix, M. A., Luallen, R. J. Selective cleaning of wild Caenorhabditis nematodes to enrich for intestinal microbiome bacteria. Journal of Visualized Experiments. (174), e62937 (2021).
- Sowa, J. N., et al. The Caenorhabditis elegans RIGI homolog DRH-1 mediates the intracellular pathogen response upon viral infection. Journal of Virology. 94 (2), 01173 (2020).
- Bakowski, M. A., et al. Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLOS Pathogens. 10 (6), 1004200 (2014).
- Reddy, K. C., et al. Antagonistic paralogs control a switch between growth and pathogen resistance in C. elegans. PLoS Pathogens. 15 (1), 1007528 (2019).
- Reddy, K. C., et al. An intracellular pathogen response pathway promotes proteostasis in C. elegans. Current Biology. 27 (22), 3544-3553 (2017).
- Balla, K. M., Lažetić, V., Troemel, E. R. Natural variation in the roles of C. elegans autophagy components during microsporidia infection. PloS One. 14 (4), 0216011 (2019).
- Dirksen, P., et al. The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biology. 14 (1), 38 (2016).
- Fu, R., Gong, J. Single cell analysis linking ribosomal (r)DNA and rRNA copy numbers to cell size and growth rate provides insights into molecular protistan ecology. The Journal of Eukaryotic Microbiology. 64 (6), 885-896 (2017).
- Willis, A. R., et al. A parental transcriptional response to microsporidia infection induces inherited immunity in offspring. Science Advances. 7 (19), (2021).
- Cuomo, C. A., et al. Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Research. 22 (12), 2478-2488 (2012).
- Reinke, A. W., Balla, K. M., Bennett, E. J., Troemel, E. R. Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nature Communications. 8 (1), 14023 (2017).
- Rivera, D. E., Lažetić, V., Troemel, E. R., Luallen, R. J. RNA fluorescence in situ hybridization (FISH) to visualize microbial colonization and infection in the Caenorhabditis elegans intestines. bioRxiv. , (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved