
Method Article
Daily Transfers, Archiving Populations, and Measuring Fitness in the Long-Term Evolution Experiment with Escherichia coli
In This Article
Summary
This protocol describes how to maintain the Escherichia coli Long-Term Evolution Experiment (LTEE) by performing its daily transfers and periodic freeze-downs and how to conduct competition assays to measure fitness improvements in evolved bacteria. These procedures can serve as a template for researchers starting their own microbial evolution experiments.
Abstract
The Long-Term Evolution Experiment (LTEE) has followed twelve populations of Escherichia coli as they have adapted to a simple laboratory environment for more than 35 years and 77,000 bacterial generations. The setup and procedures used in the LTEE epitomize reliable and reproducible methods for studying microbial evolution. In this protocol, we first describe how the LTEE populations are transferred to fresh medium and cultured each day. Then, we describe how the LTEE populations are regularly checked for possible signs of contamination and archived to provide a permanent frozen "fossil record" for later study. Multiple safeguards included in these procedures are designed to prevent contamination, detect various problems when they occur, and recover from disruptions without appreciably setting back the progress of the experiment. One way that the overall tempo and character of evolutionary changes are monitored in the LTEE is by measuring the competitive fitness of populations and strains from the experiment. We describe how co-culture competition assays are conducted and provide both a spreadsheet and an R package (fitnessR) for calculating relative fitness from the results. Over the course of the LTEE, the behaviors of some populations have changed in interesting ways, and new technologies like whole-genome sequencing have provided additional avenues for investigating how the populations have evolved. We end by discussing how the original LTEE procedures have been updated to accommodate or take advantage of these changes. This protocol will be useful for researchers who use the LTEE as a model system for studying connections between evolution and genetics, molecular biology, systems biology, and ecology. More broadly, the LTEE provides a tried-and-true template for those who are beginning their own evolution experiments with new microbes, environments, and questions.
Introduction
In February of 1988, Richard Lenski inoculated twelve flasks containing a defined glucose-limited growth medium with clonal cultures of Escherichia coli at the University of California, Irvine1. The following day, he transferred 1% of the culture from each flask to a set of new flasks containing fresh growth medium. This 1:100 dilution allowed the bacterial populations to expand 100-fold before exhausting the available glucose, corresponding to roughly 6⅔ generations of cell divisions. This procedure was repeated the following day and has been every day since, with a few interruptions. These daily transfers have continued, even as the experiment was relocated, first to Michigan State University in 1992, and then to The University of Texas at Austin in 2022. All the while, new mutations have continuously generated genetic variation in these E. coli populations and natural selection has led to evolved cells outcompeting their ancestors.
Lenski designed this experiment, now known as the Long-Term Evolution Experiment (LTEE), to investigate the dynamics and repeatability of evolution. In order to answer these questions, he included several important features in the design of the experimental setup and its protocols2. One of these features was the careful choice of a model organism. The original twelve populations were all started from single colonies that shared an immediate common ancestor, Escherichia coli B strain REL606. This strain was chosen because it had already been commonly used in lab settings, reproduced completely asexually, and contained no plasmids or intact prophages3,4 — all of which make studying its evolution simpler. Another choice that simplified the experiment was to use a very low concentration of glucose in the growth medium to limit the density of cells in each flask after growth. Using a low cell density was intended to make it easier to analyze changes in population fitness by reducing the potential for the evolution of ecological interactions within populations (e.g., by cross-feeding)5.
REL606 is unable to use ʟ-arabinose as a carbon and energy source (Ara−) due to a point mutation in the araA gene. Prior to starting the LTEE, a spontaneous mutant with a restored araA sequence, designated REL607, was isolated from REL6066. REL607 is capable of growing on ʟ-arabinose (Ara+). REL606 was used to start six of the LTEE populations, and REL607 was used to start the other six. Arabinose is not present in the growth medium used during the LTEE, so REL607 behaves the same as REL606 under these conditions. However, when plated on tetrazolium arabinose (TA) agar, Ara− and Ara+ cells form red and white colonies, respectively. This method for discriminating between the two ancestral E. coli strains and their descendants is quite useful. It can be used to detect cross-contamination between LTEE populations. It also aids in measuring the fitness of an Ara− strain or population relative to an Ara+ one when they are competed against one another. Fitness is measured by setting up a co-culture of oppositely marked competitors and then monitoring how the frequencies of red and white colonies (obtained by spreading dilutions of the culture on TA plates) change between when the competitors are initially mixed and after one or more growth cycles under the same conditions as the LTEE. The representation of the more-fit cell type will increase during each growth cycle.
Another critical feature of the LTEE is that samples of the evolving populations are periodically archived. When mixed with a cryoprotectant such as glycerol, E. coli cells can be frozen and later revived7. As part of the LTEE protocol, every 75th day (which equates to roughly 500 generations), a portion of each population that was not transferred to a new flask is mixed with glycerol, split up between multiple vials, and stored in a freezer. This frozen "fossil record" enabled researchers to perform the first studies of the LTEE, in which they revived the evolved E. coli populations from various timepoints and competed them versus the ancestral strains to track how rapidly fitness was increasing1. Fitness evolution has been remeasured periodically as more "strata" of the frozen "fossil record" have been preserved. The overall conclusion from these measurements is that fitness continues to improve in the LTEE to this day, even after so many generations of evolution in the same environment8,9,10.
What has allowed the LTEE to continue for so long? Many of the same features that enabled its original questions to be asked and answered have also served as safety measures and fail-safes against inevitable disruptions due to bad luck, human error, and world events. Every day, when the cultures are transferred to fresh growth medium, the researcher performing the transfers alternates between Ara− and Ara+ populations. Then, when the populations are frozen, they can be plated on selective and indicator agar to check if any "neighboring" populations have been accidentally cross-contaminated or mixed up (e.g., white colonies are in a population that should only form red colonies) or contaminated with foreign microbes (e.g., unexpected colony morphologies or cell densities). In the event that a population has been compromised, its progenitor can be revived from the freezer and carried forward in its place. The Ara markers and the frozen archive thus serve dual purposes as both experimental resources and safety measures.
Because its history is so well preserved and easily accessible, LTEE samples have been studied using technologies that did not exist when the experiment began. For example, whole-genome sequencing has been used to examine the dynamics of mutations in the LTEE populations11,12,13,14,15, and transcriptomics and ribosomal profiling have been used to examine changes in gene expression16,17. Genetic tools have been used to reconstruct strains that differ by single mutations or combinations of several evolved mutations to understand their effects on fitness and various phenotypes18,19,20,21. Samples from the frozen "fossil record" are easily replenished so that parts of or entire copies of the experiment's history can be shipped to other laboratories. LTEE samples now exist on all continents except Antarctica, and they are being studied by researchers who are younger than the experiment itself. The robust methods of the LTEE and evolved E. coli samples and strains from its historical record have also served as starting points for evolution experiments examining other questions and environments22,23,24,25,26,27,28,29.

Figure 1: Overview of LTEE procedures. Please click here to view a larger version of this figure.
{kind=link}
Here, we demonstrate three core protocols used in the E. coli Long-Term Evolution Experiment (Figure 1). We describe: (1) how to perform the daily transfers, (2) how to archive population samples and clonal isolates, and (3) how to perform and analyze co-culture competition assays to measure fitness differences. Our hope is that these protocols foster the continued use of LTEE resources and inform the design of new microbial evolution experiments.
Protocol
1. Daily transfers of LTEE populations
NOTE: The twelve LTEE populations are transferred daily by inoculating fresh medium with 1% of the cultures from the prior day's flasks. The steps in this process are summarized in Figure 1. The six Ara− populations started from strain REL606 are designated A−1 to A−6, and the six Ara+ populations started from strain REL607 are designated A+1 to A+6. Strict adherence to aseptic technique and to a schedule and order for transferring the populations minimizes the risk of contamination and other disruptions.
- Disinfect the surface on which the LTEE transfers will be conducted by wiping it with either 70% ethanol or a 10% bleach solution. Light a Bunsen burner to create a local updraft and enable the flaming of glassware.
NOTE: Wear laboratory gloves to prevent contamination. For safety around an open flame, it is critical to only use gloves that are made of a material like nitrile that is not flammable. - Prepare thirteen 50 mL borosilicate Erlenmeyer flasks capped with 20 mL borosilicate or polypropylene beakers that have been washed and sterilized by autoclaving. Check the flasks for visible debris and replace any that are not perfectly clean.
- Label six flasks A−1 through A−6 using a red marker and the other six flasks A+1 through A+6 using a black marker. Label the last remaining flask, which will be blank, with the date in month/day format and the day of the week.
- Fill each of the 13 flasks with 9.9 mL of DM25 medium using a sterile 10 mL serological pipette. Flame the mouth of each flask after removing the beaker serving as a lid and before replacing the beaker. Flame the tip of the pipette between filling each flask.
NOTE: Instructions for making DM25 are available online30. If using a plastic serological pipette, forego flaming its tip or limit the time in the flame to avoid melting the plastic. - Remove the prior day's LTEE flasks from the shaking incubator.
- Examine each flask by holding it up to the light to assess its turbidity and color, check flask integrity, and look for the presence of foreign matter.
NOTE: To the naked eye, all Ara− and Ara+ cultures will look slightly turbid compared to the blank, except for A−3, which will be ~10-fold more turbid than the others owing to growth on citrate in the medium. Many outside microbial contaminants are also able to grow on citrate, so increased turbidity in populations other than A−3 likely indicates contamination. See the Representative Results section for images of the LTEE cultures before a transfer. - OPTIONAL: Confirm that each LTEE culture has the expected turbidity by pipetting 1 mL of the blank and 1 mL of each culture into 1 cm plastic cuvettes and taking optical density readings at 600 nm (OD600) using a spectrophotometer after blanking the instrument.
NOTE: This extra step can be useful for researchers who are new to working with the LTEE and are unsure about judging the turbidity by eye, as well as for documenting and investigating suspected anomalies. Take samples for measuring OD600 from the prior day's flasks only after completing the day's normal transfers to new flasks (the following steps) to minimize the risk of contaminating the cell populations that will continue to be propagated if the OD600 values are as expected. See the Representative Results section for typical OD600 values for LTEE cultures. - Using a P200 micropipettor with a sterile filter tip, transfer 100 µL of culture from each LTEE flask into the corresponding flask containing fresh DM25. Begin with A−1, then transfer A+1. After that, continue alternating between the − and + populations. To keep track of which cultures have been transferred, shift flasks to the left after pipetting from or to them.
NOTE: The strict order of transfers and alternation between Ara− and Ara+ populations aids in preventing and detecting cross-contamination and mix-ups. Observe strict aseptic technique: use a fresh pipette tip for each transfer, flame the mouths of flasks immediately after uncapping and before recapping, and wipe down the barrel and ejector of the micropipettor with a lint-free paper wipe moistened with 70% ethanol between each transfer. Bleach should never be used to disinfect micropipettors, as even trace amounts can kill the cultures. - Incubate the newly inoculated flasks at 37 °C for 24 ± 1 h with 120 rpm orbital shaking over a 1-inch diameter.
- Store the cultures from the previous day at 4 °C. Retain these backup cultures for two days. Discard older cultures that were saved at 4 °C three days before at this time.
NOTE: The previous two days' cultures provide two complete sets of backups with which to restart the experiment, if necessary, should any problem or accident occur, or if contamination of the previous day's cultures is discovered prior to transferring (e.g., odd coloration or unexpected particulates). - Enter the time, date, transfer number, name or initials of the researcher who did the transfers, whether or not the cultures were okay, and any other relevant information in the transfer log notebook. Move on to steps 1.12-1.14 if any of the following situations occurs: (1) the blank from the previous day is contaminated, (2) a flask or its lid is cracked or broken, (3) a flask contains foreign material, (4) a flask is tipped over or dropped during transfers, or (5) there is any other event or observation that makes continuing from these flasks questionable.
- If there are any problems, accidents, or suspicions of contamination with the previous day's LTEE cultures, do not transfer from them. Instead, store the entire set of twelve cultures at 4 °C for later examination and further characterization.
- Retrieve the flasks containing the backup cultures that were transferred from the day before and stored at 4 °C. Place them on the benchtop to warm to room temperature. Gently swirl each flask to resuspend the cells.
- Transfer from the backup flasks to the new set of flasks containing fresh medium and continue the experiment normally as described in steps 1.6-1.11. Make a note in the transfer log that the backup cultures were used and record the same transfer number as the day before.
NOTE: Even if a problem is noted in only one population's flask, transfer all twelve populations from the backup flasks so that the number of generations that have elapsed in all populations stays in phase. If contamination is noted in the backup flasks stored at 4 °C, then the affected LTEE populations must be restarted from frozen stocks using the procedure described in steps 3.1-3.2 for population samples. The transfer number for the LTEE should not be incremented until the growth of the first cultures in DM25 following revival.
2. Archiving the LTEE populations
NOTE: Samples of the LTEE populations are frozen every 75 transfers. The populations grow ~6 ⅔ generations each day following the 100-fold transfer dilution, so this period corresponds to ~500 generations. During archiving, the LTEE populations are also plated on different types of agar media to check for contamination. Optionally, representative clones can be picked from these plates and archived at this time. These steps are summarized in Figure 1.
- On the day before the planned freeze-down or a few days before, prepare three types of agar plates: Minimal Glucose (MG), Minimal Arabinose (MA), and Tetrazolium Arabinose (TA). Make twelve plates of each type of agar, plus a few extras. Also prepare at least 250 mL of 0.85% (w/v) sterile saline and 50 mL of 80% (v/v) sterile glycerol.
NOTE: Recipes for all media and solutions are available online30. The day before the LTEE reaches a generation that is a multiple of 500 for the regular archiving schedule is the 74th day since the last freeze-down plus any days that were added owing to transfers from the 4 °C backup flasks when problems were detected or suspected. - OPTIONAL: If archiving clonal isolates from the LTEE populations, prepare additional supplies: isolating three clones from each population requires 72 MG plates, 80 mL of 80% (v/v) glycerol, and 370 mL of DM1000.
- Prepare an extra set of twelve flasks when performing step 1.2 of the daily LTEE transfers on the day before the planned freeze-down. Label six of the additional flasks xA−1 through xA−6 using a red marker, and the other six xA+1 through xA+6 using a black marker.
NOTE: The "x" indicates that the extra set of flasks will be used for archiving and differentiates them from the other set of flasks that will be used to continue the daily transfers of the LTEE in parallel. - Fill each of the additional flasks that will be used for archiving with 14.85 mL of DM25 using a 25 mL serological pipette when performing step 1.4 of the daily LTEE transfers.
- Complete the normal LTEE transfer as described in steps 1.5−1.11. Then, repeat the instructions for step 1.8, but this time transfer 150 µL from each of the previous day's LTEE cultures to the additional flasks of 14.85 mL of fresh DM25 that will be used for archiving.
NOTE: In this and all subsequent steps, avoid contamination and mixups by following these guidelines. Begin with population A−1, then transfer A+1, and then continue alternating − and + populations. Wipe down the barrel and ejector of the micropipettor with a lint-free paper wipe moistened with 70% ethanol when switching populations. Shift flasks and test tubes over in their trays or racks after pipetting from or to them to keep track of which transfers have been completed. - Incubate the set of twelve flasks for archiving at 37 °C for 24 ± 1 h with 120 rpm orbital shaking over a 1-inch diameter alongside the twelve LTEE cultures and the blank as described in step 1.9.
- Prepare supplies for plating the LTEE populations at least one hour before the LTEE transfers are to be performed on the day of the freeze-down.
- Select twelve MG, twelve MA, and twelve TA agar plates. Visually inspect each one to be sure it does not have any obvious contamination.
- Label one of each type of plate for each of the twelve LTEE populations (A−1 through A+6).
NOTE: When labeling plates, write on the sides of the bottom of the petri dish. This is important for not obscuring colonies when one wants to examine or photograph them from below the agar. Do not write on the lids, as these can be mixed up. - Place the agar plates in a 37 °C incubator for at least 20 min to warm them before using them in step 2.10.
- Prepare 24 test tubes containing 9.9 mL of saline. Arrange them in twelve sets of two tubes each.
- Label each of the two sets of twelve test tubes in the same way as the plates, adding a "1" or a "2" below the LTEE population identifier to designate the order in which they will be used for making dilutions of that population.
- Perform steps 1.1-1.11 using the flasks that will continue the daily transfers of the LTEE as usual. During step 1.5, also remove the twelve flasks containing the extra cultures for archiving from the shaking incubator.
- Pipette 100 µL of the culture from each of the twelve additional flasks for archiving into the first test tube of saline in the pair for that LTEE population. Vortex the tubes with these 100-fold dilutions thoroughly. Then, pipette 100 µL from each one to the corresponding second tube of saline. Vortex the final 10,000-fold culture dilutions thoroughly.
- Pipette 80 µL from each of the tubes containing a 10,000-fold culture dilution to the labeled TA, MG, and MA plates for that population. Spread the liquid uniformly across the agar surface using either a sterile spreading rod or sterile spreading beads, as preferred. Repeat until all twelve populations have been plated on all three types of media.
- If necessary, allow the plates to dry until no liquid is visible on the agar. Place the plates upside down (with the agar side up) in a gravity convection incubator set to 37 °C.
NOTE: Incubating plates upside down keeps the agar from drying out and prevents condensation from dripping onto the agar surface. Movement of cells in liquid on the agar surface during incubation can smear colonies and yield incorrect colony counts. - Add 3 mL of sterile 80% (v/v) glycerol to each of the twelve extra flasks earmarked for archiving. Mix thoroughly by swirling and gently vortexing.
- Distribute the mixture from each flask to sterile cryovials that have been labeled with a unique identifier for the sample, the LTEE population to which the sample belongs, the generation at which it was frozen, that it is a mixed (population) sample, and the date. Pipette 6 mL into one large vial and 1.25 mL into each of six small vials.
NOTE: The large vial is the working stock. One small vial is a backup in case the working stock is exhausted or becomes contaminated. The other five small vials are copies that can be sent to other labs. - Freeze the filled vials at −80 °C.
- Examine and document the growth and morphologies of colonies on the TA, MG, and MA plates after 24 h and 48 h of incubation.
NOTE: See the Representative Results section for images and descriptions of colonies formed by the REL606 and REL607 ancestors and each of the twelve LTEE populations when they were plated at 76,000 generations. - OPTIONAL: Perform the following steps when archiving clonal isolates.
- Pick three clonal isolates (colonies) for each LTEE population from the MG plates, streak each one separately on a new MG plate, and incubate these plates for 16-24 h at 37 °C.
NOTE: If colonies with different morphologies are present, standard practice in the LTEE is to sample for maximum diversity by first picking the most common type, then selecting further colonies from minority types. One can also use a random sampling strategy by marking dots on the underside of the bottom of the petri dish before spreading cells and then picking the isolated colony nearest each mark after growth. - The next day, streak a representative colony from each plate on a new MG plate and incubate these plates for 16-24 h at 37 °C.
- The following day, inoculate one isolated colony from each MG plate into a flask containing 10 mL of fresh DM1000. Also, fill one additional flask with 10 mL of DM1000 to serve as an uninoculated blank to test for media contamination.
- Incubate the flasks at 37 °C for 16-24 h with 120 rpm orbital shaking over a 1-inch diameter.
- After incubation, add 2 mL of sterile 80% (v/v) glycerol to each flask and swirl to mix.
- Distribute 1.25 mL aliquots from each flask into small, sterile vials labeled with a unique identifier for each clone, its LTEE population and generation of origin, that it is a clonal sample, and the date.
- Freeze the filled vials at −80 °C.
- Pick three clonal isolates (colonies) for each LTEE population from the MG plates, streak each one separately on a new MG plate, and incubate these plates for 16-24 h at 37 °C.
3. Competitive fitness assays
NOTE: In the LTEE, reproductive fitness is quantified in terms of the relative number of doublings that different bacteria achieve over one or more 24 h culture cycles under the same conditions as the daily transfers. Specifically, the relative fitness of one competitor to another is the ratio of their realized doubling rates when they compete head-to-head in a co-culture. Each competitor in a pair may be a full population or a clonal isolate that was previously archived as part of the frozen "fossil record" of the LTEE. Alternatively, one or both competitors may be a clone that has been genetically modified to add or remove specific mutations to test their effects. The two competitors must have opposite Ara+/Ara− states because this genetic marker is used to differentiate them during this assay. The overall workflow for a competition assay is shown in Figure 2. The duration of the co-culturing phase can be extended from one to three (or more) days to improve the precision of fitness estimates when testing for differences between competitors that are nearly evenly matched. See the Disussion for other critical considerations and possible modifications of this protocol.

Figure 2: Competition assay flowchart. The full procedure for a one-day competition assay is shown. The three-day procedure continues with the alternative pathway on Day 1 and Day 2 until plating on Day 3 in the same way that is pictured for Day 1 of the one-day competition. Please click here to view a larger version of this figure.
{kind=link}
- Prepare supplies
- Decide how many competitor LTEE strains and/or populations will be used and how many replicate competition assays will be performed for each pair of competitors. Prepare the necessary supplies as described in the following steps.
NOTE: Recipes for all media and solutions are available online30. The flasks and test tubes required for all days of a competition experiment can be filled beforehand or as needed on the days they will be used. If flasks and test tubes are filled ahead of time, store them at room temperature in the dark to minimize evaporation. TA plates need to be prepared at least two days in advance of when they will be used so they can dry enough after being poured to allow for spreading culture dilutions. Always prepare a few extra flasks, test tubes, and TA plates so that an experiment can continue if there are pipetting mistakes, contaminated plates, or other minor mishaps. - For the revival day (Day −2), fill one sterile 50 mL Erlenmeyer flask capped with a 20 mL beaker with 9.9 mL of either DM1000 or Lysogeny Broth (LB) per strain or population of E. coli that will be used as a competitor. Fill one more flask with 9.9 mL of the same medium to serve as an uninoculated blank.
- For the preconditioning day (Day −1), fill one test tube with 9.9 mL of 0.85% (w/v) sterile saline per competitor, two flasks with 9.9 mL of DM25 per replicate assay between a pair of competitors, and one more flask with 9.9 mL of DM25 for a blank.
- For the day the competition begins (Day 0), fill one flask with 9.9 mL of DM25, fill one test tube with 9.9 mL of 0.85% (w/v) sterile saline, and prepare one TA plate per competition assay replicate. Fill one more flask with 9.9 mL of DM25 to serve as a blank.
- ALTERNATIVE: For each day of a multi-day competition past the first, fill one flask with 9.9 mL DM25 per competition replicate and fill one more flask with 9.9 mL of DM25 for a blank.
- For the final day of the competition (e.g., Day 1 or Day 3), fill two test tubes with 9.9 mL of 0.85% (w/v) sterile saline and prepare one TA plate per competition replicate.
- Decide how many competitor LTEE strains and/or populations will be used and how many replicate competition assays will be performed for each pair of competitors. Prepare the necessary supplies as described in the following steps.
- Day −2: Revive competitors separately in DM1000 or LB
- For each of the competitors, label a flask filled with 9.9 mL of either DM1000 or LB. Label an additional flask filled with 9.9 mL from the same batch of medium to serve as an uninoculated blank to test for contamination.
NOTE: Frozen stocks are revived in LB or DM1000 for more uniform and predictable recovery of cryopreserved cells. The glycerol used as a cryoprotectant can be metabolized by E. coli, which will lead to higher cell densities than expected if samples are revived in DM25. LB and DM1000 support growth to such high cell densities that this complication becomes negligible. - Take the cryovials containing the frozen stocks of the competitor strains out of the −80 °C freezer. Keep the vials chilled in an ice bucket while using them.
- After each frozen stock has thawed, vortex it thoroughly to resuspend the E. coli cells. If reviving a clone, inoculate the flask containing fresh medium with 12 µL of the frozen stock. If reviving a population, inoculate the flask with 120 µL of the frozen stock.
NOTE: The 120 µL volume of the frozen stock is used for populations so that the number of cells that is revived is approximately the same as the daily bottleneck when 1% of the LTEE population is transferred to a new flask. Thawing and vortexing frozen stocks multiple times can stress cells and reduce the viability of stocks over time. If a given LTEE population or clone is going to be used in competitions multiple times, it is good practice to regrow and freeze multiple copies of the stock so that no single one is thawed and re-frozen many times. - Incubate the revival flasks and the blank at 37 °C overnight (16-24 h) with 120 rpm orbital shaking over a 1-inch diameter.
- For each of the competitors, label a flask filled with 9.9 mL of either DM1000 or LB. Label an additional flask filled with 9.9 mL from the same batch of medium to serve as an uninoculated blank to test for contamination.
- Day −1: Precondition competitors separately in DM25
- For each competitor, label a test tube filled with 9.9 mL of saline. For each replicate competition assay between a pair of competitors, label two 50 mL flasks filled with 9.9 mL of DM25, each with the replicate number and the name of one of the competitors. Label one additional flask filled with 9.9 mL of DM25 to serve as a blank.
- Take the flasks containing the cultures of revived competitors out of the incubator. Examine their turbidity by eye to confirm that they grew and that there is no obvious contamination.
- Pipette 100 µL from each flask into the test tube of saline for that competitor.
NOTE: This step dilutes the culture 100-fold, which is necessary because the density of cells is much higher in LB and DM1000 than it is in the DM25 environment used in the LTEE (see Representative Results). - Vortex each dilution tube thoroughly right before pipetting 100 µL from the diluted culture into a flask with fresh DM25. Inoculate two of these preconditioning flasks for each replicate assay, one for each of the competitors.
- Incubate the preconditioning flasks and the blank at 37 °C for 24 ± 1 h with 120 rpm orbital shaking over a 1-inch diameter.
- Day 0: Begin competition by mixing competitors and plate for initial counts
- For each competition assay replicate, label one flask filled with 9.9 mL of DM25 and one test tube filled with 9.9 mL of saline. Label the flasks and tubes in a way that uniquely identifies each pair of competitors and the replicate number of the competition assay. Label one additional flask filled with 9.9 mL of DM25 to serve as a blank.
- Take the preconditioning flasks out of the incubator. Examine their turbidity by eye to confirm that they grew and that there is no obvious contamination.
- Transfer 50 µL of the Ara− competitor into the first replicate competition flask filled with fresh DM25. Immediately, transfer 50 µL of the Ara+ competitor into the same competition flask and mix it by gently swirling.
- Repeat step 3.4.3 for all replicates of all pairs of competitors.
NOTE: The competition flasks now have an overall 100-fold dilution of E. coli cultures grown in DM25, the same condition cells in the LTEE experience after each daily transfer. The order of performing transfers and mixing is important. Add both competitors to each flask immediately one-after-the-other so that neither gets a head start growing in the fresh medium. For example, do not add the Ara− cultures to all competition flasks and then go back and add all of the Ara+ strains. - Pipette 100 µL from each newly inoculated competition flask into the test tube of saline labeled for that competition assay replicate so that each of these tubes contains an overall 10,000-fold dilution of the preconditioned DM25 cultures that were combined.
- Place the competition flasks and blank into the shaking incubator. Incubate the competition flasks at 37 °C for 24 ± 1 h with 120 rpm orbital shaking over a 1-inch diameter.
- On the same day, immediately after placing the competition flasks into the incubator, vortex each test tube from step 3.4.5 thoroughly and spread 80 µL of these 10,000-fold dilutions on TA plates as described in step 2.10. Label the side of the bottom of each plate with the pair of strains that were mixed, the replicate number, and "Day 0" to indicate it will be used to determine the initial representation of each competitor.
- Incubate the TA plates upside down in a gravity convection incubator at 37 °C until colonies of both the Ara− and Ara+ competitors are visible and distinguishable. Generally, this occurs within 16-24 h, but it may take longer for some evolved strains. Count the numbers of Ara− (red) and Ara+ (white) colonies on each plate and record the results.
NOTE: Differences between the colors of Ara- and Ara+ colonies on TA plates become less distinct over time, even when plates are stored at 4 °C, so they need to be counted as quickly as possible once they are removed from the incubator. Images of TA plates showing the typical appearances of colonies formed by Ara− and Ara+ cells are included in the Representative Results. That section also has images of common "edge case" colonies (e.g., overlap or outgrowth of different colony types), and explains how to count them. If the growth rates and morphologies of colonies formed on TA plates by any of the competitors have not been previously characterized, spread 80 µL of a 10,000-fold dilution in saline from the preconditioning flasks on Day 0, when the competitors are still separate from one another. Then, examine colonies on these control plates after incubation at 37 °C for 16-24 h or longer.
- ALTERNATIVE: Days 1 and 2: Continue three-day competition
- For each competition assay replicate, label one flask filled with 9.9 mL of DM25. Label the flasks in a way that uniquely identifies each pair of competitors, the replicate number, and the day of the competition assay. Label one additional flask filled with 9.9 mL of DM25 to serve as a blank.
- Take the competition flasks from the previous day out of the incubator. Examine their turbidity by eye to verify expected growth and detect contamination.
- Transfer 100 µL from each competition flask into the corresponding flask of fresh medium for the next day of the competition.
- Place the new competition flasks and blank into the shaking incubator. Incubate them at 37 °C for 24 ± 1 h with 120 rpm orbital shaking over a 1-inch diameter.
- Repeat steps 3.5.1-3.5.4 on Day 2 of the competition before proceeding.
- Day 1 or 3: Finish competition and plate for final counts
- For each competition flask, prepare two test tubes filled with 9.9 mL of saline. Label them in a way that uniquely identifies each pair of competitors, the replicate number, and whether they are for the first or second dilution.
- Take the competition flasks out of the incubator. Examine their turbidity by eye to detect that they grew and there was no obvious contamination.
- Pipette 100 µL from each competition flask into the first tube of saline for that replicate. The resulting tubes contain 100-fold dilutions of the DM25 cultures.
- Vortex each 100-fold dilution tube to mix it thoroughly and pipette 100 µL to the second tube of saline for that replicate. The resulting tubes contain 10,000-fold dilutions of the DM25 cultures.
- Vortex each test tube containing a 10,000-fold dilution thoroughly and spread 80 µL of it on a TA plate as described in step 2.10. Label the side of the bottom of each plate with the pair of strains that were mixed, the replicate number, and "Day 1" for a one-day competition or "Day 3" for a three-day competition to indicate it will be used to determine the final representation of each competitor.
- Incubate TA plates at 37 °C and count the Ara− and Ara+ colonies after growth as described in step 3.4.8.
NOTE: Keep track of the replicate number of each competition assay throughout all transfers and plating steps. Confusing which final and initial counts correspond between different replicate assays — even when the same two competitors were mixed in each one — will result in incorrect fitness estimates.
- Calculation and plot fitness
- If using Excel for calculating and plotting relative fitness, download the XLS spreadsheet (Supplemental File 1). If using R, install the fitnessR package31 and download the comma-separated values (CSV) template (Supplemental File 2) or generate a new copy of this file following the directions in its vignette.
- Enter a "transfer dilution" of 100 for the competition assays conducted in the designated cell or column in the downloaded file. Enter the total number of daily growth cycles during which the competitors were co-cultured as the "number of transfers" (e.g., 3 for a three-day competition).
- Enter the names of each pair of competitors into the designated cells or columns with the reference strain as "competitor1" and the test strain or population as "competitor2".
- For each competition assay replicate, enter the respective initial and final colony counts into the designated columns of the downloaded file.
- If using the Excel spreadsheet, it will now display the mean relative fitness value and 95% confidence limits on this estimate. Copy the results for different combinations of competitors to another sheet and create a chart that summarizes the results. If using R to analyze the data, follow the directions in the vignette for the fitnessR package to perform these calculations, output a CSV file with the calculated values, and plot the results.
Results
Appearance and turbidity of LTEE cultures
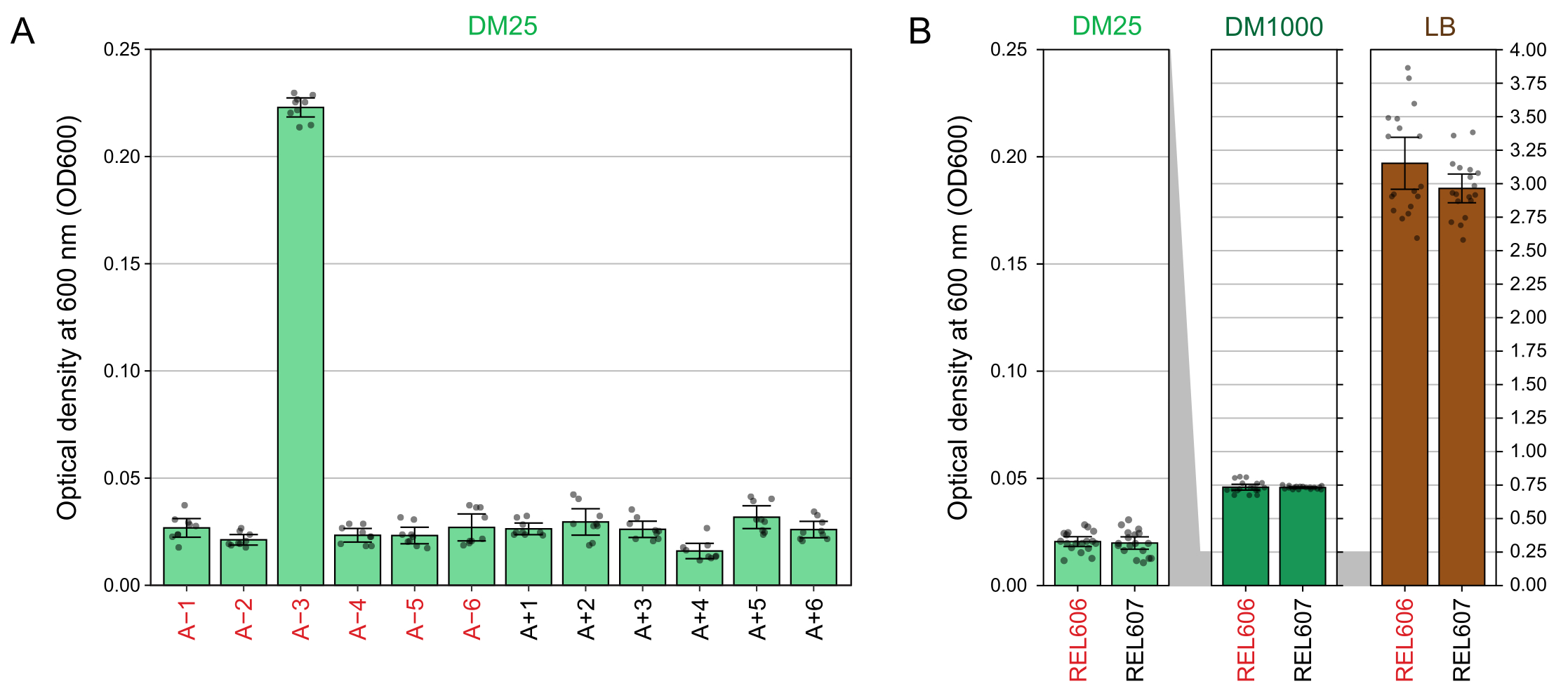
Due to the low glucose concentration in DM25, the turbidity of fully grown LTEE populations is only barely visible in eleven of the twelve flasks. When examining the LTEE cultures by eye for normal growth and signs of contamination (step 1.6), each flask containing an LTEE population should be compared side-by-side to the blank (Figure 3A). The exception is population A−3, which evolved to use citrate as an additional carbon and energy source and therefore reaches a higher cell density32. The turbidity of DM25 cultures of the REL606 and REL607 ancestor strains is similar to that of a typical evolved population (Figure 3B). LTEE strains and populations grow to a higher density in DM1000 owing to the higher concentration of glucose, and a much higher density in LB (Figure 3B). The density of DM25 cultures of the A−3 LTEE population is intermediate between the densities of cultures of REL606 in DM25 and DM1000 (Figure 3C).

Figure 3: Appearance of LTEE cultures. (A) Flasks containing the twelve LTEE populations after 24 h of growth in DM25 on the day when the experiment reached 76,253 ⅓ generations are pictured alongside the blank. (B) Flasks containing cultures of the REL606 and REL607 ancestors grown for 24 h in DM25, DM1000, and LB are pictured alongside media blanks. (C) Zoomed in pictures of the same flasks side-by-side showing how the turbidity of the A−3 population flask in DM25 compares to the REL606 ancestor in DM25 and DM1000. Please click here to view a larger version of this figure.
{kind=link}
Spectrophotometer readings of the optical densities at 600 nm (OD600) of cultures grown in DM25 (step 1.7) match these visual observations for both the LTEE populations (Figure 4A) and their ancestors (Figure 4B). These readings can be used to quantitatively compare and document growth when contamination or a mistake is suspected. For measurements of the LTEE populations between 76,000 and 76,500 generations, we found that the OD600 of A−3, the population that evolved to grow on citrate, was 0.223 on average (0.218-0.227, 95% confidence interval). The OD600 of the other eleven populations was 0.0252 on average (0.0239-0.0265, 95% confidence interval). There was slight, but significant variation in OD600 readings among the eleven normal populations (F10,88 = 5.1035, p = 7.5×10−6). The LTEE populations reach stationary phase after roughly 5-6 hours of incubation. If they are transferred in the morning, growth will be visible by mid- to late afternoon of the same day. Many species of microbes are able to grow aerobically on citrate. Therefore, increased turbidity in populations other than A−3 is likely a sign of outside contamination.

Figure 4: Turbidity of LTEE cultures. (A) Optical density at 600 nm (OD600) of the twelve LTEE populations after the 24 h growth cycle on three different days between 76,000 and 76,500 generations of the experiment. The OD600 values of three 1-mL aliquots on each of the three different days are plotted as points. The mean OD600 value of three different aliquots of the blank from the same day was subtracted from these values. Filled bars show averages. Error bars are 95% confidence limits. (B) OD600 of cultures of the REL606 and REL607 ancestors in DM25, DM1000, and LB. The OD600 values of three 1-mL aliquots on each of three different days of two separate cultures for each condition and strain are plotted as points. The mean OD600 value of three different aliquots of the blank from the same day was subtracted from these values. Filled bars show averages and error bars are 95% confidence limits. Grey shaded areas between the panels show how the OD600 axis is rescaled between the DM25 panel and the DM1000 and LB panels. Please click here to view a larger version of this figure.
{kind=link}
Growth and morphology of LTEE colonies
When checking the populations for contamination by plating them on different media (step 2.15), the REL606 and REL607 ancestors and all evolved populations form white colonies with translucent and somewhat irregular edges on minimal glucose (MG) agar plates (Figure 5A). The composition of MG agar is the same as that of the DM25 used in the daily LTEE transfers, except with a higher concentration of glucose, so the evolved LTEE populations often form larger colonies on MG than the ancestors. Owing to its higher cell density in DM25, the A−3 population will have several-fold more colonies if the same volume is plated for it as for the other populations, and this may limit the size of the colonies. The most common types of contaminating microbes form stark white, opaque, and perfectly circular colonies on MG.
On minimal arabinose (MA) agar, the REL607 ancestor and the Ara+ populations typically all form slightly translucent white colonies. This typical growth pattern has persisted for the Ara+ populations through 76,000 generations, except A+6, which has evolved a defect in growth on arabinose and no longer forms colonies on MA (Figure 5B). There is no selection to maintain growth on arabinose during the LTEE transfers in DM25, so other Ara+ populations may also eventually stop forming colonies on MA agar plates as the experiment continues. With the exception of A−3, the Ara− populations do not form colonies on MA agar, though close examination may reveal microcolonies owing to trace nutrients in the agar. The A−3 population forms numerous small colonies on MA, as these cells can grow on the citrate that is also present in this medium. Contaminant colonies on MA are rare.

Figure 5: Plating LTEE populations to detect contamination. Dilutions of the REL606 and REL607 ancestors and the twelve LTEE populations on the day when the experiment reached 76,026 ⅔ generations were plated on (A) MG, (B) MA, and (C) TA agar plates and photographed after 24 h and 48 h. The same dilutions were made for all cultures, but half as much volume was plated for the ancestors as is described in the protocol for the LTEE populations, to account somewhat for their higher cell densities. Please click here to view a larger version of this figure.
{kind=link}
On tetrazolium arabinose (TA) agar, the REL606 ancestor and all Ara− populations are expected to form red colonies, while the REL607 ancestor and all Ara+ populations should generally form colonies that are white (which can include light pink or peach shades) (Figure 5C). The LTEE ancestors form robust colonies, which are easily identifiable as Ara− and Ara+ on TA agar within 16-24 hours. Originally, this difference could be used to detect cross-contamination between Ara− and Ara+ populations. However, TA agar has a more complex nutrient composition than the chemically defined DM25 medium used in the daily transfers, and there has not been an evolutionary pressure for E. coli in the LTEE to maintain an ability to robustly grow under these conditions. Consequently, some evolved LTEE populations now exhibit poor growth on TA plates, taking 48 hours to form colonies or not reliably growing at all. The colors and morphologies of colonies formed on TA by the evolved LTEE populations have also changed relative to the ancestors and diverged from one another. The presence of a few aberrant colonies is not always an indication of contamination. Spontaneous mutations can occur that switch the Ara marker state of LTEE strains, especially from Ara+ to Ara− due to the higher likelihood of loss-of-function mutations affecting arabinose utilization versus reversion mutations that restore araA activity. Mutations switching Ara marker states are more common in populations that have evolved hypermutation (A−1, A−2, A−3, A−4, A+3, and A+6)13. On TA agar, contaminating microbes of other species often (but not always) form small, perfectly circular colonies with red centers ringed by distinct white boundaries that are unlike those formed by any LTEE strains or populations.
Co-culture competition results
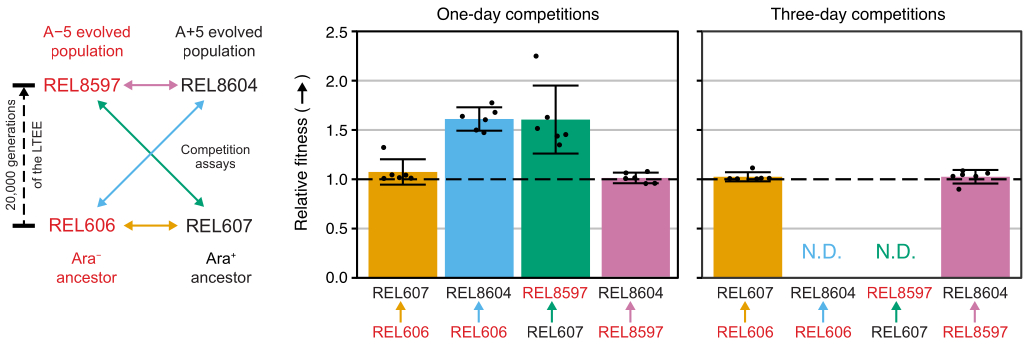
Competitions between all Ara− and Ara+ pairs of the two LTEE ancestors (REL606 and REL607, respectively) and the A−5 and A+5 population samples archived at 20,000 generations (REL8597 and REL8604, respectively) show how colonies with different Ara marker states can be differentiated and counted on TA agar (steps 3.4.8 and 3.6.6) (Figure 6). Colonies were counted for six replicate flasks for each pair of competitors before and after one-day and three-day assays that began with revival in DM1000 (Table 1). The total numbers of colonies observed for the same dilution and volume plated vary with which competitors were mixed because cultures of evolved LTEE populations reach lower cell densities than cultures of the ancestor strains in DM25. This difference is a consequence of the evolution of increased cell size, which occurred in all LTEE populations during the first few thousand generations of the experiment8, 33.

Figure 6: Competition assays plated on TA agar plates. Examples of TA agar plates from competition assays. REL606 and REL607 are the Ara− and Ara+ ancestors of the LTEE, respectively. REL8597 and REL8604 are the 20,000 generation A−5 and A+5 populations, respectively, from the frozen "fossil record" of the LTEE. TA plates corresponding to one replicate assay between each pair of strains are shown for Day 0, Day 1, and Day 3 of the competition. Plates were photographed after 24 h of growth at 37°C. Cells of the REL606 and REL8597 competitors are Ara− and form red colonies. Cells of the REL607 and REL8604 competitors are Ara+ and form white colonies. Please click here to view a larger version of this figure.
{kind=link}
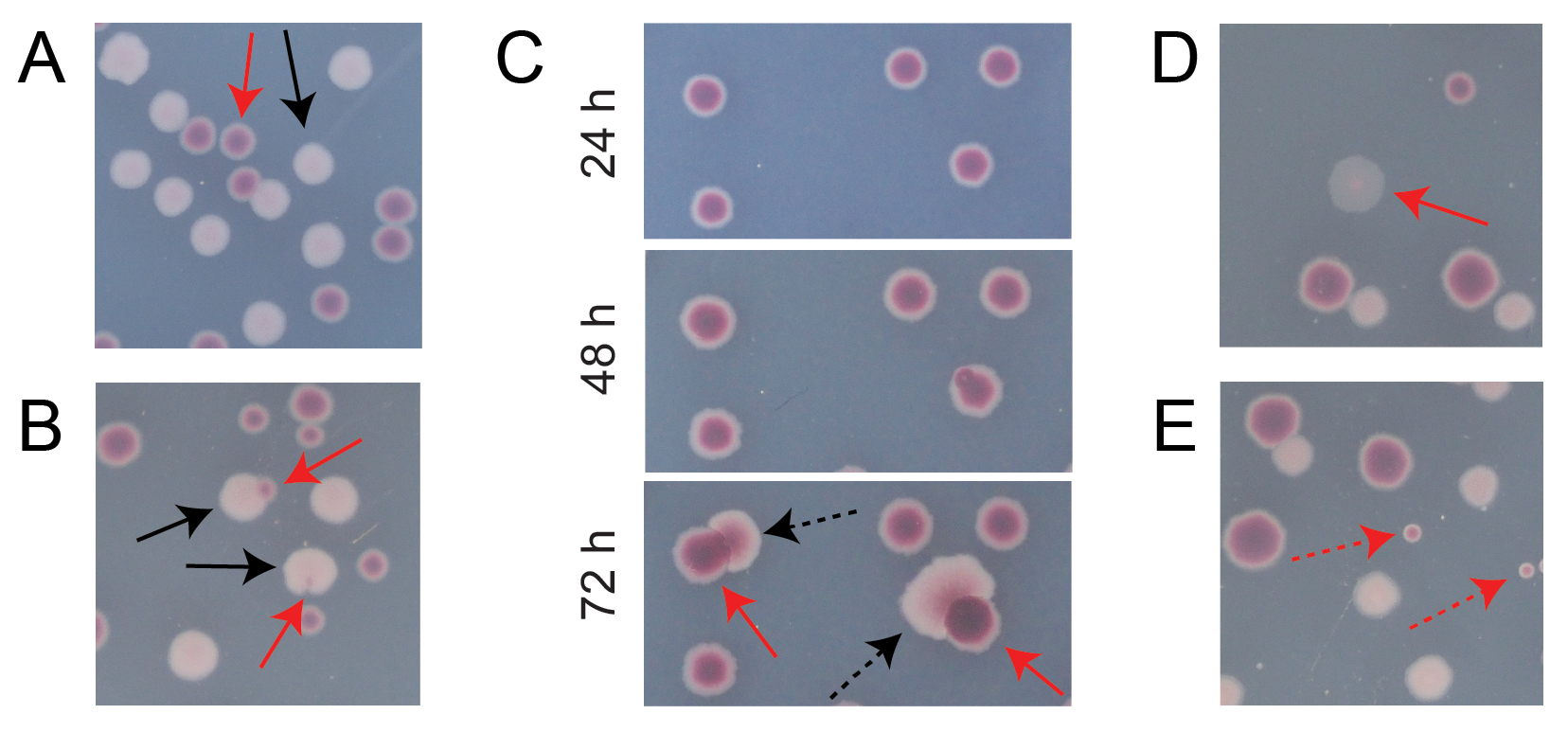
Most colonies on a typical competition TA plate will be well-separated or overlap in ways for which it is easy to count how many initially circular colonies of different types grew together (Figure 7A). However, some situations may arise in which it is not obvious how to count an atypical colony or growth that is a mixture of the two colors. First, when a white Ara+ colony and a red Ara− colony overlap, the Ara+ colony tends to overgrow and envelope the Ara− colony. In this situation, one should count a small red patch or translucent "gap" in the larger Ara+ colony as an Ara− colony (Figure 7B). Second, spontaneous Ara+ mutants will occasionally arise in Ara− colonies. These mutants typically appear as white sectors (papillae) spreading more quickly out of the interior of a red colony because they grow more quickly once they gain access to arabinose as an additional nutrient (Figure 7C). These white-sectored colonies are counted as one Ara− colony and no Ara+ colonies. This situation becomes more common if plates are incubated for 48 h or longer. Third, sometimes translucent pinkish colonies are observed (Figure 7D). These are formed by the Ara- competitor. Finally, a small number of circular colonies with interiors that are a slightly different shade of red sometimes grow on TA plates when they are contaminated by a few outside microbial cells during preparation of the agar or when spreading culture dilutions on their surfaces (Figure 7E). These contaminant colonies should not be counted. If contamination of a competition culture is suspected because there are many atypical colonies on any of its TA plates, that replicate should be excluded.

Figure 7: Edge cases encountered when counting Ara− and Ara+ colonies on TA agar. In each panel, some Ara− and Ara+ colonies that should be counted are marked with solid red and black arrows, respectively. Colonies that should not be counted are indicated with dashed arrows corresponding to the type that they appear to be. All photos were taken after 24 h of incubation except in panel C. (A) Examples of normal Ara− and Ara+ colonies. (B) Examples of Ara+ colonies overgrowing nearby Ara− colonies, including one that is only barely visible as a transparent gap in the outside of the white colony. Count each of these cases as two colonies, one of each type. (C) Examples of Ara− colonies giving rise to Ara+ mutant sectors. Count each case as only a single Ara− colony. The white sector (papilla) that arises is due to an Ara+ mutant arising within the colony. The same field of colonies is shown following 24 h, 48 h, and 72 h of growth. (D) Example of a translucent pink colony. Count it as Ara−. (E) Examples of colonies formed by outside contamination by a microbe that is not E. coli. These are red but smaller and perfectly circular with a distinct white boundary. Please click here to view a larger version of this figure.
{kind=link}
Analyzing the colony counts from these competitions using the Excel spreadsheet (Supplemental File 1) or by running the fitnessR package functions in R on colony counts entered into the CSV template (Supplemental File 2) shows that the two ancestors are indistinguishable in terms of their fitness within the precision of the assay, that both the 20,000-generation A−5 and A+5 populations are significantly more fit than the ancestors, and that neither evolved population is significantly more fit than the other (Welch's t-tests, p > 0.05) (Figure 8). The precision of the relative fitness estimate improves in the three-day competitions versus the one-day competitions for one of the closely matched pairs (REL606 vs. REL607). The precision of these measurements could be increased further by conducting longer competitions with more growth cycles, if so desired. However, the results from multi-day competitions are not informative once one competitor becomes so abundant relative to the other after the additional days of competition that the ratio of the two strains cannot be accurately determined because there are very few to no colonies of the less-fit type to count. This is the case for the three-day competitions of the ancestors against the evolved 20,000 generation populations (REL606 vs. REL8604 and REL607 vs. REL8597) (Figure 6 and Table 1).
Table 1: Colony counts from competitive fitness assays. One-day and three-day competition assays with six replicates were performed for all pairwise combinations of two Ara− and the two Ara+ competitors. REL606 and REL607 are the Ara− and Ara+ ancestors of the LTEE, respectively. REL8597 and REL8604 are the 20,000-generation A−5 and A+5 populations, respectively, from the frozen "fossil record" of the LTEE. Please click here to download this Table.

Figure 8: Relative fitness measured using competition assays. Results of one- and three-day competition assays between LTEE ancestors and the 20,000 generation A−5 and A+5 LTEE populations. The diagram on the left shows the four pairwise competitions as color-coded double-headed arrows. Each combination of the two Ara− (red labels) and the two Ara+ (black labels) competitors was tested with six-fold replication. Colony counts from Table 1 were analyzed in R using the fitnessR package31, and the results were plotted using the ggplot2 package (version 3.4.0)34. Fitness is displayed as the competitor the arrow in the label is going toward relative to the competitor the arrow is coming from (e.g., REL8604 relative to REL606). Relative fitness values estimated from the colony counts for each competition assay replicate (points), mean relative fitness values for the pair of competitors (bars filled with the same color-coding as the diagram), and 95% confidence intervals (error bars) are shown. Relative fitness values could not be determined (N.D.) for the three-day competitions between the ancestors and the evolved populations because there were zero or very few colonies of the ancestors on the Day 3 plates (see Table 1). Please click here to view a larger version of this figure.
{kind=link}
Supplemental File 1. Excel spreadsheet file for calculating relative fitness. Please click here to download this File.
Supplemental File 2. Comma-separated values input file template for calculating relative fitness in R using the fitnessR package. Please click here to download this File.
Discussion
Long-term resilience of the LTEE and its methods
The E. coli Long-Term Evolution Experiment (LTEE) is now in its fourth decade. For a microbial evolution experiment of any duration, it is critical to maintain a reproducible environment, avoid contamination, archive samples, and accurately measure fitness. The LTEE demonstrates several time-tested strategies for achieving these objectives, including the use of well-shaken flasks that create a homogenous environment and a chemically defined growth medium that supports a low cell density. Moreover, the LTEE employs ancestor strains that differ in a genetic marker that gives a phenotype (colony color) that is both easily screened and selectively neutral in the evolution environment. This experimental design feature provides a means of identifying internal and external contamination and facilitates measuring fitness. However, not all of the procedures and safeguards that have been used by the LTEE since 1988 have proven equally robust. Some methods that were reliable when the LTEE began have become less effective as the E. coli populations have evolved. Fortunately, these problematic methods can now be augmented or replaced using technologies developed since the experiment's inception.
Detecting contamination
Detection of contamination is critical to the LTEE. Contamination can be of two sorts: between LTEE populations (cross-contamination) and with microbes from the environment (outside contamination). For the most part, careful use of aseptic techniques and close attention during media preparation and the daily transfers prevent both types of contamination, but they do happen. Early in the experiment, plating on TA agar could be used to detect instances of cross-contamination because transfers have always alternated between Ara− and Ara+ populations. The fingerprint of sensitivity and resistance of these E. coli to certain bacteriophages was also intended to be a design feature that could differentiate the LTEE populations from commonly used E. coli laboratory strains that might contaminate them4. However, these genetic markers have become unreliable as the experiment has progressed (e.g., some populations no longer form colonies on TA agar)10,35. Fortunately, the populations have genetically diverged as they have experienced separate evolutionary histories during the experiment, which has created new genetic markers that can now be used to detect cross-contamination. For instance, each population has evolved a unique combination of mutations in the pykF and nadR genes14,36,37. We sometimes PCR amplify and Sanger sequence these two genes to test whether colonies with unusual morphologies or colors are due to cross-contamination. As the costs of whole-genome and whole-population sequencing continue to drop, routine sequencing of the LTEE populations may soon be possible, thereby presenting new opportunities to monitor them for signs of contamination.
Measuring competitive fitness
Another case in which the LTEE has outgrown its original methods is that the fitness of the evolved E. coli has increased in the experimental environment to such a degree that one can no longer directly measure the fitness of the populations of today relative to their ancestors using the protocol described here. The evolved populations outcompete the ancestors to such an extent that few to no ancestor colonies remain to count after a one-day competition. One approach for dealing with this large fitness difference is to use unequal starting ratios of the strains, weighting the initial volumes that are mixed toward the less-fit competitor (e.g., 90 µL ancestor and 10 µL evolved competitor). A second approach is to identify an evolved Ara− clone that has a higher fitness than the LTEE ancestor, isolate a spontaneous Ara+ revertant mutant of it by selection on MA agar, and then verify that the revertant strain has the same fitness as its parent using a competition assay6,38. This new Ara−/Ara+ pair can then be used as a set of common competitor strains in lieu of REL606/REL607. Ideally, the evolved Ara− clone chosen as a common competitor (and its Ara+ revertant) will have intermediate fitness relative to all strains of interest in an experiment. Over the first 50,000 generations of the LTEE, these two approaches (using unequal starting ratios or a common competitor) did not produce meaningfully different fitness measurements versus the typical approach39.
These modifications to the competition protocol make certain simplifying assumptions that may not always be true. One is that fitness measurements are transitive. That is, if we compete two populations each versus a common competitor strain separately, then we can infer the relative fitness of the two populations to one another. This relationship has been found to be true for the LTEE40, for the most part, but it is not for other experiments41. One reason for this discrepancy can be the evolution of negative frequency-dependent fitness effects. This situation occurs when strains isolated from two different diverged lineages from population A−2 of the LTEE are competed against one another19,42. Each has an advantage when rare, due to cross-feeding, which stabilizes their co-existence. Sequencing data showing long-term co-existence of lineages with different sets of mutations suggests that similar interactions may also have arisen in other LTEE populations14,43, though it is not clear whether they are strong enough to noticeably alter fitness estimates. Finally, the evolution of aerobic growth on citrate in population A−3 of the LTEE32 means that the fitness of these cells now incorporates the use of a "private" resource when they are competed against cells that cannot use citrate, which complicates interpreting these results. Despite these exceptions, the use of a low glucose concentration and well-shaken environment has undoubtedly simplified making fitness comparisons between LTEE strains and populations.
At later generations, some of the LTEE populations no longer form colonies on TA agar, which makes performing competition experiments using even modified protocols difficult or impossible10. Alternative methods that do not require colony growth can potentially be used to determine the relative representation of two competitors, such as FREQ-seq which uses next-generation sequencing to count the proportion of reads containing two alternative alleles in an amplicon44. This method or a similar one could potentially be used with the Ara alleles or with newly evolved mutations, such as those in pykF and nadR, versus the ancestral sequence. Performing genetic modifications that introduce other types of neutral markers can also be used to measure relative fitness. For example, fluorescent protein genes have been inserted into the chromosomes of cells in LTEE offshoot experiments so that competitors can be counted using flow cytometry45. Another approach, which opens up the possibility of mixing together more than two strains in the same competition flask, is to insert barcodes that can be PCR amplified and sequenced into the genomes of different competitors. This approach has been used for lineage tracing in evolution experiments46. Both flow cytometry and barcode sequencing can accurately measure much more extreme ratios of two strains versus colony counting (because they can query > 10,000 cells/genomes versus the < 500 that can be counted on an agar plate), so using these methods also promises to increase the dynamic range in terms of fitness differences that can be measured relative to a common competitor.
Alternative designs for long-term microbial evolution experiments
For all its virtues, the LTEE is not perfect. Certain aspects of its design make it labor intensive and susceptible to human error. For instance, each day a researcher must come into lab and pipette between Erlenmeyer flasks to continue the experiment. Competition experiments can also pose daunting logistical hurdles, given that requirements for sterile glassware, media, incubator space, and colony counting rapidly escalate when even a small number of competitors are being tested with modest replication. We are often asked why we don't take advantage of laboratory automation systems, such as pipetting robots that operate on 96-well microplates, or continuous culture systems, such as chemostats or turbidostats. The answer is simple: the LTEE is, in a sense, a prisoner of its own long history. We dare not deviate from 10 mL cultures shaking at a specific speed in 50 mL Erlenmeyer flasks because this would risk fundamentally changing the experiment. Subtle aspects of the environment to which these populations have been adapting for decades (e.g., the amount of aeration), would be altered in microplates or continuous culture systems. The population bottleneck at each transfer might also be different (smaller in microplates, for example), changing the evolutionary dynamics. In short, deviating from the methods described here would make the LTEE a different experiment, or at the very least risk introducing a discontinuity that would disrupt evolutionary trajectories.
Researchers designing new evolution experiments should consider these other ways of propagating microbial populations, while being aware of their potential benefits and drawbacks. Using pipetting robots to transfer populations in microwell plates is logistically simpler in some ways and can prove quite powerful due to the high numbers of replicate populations that can be propagated in this way47,48,49. However, automated transfers in most current setups do not take place under completely sterile conditions, which increases the likelihood of outside contamination. To prevent contamination, the growth medium is often supplemented with antibiotics, which become a feature of the environment that affects evolution. Transfers in microwell plates are also more prone to cross-contamination events. Finally, the environment of microwell plates — particularly if they are not shaken — tends to select for wall growth, aggregation, and other phenomena that can complicate evolution by creating multiple niches in one well. Using rich media or high concentrations of nutrients to keep population sizes large in small wells is likely to exacerbate these complexities. If such interactions arise, they can make measuring and interpreting fitness much more difficult.
Continuous culture systems for microbial evolution include chemostats, in which fresh medium is constantly pumped in and culture is pumped out, and turbidostats, in which cultures are periodically diluted through automated sensing and pumping to maintain cells in a state of constant growth. These systems are very useful when one wants to model microbial physiology and evolution because they avoid having microbes transition between growth and starvation by keeping them in an environment that always has nutrients50. One can even add sensors that make real-time measurements of optical density, O2 consumption, pH, and other aspects of a culture's environment and growth. However, current continuous culture systems either require expensive equipment purchases or specialized expertise to build custom setups51,52,53,54. Also, wall growth, in which cells escape dilution by adhering to the culture chamber, bedevils evolutionary dynamics in continuous culture systems unless they are periodically sterilized. Due to these constraints, most chemostat and turbidostat evolution experiments to date have been of limited duration and/or involved relatively few independently evolving populations compared to serial transfer evolution experiments.
Conclusion
The methods we demonstrate here for the LTEE are critical for studying its unique historical record and continuing the open-ended evolution of these E. coli populations. They also provide a starting point for others who are considering new evolution experiments that may take advantage of laboratory automation or add back various elements of the complexity found in natural environments that were purposefully omitted from the LTEE. Since 1988, experimental evolution has flourished as a field. During this time, researchers in laboratories across the globe have demonstrated the immense flexibility of this approach for studying evolution, innovating by introducing creative experimental designs and monitoring the results using new technologies. The methods of the LTEE do not represent an endpoint, but we hope they will continue to inspire and provide a foundation for the field far into the future.
Disclosures
No conflicts of interest declared.
Acknowledgements
We thank Richard Lenski and the many researchers who have studied and contributed to maintaining the Long-Term Evolution Experiment with E. coli, including especially Neerja Hajela. The LTEE is currently supported by the National Science Foundation (DEB-1951307).
Materials
| Name | Company | Catalog Number | Comments |
| 2,3,5-Triphenyltetrazolium chloride (TTC) | Sigma-Aldrich | T8877 | |
| 20 mL Glass Beaker | Sigma-Aldrich | CLS100020 | |
| 50 mL Erlenmeyer Flasks | Sigma-Aldrich | CLS498050 | |
| Agar | Sigma-Aldrich | A1296 | |
| Ammonium Sulfate | Sigma-Aldrich | AX1385 | |
| Antifoam | Sigma-Aldrich | A5757 | |
| Arabinose | Sigma-Aldrich | A3256 | |
| Freezer Box (2") | VWR | 82007-142 | |
| Freezer Box (3") | VWR | 82007-144 | |
| Freezer Box Cell Divider (49-place) | VWR | 82007-150 | |
| Freezer Box Cell Divider (81-place) | VWR | 82007-154 | |
| Freezer Vials (1/2-Dram) | VWR | 66009-816 | |
| Freezer Vials (2-Dram) | VWR | 66010-560 | |
| Glucose | Sigma-Aldrich | G8270 | |
| Glycerol | Fisher Scientific | G33 | |
| Magnesium Sulfate | Sigma-Aldrich | M7506 | |
| Metal Tray | Winco | SPJP-202 | |
| Petri Dish | Fisher Scientific | FB0875712 | |
| Potassium Phosphate Dibasic Trihydrate | Sigma-Aldrich | P5504 | |
| Potassium Phosphate Monobasic | Sigma-Aldrich | P5379 | |
| Sodium Chloride | Sigma-Aldrich | M7506 | |
| Sodium Citrate Tribasic Dihydrate | Sigma-Aldrich | C7254 | |
| Test Tube Cap (18mm) | VWR | 10200-142 | |
| Test Tube Rack (18mm, steel) | Adamas-Beta | N/A | Test Tube Racks Stainless Steel Grid Arrangement 72 Holes (17-19 mm) |
| Test Tubes (18 x 150 mm) | VWR | 47729-583 | |
| Thiamine, Hydrochloride | Millipore | 5871 | |
| Tryptone | Gibco | 211705 | |
| Yeast Extract | Gibco | 212750 |
References
- Lenski, R. E., Rose, M. R., Simpson, S. C., Tadler, S. C. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. The American Naturalist. 138 (6), 1315-1341 (1991).
- Fox, J. W., Lenski, R. E. From here to eternity-the theory and practice of a really long experiment. PLoS Biology. 13 (6), e1002185(2015).
- Daegelen, P., Studier, F. W., Lenski, R. E., Cure, S., Kim, J. F. Tracing ancestors and relatives of Escherichia coli B, and the derivation of B strains REL606 and BL21(DE3). Journal of Molecular Biology. 394 (4), 634-643 (2009).
- Studier, F. W., Daegelen, P., Lenski, R. E., Maslov, S., Kim, J. F. Understanding the differences between genome sequences of Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. Journal of Molecular Biology. 394 (4), 653-680 (2009).
- Barrick, J. E., Lenski, R. E. Genome dynamics during experimental evolution. Nature Reviews Genetics. 14 (12), 827-839 (2013).
- Lenski, R. E. Experimental studies of pleiotropy and epistasis in Escherichia coli. II. Compensation for maladaptive pleiotropic effects associated with resistance to virus T4. Evolution. 42 (3), 425-432 (1988).
- Calcott, P. H., Gargett, A. M. Mutagenicity of freezing and thawing. FEMS Microbiology Letters. 10 (2), 151-155 (1981).
- Lenski, R. E., Travisano, M. Dynamics of adaptation and diversification: a 10,000-generation experiment with bacterial populations. Proceedings of the National Academy of Sciences of the United States of America. 91 (15), 6808-6814 (1994).
- Wiser, M. J., Ribeck, N., Lenski, R. E. Long-term dynamics of adaptation in asexual populations. Science. 342 (6164), New York, N.Y. 1364-1367 (2013).
- Lenski, R. E., et al. Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with Escherichia coli. Proceedings of the Royal Society B: Biological Sciences. 282 (1821), 20152292(2015).
- Barrick, J. E., et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 461 (7268), 1243-1247 (2009).
- Blount, Z. D., Barrick, J. E., Davidson, C. J., Lenski, R. E. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature. 489 (7417), 513-518 (2012).
- Tenaillon, O., et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature. 536 (7615), 165-170 (2016).
- Good, B. H., McDonald, M. J., Barrick, J. E., Lenski, R. E., Desai, M. M. The dynamics of molecular evolution over 60,000 generations. Nature. 551 (7678), 45-50 (2017).
- Consuegra, J., et al. Insertion-sequence-mediated mutations both promote and constrain evolvability during a long-term experiment with bacteria. Nature Communications. 12 (1), 980-980 (2021).
- Cooper, T. F., Rozen, D. E., Lenski, R. E. Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 100 (3), 1072-1077 (2003).
- Favate, J. S., Liang, S., Cope, A. L., Yadavalli, S. S., Shah, P. The landscape of transcriptional and translational changes over 22 years of bacterial adaptation. eLife. 11, e81979(2022).
- Khan, A. I., Dinh, D. M., Schneider, D., Lenski, R. E., Cooper, T. F. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 332 (6034), 1193-1196 (2011).
- Plucain, J., et al. Epistasis and allele specificity in the emergence of a stable polymorphism in Escherichia coli. Science. 343 (6177), 1366-1369 (2014).
- Quandt, E. M., Deatherage, D. E., Ellington, A. D., Georgiou, G., Barrick, J. E. Recursive genomewide recombination and sequencing reveals a key refinement step in the evolution of a metabolic innovation in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 111 (6), 2217-2222 (2014).
- Leon, D., D'Alton, S., Quandt, E. M., Barrick, J. E. Innovation in an E. coli evolution experiment is contingent on maintaining adaptive potential until competition subsides. PLoS Genetics. 14 (4), e1007348(2018).
- Bennett, A. F., Lenski, R. E., Mittler, J. E. Evolutionary adaptation to temperature. I. Fitness responses of Escherichia coli to changes in its thermal environment. Evolution. 46 (1), 16-30 (1992).
- Kibota, T. T., Lynch, M. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature. 381 (6584), 694-696 (1996).
- Friesen, M. L., Saxer, G., Travisano, M., Doebeli, M. Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution. 58 (2), 245-260 (2004).
- Cooper, T. F. Recombination speeds adaptation by reducing competition between beneficial mutations in populations of Escherichia coli. PLoS Biology. 5 (9), e225(2007).
- Cooper, T. F., Lenski, R. E. Experimental evolution with E. coli in diverse resource environments. I. Fluctuating environments promote divergence of replicate populations. BMC Evolutionary Biology. 10, 11(2010).
- Quan, S., et al. Adaptive evolution of the lactose utilization network in experimentally evolved populations of Escherichia coli. PLoS Genetics. 8 (1), e1002444(2012).
- Deatherage, D. E., Kepner, J. L., Bennett, A. F., Lenski, R. E., Barrick, J. E. Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures. Proceedings of the National Academy of Sciences of the United States of America. 114 (10), E1904-E1912 (2017).
- Izutsu, M., Lake, D. M., Matson, Z. W. D., Dodson, J. P., Lenski, R. E. Effects of periodic bottlenecks on the dynamics of adaptive evolution in microbial populations. BioRixv. , 4457(2021).
- Chavarria-Palma, J. E., Blount, Z. D., Barrick, J. E. LTEE Media Recipes. , (2022).
- Barrick, J. E., Lake, D. M. fitnessR: fitnessR-v1.0.0. barricklab. , (2023).
- Blount, Z. D., Borland, C. Z., Lenski, R. E. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 105 (23), 7899-7906 (2008).
- Grant, N. A., Magid, A. A., Franklin, J., Dufour, Y., Lenski, R. E. Changes in cell size and shape during 50,000 generations of experimental evolution with Escherichia coli. Journal of Bacteriology. 203 (10), 22(2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. , Springer-Verlag. New York. (2016).
- Meyer, J. R., et al. Parallel changes in host resistance to viral infection during 45,000 generations of relaxed selection. Evolution. 64 (10), 3024-3034 (2010).
- Woods, R., Schneider, D., Winkworth, C. L., Riley, M. A., Lenski, R. E. Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 103 (24), 9107-9712 (2006).
- Barrick, J. E., Deatherage, D. E., D'Alton, S. LTEE-Ecoli: genomics resources for the Long-Term Evolution Experiment with Escherichia coli. , Available from: https://github.com/barricklab/LTEE-Ecoli (2022).
- Izutsu, M., Lenski, R. E. Experimental test of the contributions of initial variation and new mutations to adaptive evolution in a novel environment. Frontiers in Ecology and Evolution. 10, 958406(2022).
- Wiser, M. J., Lenski, R. E. A comparison of methods to measure fitness in Escherichia coli. PLoS One. 10 (5), 0126210(2015).
- de Visser, J. A. G. M., Lenski, R. E. Long-term experimental evolution in Escherichia coli. XI. Rejection of non-transitive interactions as cause of declining rate of adaptation. BMC Evolutionary Biology. 2 (1), 19(2002).
- Paquin, C. E., Adams, J. Relative fitness can decrease in evolving asexual populations of S. cerevisiae. Nature. 306 (5941), 368-371 (1983).
- Rozen, D. E., Lenski, R. E. Long-Term Experimental Evolution in Escherichia coli. VIII. Dynamics of a balanced polymorphism. The American Naturalist. 155 (1), 24-35 (2000).
- Quandt, E. M., Gollihar, J., Blount, Z. D., Ellington, A. D., Georgiou, G., Barrick, J. E. Fine-tuning citrate synthase flux potentiates and refines metabolic innovation in the Lenski evolution experiment. eLife. 4, e09696(2015).
- Chubiz, L. M., Lee, M. -C., Delaney, N. F., Marx, C. J. FREQ-Seq: a rapid, cost-effective, sequencing-based method to determine allele frequencies directly from mixed populations. PLoS One. 7 (10), e47959(2012).
- Gallet, R., Cooper, T. F., Elena, S. F., Lenormand, T. Measuring selection coefficients below 10-3: method, questions, and prospects. Genetics. 190 (1), 175-186 (2012).
- Levy, S. F., et al. Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature. 519 (7542), 181-186 (2015).
- Lang, G. I., Botstein, D., Desai, M. M. Genetic variation and the fate of beneficial mutations in asexual populations. Genetics. 188 (3), 647-661 (2011).
- Frenkel, E. M., et al. Crowded growth leads to the spontaneous evolution of semistable coexistence in laboratory yeast populations. Proceedings of the National Academy of Sciences. 112 (36), 11306-11311 (2015).
- Jordt, H., et al. Coevolution of host-plasmid pairs facilitates the emergence of novel multidrug resistance. Nature Ecology and Evolution. 4 (6), 863-869 (2020).
- Gresham, D., Dunham, M. J. The enduring utility of continuous culturing in experimental evolution. Genomics. 104 (6), 399-405 (2014).
- Miller, A. W., Befort, C., Kerr, E. O., Dunham, M. J. Design and use of multiplexed chemostat arrays). Journal of Visualized Experiments. (72), e50262(2013).
- Toprak, E., et al. Building a morbidostat: an automated continuous-culture device for studying bacterial drug resistance under dynamically sustained drug inhibition. Nature Protocols. 8 (3), 555-567 (2013).
- Wong, B. G., Mancuso, C. P., Kiriakov, S., Bashor, C. J., Khalil, A. S. Precise, automated control of conditions for high-throughput growth of yeast and bacteria with eVOLVER. Nature Biotechnology. 36 (7), 614-623 (2018).
- Ekkers, D. M., Branco Dos Santos, F., Mallon, C. A., Bruggeman, F., Van Doorn, G. S. The omnistat: A flexible continuous-culture system for prolonged experimental evolution. Methods in Ecology and Evolution. 11 (8), 932-942 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved