
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
توليد كريسبر / Cas9 ساطة Monoallelic الحذف لدراسة وظيفة محسن في الخلايا الجذعية الجنينية الماوس
In This Article
Summary
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Abstract
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introduction
العناصر التنظيمية النسخي حاسمة لضبط المكانية والزمانية في التعبير الجيني خلال تنمية (1) وتعديل هذه العناصر يمكن أن يؤدي إلى المرض بسبب التعبير الجيني الشاذة 2. العديد من المناطق الأمراض المرتبطة حددها الجينوم الدراسات جمعية واسعة هي في مناطق غير الترميز ولها ميزات معززات النسخي 3-4. تحديد القدرة ومضاهاتها مع الجينات التي تنظم معقدة لأنها غالبا ما تقع عدة kilobases بعيدا عن الجينات التي تنظم ويمكن تفعيلها بطريقة الأنسجة محددة 5-6. وتستند توقعات محسن عادة على علامات هيستون تعديل، مجمعات للوسيط cohesin وملزمة من نوع معين الخلية عوامل النسخ 7-10. وغالبا ما يتم التحقق من القدرة على التنبؤ من خلال فحص ناقلات القائم فيها محسن ينشط التعبير عن الجينات مراسل 11-12. وتوفر هذه البيانات الخامسمعلومات aluable حول إمكانية التنظيمية للتسلسل محسن المفترضة ولكن لا تكشف عن وظيفتها في سياق الجيني الذاتية أو التعرف على الجينات التي تنظم. يخدم تحرير الجينوم كأداة قوية لدراسة وظيفة العناصر التنظيمية النسخي في سياقها الذاتية عن طريق تحليل الخسارة من وظيفة.
التطورات الحديثة في تحرير الجينوم، وهما / Cas9 تحرير النظام الجينوم كريسبر، تسهيل التحقيق في وظيفة الجينوم. النظام / Cas9 كريسبر سهلة الاستخدام وقابلة للتكيف لكثير من النظم البيولوجية. ويستهدف هذا البروتين Cas9 إلى موقع معين في الجينوم عن طريق الحمض النووي الريبي دليل (gRNA) 13. مجمع SpCas9 / gRNA بفحص جينوم للتسلسل الجيني هدفه الذي يجب أن يكون 5 'إلى protospacer عزر المجاور (PAM) تسلسل، NGG 14-15. الاقتران قاعدة gRNA إلى هدفه، 20 النوكليوتيدات (الإقليم الشمالي) تسلسل مكملة لgRNA، ينشط النشاط نوكلياز SpCas9 مما أدى إلى عزل مزدوكسر ه حبلا (DSB) 3 بي بي المنبع من سلسلة حزب الأصالة والمعاصرة. ويتحقق خصوصية من خلال قاعدة الاقتران الكامل في المنطقة المصنفة gRNA، 6-12 الإقليم الشمالي المحاذي لحزب الأصالة والمعاصرة. على العكس، عدم التطابق 5 'من البذور وعادة ما يتم التغاضي 16-17. جهاز تسوية المنازعات قدم يمكن إصلاحه إما بنهاية غير المتجانسة الانضمام (NHEJ) إصلاح الحمض النووي أو التماثل الموجه إصلاح (HDR) mechanisms.NHEJ إصلاح الحمض النووي غالبا ما يخلق الإدراج / حذف (indels) من عدد قليل من الغليان في الموقع المستهدف التي يمكن أن تعطل في إطار مفتوح للقراءة (ORF) من الجين. لتوليد الحذف أكبر في الجينوم اثنين gRNAs، الذي تطويق المنطقة من اهتمام، ويمكن استخدامها 18-19. هذا النهج هو مفيدة بشكل خاص لدراسة معززات النسخي تتجمع في مناطق سيطرة موضع أو القدرة الفائقة التي هي أكبر من القدرة التقليدية 9،18،20-22.
الحذف Monoallelic هي نموذجا قيما لدراسة -regulation رابطة الدول المستقلة من النسخ. تشانغ لاحظه في مستوى النص بعد حذف monoallelic من محسن يرتبط إلى دور تلك محسن في تنظيم الجينات دون آثار الخلط التي يمكن أن تحدث عندما يتأثر النسخ من كل من الأليلات المحتمل أن تؤثر على اللياقة البدنية الخلوية. تقييم خفض التعبير من الصعب ولكن من دون القدرة على التمييز بين حذفها من نوع أليل البرية. وعلاوة على ذلك، التنميط الجيني الحذف في كل أليل دون القدرة على التمييز بين الاليلين يشكل تحديا، خاصة بالنسبة للالحذف كبيرة من> 10 كيلو بايت إلى 1 ميغابايت 23 التي من الصعب تضخيم كامل المنطقة نوع البرية من قبل PCR. استخدام الخلايا F1 ES الناتجة عن عبور المصحف العضلة 129 مع المصحف castaneus يسمح الاليلين لتكون متباينة من جانب أليل محددة PCR 18،24. الجينوم الهجين في هذه الخلايا يسهل أليل معين فحص الحذف وتحليل التعبير. في المتوسط هناك SNP كل 125 سنة مضت بين هذه الجينومات اثنين، وتوفير المرونة في التصميم التمهيدي للتعبير والتنميط الجيني يحلل. وجود واحد SNP يمكن أن تؤثر على درجة حرارة التمهيدي ذوبان (T م)، وتستهدف التحديد في الوقت الحقيقي الكمي التضخيم PCR (QPCR) مما يسمح للتمييز من اثنين من الأليلات 25. وعلاوة على ذلك عدم تطابق في نهاية 3 'من التمهيدي يؤثر بشكل كبير على قدرة البلمرة DNA لتمتد من التمهيدي منع التضخيم من الهدف الأليل غير المرغوب فيه (26). هو موضح في البروتوكول التالي هو استخدام الخلايا F1 ES لأليل معين الحذف محسن أكبر من 1 كيلو بايت وتحليل التعبير لاحق باستخدام / Cas9 تحرير النظام الجينوم كريسبر (الشكل 1).

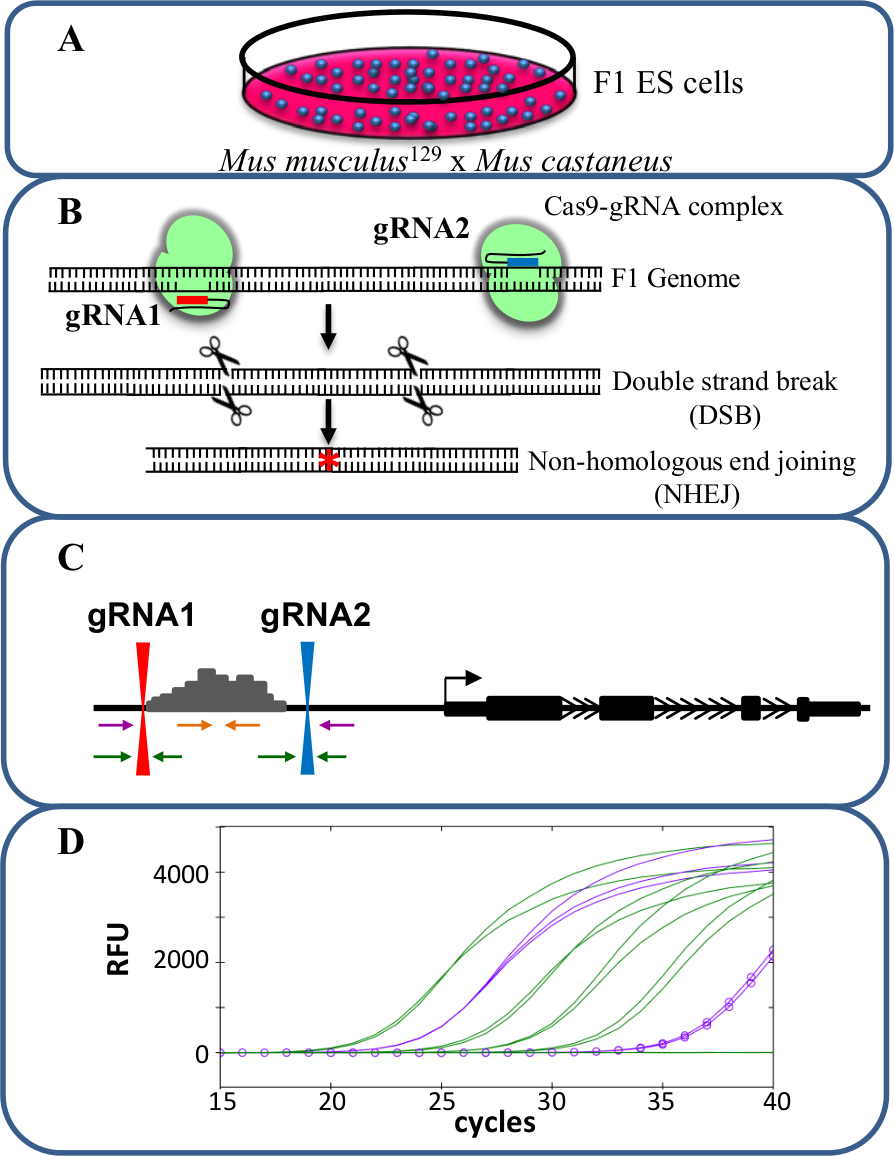
الشكل 1. محسن الحذف باستخدام كريسبر / Cas9 لدراسة رابطة الدول المستقلة -regوتستخدم ulation التعبير الجيني. (أ) الخلايا F1 ES الناتجة عن خليط بين المصحف العضلة 129 و المصحف castaneus للسماح للأليل حذف معين. (ب) اثنين من الرنا دليل (gRNA) وتستخدم للحث على الحذف واسع بوساطة Cas9 للمنطقة محسن. وتستخدم (C) مجموعات التمهيدي لتحديد أحادية كبيرة والحذف ثنائية أليلية. الاشعال البرتقال هي الاشعال الداخل، الاشعال الأرجواني هي الاشعال الخارجية والاشعال الخضراء والاشعال المرافقة gRNA. ويتم رصد (D) التغيرات في التعبير الجيني باستخدام-أليل معين QPCR. RFU يدل على وحدات مضان النسبية. الرجاء انقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocol
1. تصميم وبناء وgRNA
- لحذف المناطق محسن النسخي استخدام اثنين gRNAs، واحد 5 "واحدة 3" في المنطقة من الفائدة. استخدام الماوس UCSC متصفح الجينوم المسار الناتجة عن المختبر تشانغ لتحديد تسلسل gRNA فريدة (http://www.genome-engineering.org 15). التالي التحقق من هذه gRNAs وحزب الأصالة والمعاصرة المجاورة من أجل تعدد الأشكال وindels باستخدام أدوات الإنترنت التي تقدمها معهد سانجر (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. لاستهداف كل من الأليلات مع كفاءة متساوية، وتجنب gRNA متواليات / حزب الأصالة والمعاصرة التي تحتوي على SNP أو INDEL.
- في حين ان اختيار gRNA، تحقق من جدوى تصميم الاشعال-أليل معين لالتنميط الجيني الحذف. الرجوع إلى المادة 5 لتصميم التمهيدي أليل معين.
- تجميع اثنين من البلازميدات gRNA على أساس بروتوكول وصفها في مالي وآخرون. 2013 15. دمج المحدد فريد 20 نقطة أساس تسلسل الهدف كثافة العملياتس أليغنوكليوتيد 61mer كما هو مبين في الجدول 7 (يتم عرض تسلسل في 5 'إلى 3' التوجه، وقواعد الغامق هي الهدف تسلسل 20 شركة بريتيش بتروليوم التي يكمل عكس بعضها البعض).
- مزيج 10 ميكرولتر من 10 ميكرومتر gRNA Primer_F و 10 ميكرولتر من 10 ميكرومتر Primer_R مكملة في أنبوب.
- يصلب الاشعال التي يحتضنها مزيج التمهيدي في 100 درجة مئوية لمدة 5 دقائق ثم تبرد 1 ° C / ثانية إلى 25 درجة مئوية. لهذه الخطوة، واستخدام آلة PCR أو وضع أنبوب في الماء المغلي واتركه ليبرد إلى RT.
- إلى مزيج التمهيدي صلب، إضافة مزيج التفاعل التالي واحتضان عند 72 درجة مئوية لمدة 30 دقيقة لتوسيع كل التمهيدي: 18.5 ميكرولتر من الماء، و 10 ميكرولتر من العازلة 5X HF، 1 ميكرولتر من 10 ملي dNTP مزيج 0.5 ميكرولتر من عالية الإخلاص الحمض النووي بوليميريز.
- تشغيل 10 ميكرولتر من جزء الهدف على هلام الاغاروز 2٪ إلى تأكيد وقد تم إنتاج 100 شظايا دليل مضت.
- خطي ناقلات gRNA (هدية من جيوالكنيسة RGE. Addgene البلازميد # 41824) 15 مع أفل الثاني باستخدام التفاعل التالي إعداد: 5 ميكرولتر من gRNA ناقلات العمود الفقري (2-4 ميكروغرام)، 5 ميكرولتر من 10X العازلة، 3 ميكرولتر من AFL الثاني (20 وحدة / ميكرولتر) و 32 ميكرولتر من الماء. احتضان مزيج رد فعل لمدة 3 ساعة على 37 درجة مئوية.
- تشغيل المنتج الهضم على هلام الاغاروز 1٪ وتنقية الفرقة الحمض النووي المقابلة لكيلوبايت 3.5 خطي ناقلات gRNA استخدام عدة استخراج الهلام باتباع إرشادات الشركة المصنعة.
- إعداد جيبسون ردود الفعل التجمع 29-30 باستخدام ناقل gRNA خطي واستهداف جزء من الخطوة 1.2.3 على النحو التالي: 1 ميكرولتر من الخطي الموجه gRNA (50 نانوغرام / ميكرولتر)، 1 ميكرولتر من جزء الهدف، 10 ميكرولتر من 2X جيبسون مزيج الرئيسي التجمع و8 ميكرولتر من المياه. احتضان ردود الفعل عند 50 درجة مئوية لمدة 60 دقيقة.
- تحويل خلايا القولونية مع تجميعها gRNA المتجهات.
- مزيج 1 ميكرولتر من ناقلات gRNA تجميعها من 1.2.7و50 ميكرولتر من DH5α (القولونية سلالة) خلايا في أنبوب. تحويل الخلايا DH5α بطريقة الصدمة الحرارية من خلال تعريض الخلايا إلى 42 درجة مئوية لمدة 45 ثانية.
- التقط البرد أنابيب على الجليد لمدة 5 دقائق. ثم إضافة 400 ميكرولتر من المتوسطة SOC واحتضان عند 37 درجة مئوية لمدة 45 دقيقة في حاضنة تهتز.
- نشر 100 ميكرولتر من الخلايا DH5α على لوحة لاختيار إيجابية من الخلايا تحولت إلى LB-كانامايسين (50 ميكروغرام / مل)، واحتضان O / N عند 37 درجة مئوية.
- فحص إيجابي القولونية المستعمرات لgRNA إدراج.
- اختيار كانامايسين مستعمرة المقاومة وresuspend في 3 مل LB يحتوي على 50 ميكروغرام / مل من الكانامايسين. كرر الشيء نفسه بالنسبة 6-8 المستعمرات واحتضان جميع الأنابيب عند 37 درجة CO / N في حاضنة تهتز.
- استخراج البلازميدات من O / N ثقافة نمت باستخدام بلازميد عدة الإعدادية مصغرة باتباع دليل الشركة المصنعة.
- إعداد مزيج رد فعل الهضم ECOR أنا للتحقق من gRNA تسلسل إدراجفي البلازميد. لكل عينة، وإعداد مزيج رد فعل على النحو التالي: 2 ميكرولتر من العازلة انزيم، 1 ميكرولتر من ECOR الأول، 15 ميكرولتر من الماء. قسامة مزيج التفاعل إلى 1.5 مل أنابيب وإضافة 2 ميكرولتر من البلازميد. احتضان الأنابيب عند 37 درجة مئوية لمدة 2 ساعة.
- تشغيل المنتج الهضم على هلام الاغاروز 1.5٪.
ملاحظة: إن العينات مع إدراج عرض حجم الفرقة 475 الغليان الذي هو 100 نقطة أساس أعلى من الحيوانات المستنسخة دون تدرج.
ملاحظة: بدلا من ذلك، استنساخ إيجابية يمكن فرزهم من قبل مستعمرة PCR باستخدام SP6 (إلى الأمام) وT7 (عكس) الاشعال (الجدول 7) التي تربط لتسلسل ناقلات لإعطاء حجم قطعة 642 نقطة أساس في وجود gRNA إدراج. النهج PCR مستعمرة هو مفيد عندما يكون هناك موقع ECOR أنا تقييد ضمن تسلسل gRNA.
- تأكيد تسلسل من gRNA إدراج من تسلسل الحمض النووي باستخدام T7 التمهيدي.
2. ترنسفكأيشن
ملاحظة:Electroporation للهو وسيلة فعالة لtransfecting البلازميدات الى خلايا ES. الطريقة الموصوفة هنا يستخدم التكنولوجيا microporator ترنسفكأيشن.
- زراعة خلايا F1 ES في صحن 10 سم المغلفة الجيلاتين تحتوي على 10 مل من وسائل الاعلام الخلية ES (الجدول 1) عند 37 درجة مئوية / 5٪ CO 2. عندما تصل الخلايا 85٪ confluency إزالة وسائل الإعلام وإضافة 2 مل من التربسين. احتضان عند 37 درجة مئوية في حاضنة CO 2 لمدة 5 دقائق.
ملاحظة: تم الحصول على خلايا F1 ES من باربرا الطبخ 24 ومتوفرة عند الطلب. - تحييد التربسين بإضافة 10 مل من وسائل الاعلام تدور (الجدول 2). ماصة مرارا وتكرارا لفصل الخلايا تماما.
- جمع كل الخلايا في أنبوب 15 مل وتدور في 300 x ج لمدة 5 دقائق. resuspend في 3 مل برنامج تلفزيوني وعد الخلايا باستخدام عدادة الكريات أو آلية مكافحة الخلايا.
- بيليه 1 × 10 6 ES خلايا في أنبوب 1.5 مل بواسطة الطرد المركزي في 300 x ج لمدة 5 دقائق و resuspend في 100 ميكرولتر من R (إعادة تعليق) العازلة كما تم توفيره من قبل الشركة المصنعة عدة.
- إضافة 5 ميكروغرام لكل من pCas9_GFP (هدية من كيران Musunuru، Addgene البلازميد # 44719) 31، 5 'و 3' البلازميدات gRNA للحذف من المنطقة المستهدفة وتخلط بلطف مع ماصة لتجنب إدخال الفقاعات.
- استخدام غيض ماصة الالكترونية لنضح 100 ميكرولتر من مزيج Electroporation لل، والحرص على تجنب حدوث فقاعة في الطرف.
- برنامج فولت، عرض والبقول ل electroporation. لخلايا F1 ES، استخدم 1400 V، و 10 مللي ثانية لمدة 3 البقول.
- أثناء تشغيل Electroporation للمراقبة طرف لمشاهدة اي الشرر في الحل. تشير شرارة وجود فقاعة الهواء وسوف تتداخل مع ترنسفكأيشن.
- إخراج خلايا ES transfected في صحن 10 سم المغلفة الجيلاتين تحتوي على 10 مل وفاق وسائل الاعلام الخلية (الجدول 1)، واحتضان عند 37 درجة مئوية / 5٪ CO 2.
3. نظام مراقبة الأصول الميدانية الفرز خلايا Transfected
- بعد 48 ساعة، فصل الخلايا عن طريق إضافة 2 مل من التربسين واحتضان عند 37 درجة مئوية في حاضنة CO 2 لمدة 5 دقائق.
- تحييد لوحة بإضافة 10 مل من العازلة جمع (الجدول 3). جمع الخلايا في أنبوب 15 مل وتدور في 300 x ج لمدة 5 دقائق.
- تجاهل وطاف resuspend الخلايا في 1 مل من فرز عازلة (الجدول 4). عد الخلايا وتمييع التي تعتمد على منصة الفرز. تمييع الخلايا إلى 0.5-1 × 10 6 خلية / مل للفرز إلى 15 مل الأنابيب ولفرز الخلايا الفردية مباشرة إلى لوحات 96-جيدا، وتمييع الخلايا إلى 2-5 × 10 6 خلية / مل.
- نوع الخلايا Cas9-GFP + ES باستخدام تدفق FACS الكريات 32. جمع الخلايا بكميات كبيرة في الأنابيب مع 2 مل وسائط استرداد (الجدول 5) وصفيحة كما هو موضح في 3.5 لقطف مستعمرة، أو الخلايا الفردية الفرز مباشرة إلى لوحات 96-جيدا المغلفة الجيلاتين تحتوي على 100 ميكرولتر وسائل الاعلام الخلية ES / جيد ( الجدول 1).
- بذور 1-1،5 × 10 4 GFP + ES الخلايا في صحن 10 سم المغلفة الجيلاتين تحتوي على 10 وسائل الإعلام خلية مل ES (الجدول 1). والطلاء في هذه الكثافة المنخفضة تسهيل اختيار الفردية مستعمرات الخلايا ES.
4. التثقيف النسخ لالجيني، تحليل التعبير والأرصدة زنزانة شديدة البرودة
- في يوم 4-5 بعد الفرز، ويسجل كل بئر من لوحات 96-جيدا فرز المباشرة عن وجود مستعمرات الخلايا ES.
- فصل المستعمرات خلية ES عن طريق إزالة وسائل الإعلام وإضافة 30 ميكرولتر من التربسين. احتضان عند 37 درجة مئوية لمدة 5 دقائق. تحييد التربسين بإضافة 170 ميكرولتر من وسائل الاعلام الخلية ES (الجدول 1)، وماصة صعودا وهبوطا لالتفكك الكامل للمستعمرة في الخلايا وحيدة. تنمو الخلايا عند 37 درجة مئوية / 5٪ CO 2 حتى معظم الآبار هي أكثر من 70٪ متموجة (عادة 2-3 أيام).
- بدلا من اختيار الفرد المستعمرات خلية ES من 10 سم أطباق باستخداممجهر مقلوب. بعد الشفط مستعمرة في متابعة خطوة ماصة 4.1.1 وضع كل مستعمرة في بئر واحدة من لوحة 96-جيدا، سابقة التجهيز مع الجيلاتين وتحتوي على 30 ميكرولتر من التربسين.
ملاحظة: يمكن أن المستعمرات الجلوس في التربسين في RT بينما يتم اختيار واحد الصف بأكمله من المستعمرات. - مرة واحدة وقد تم القبض على جميع المستعمرات وفصلها إلى وسائل الإعلام تنمو الخلايا عند 37 درجة مئوية في حاضنة CO 2 حتى معظم الآبار هي أكثر من 70٪ متموجة (عادة 2 أيام).
- عندما لوحات 96-جيدا جاهزة للتقسيم، وإزالة وسائل الإعلام، إضافة 30 ميكرولتر من التربسين واحتضان عند 37 درجة مئوية لمدة 5 دقائق. تحييد التربسين بإضافة 180 ميكرولتر من وسائل الاعلام الخلية ES (الجدول 1) إلى كل بئر وماصة صعودا وهبوطا لالتفكك الكامل إلى الخلايا وحيدة.
- من الناتج 210 ميكرولتر، البذور 70 ميكرولتر إلى ثلاثة الجيلاتين المغلفة 96-جيدا لوحات تحتوي كل منها على 130 ميكرولتر من وفاق وسائل الإعلام خلية / جيد (الجدول 1). استخدام هذه اللوحات لشركة جنرال الكتريكnotyping، تحليل التعبير وتجميد الأرصدة خلية لكل استنساخ كما هو موضح أدناه.
- عندما تصل إلى لوحة التنميط الجيني 70-85٪ التقاء، وعلاج لوحة كما هو موضح في قسم 6 "التنميط الجيني حذف".
- عندما تصل إلى لوحة تحليل التعبير 70-85٪ التقاء، وإزالة وسائل الإعلام، وختم لوحة مع ختم الشريط ومخزن في -80 درجة مئوية حتى تم مرمزة الحيوانات المستنسخة.
ملاحظة: لوحة تحليل التعبير هي مفيدة لتحليل التغيرات في التعبير الجيني في المقاطع الأولى من الحيوانات المستنسخة. تحليل التعبير الجيني من لوحة 96-جيدا ممكن ولكن كما أرقام الهواتف المحمولة منخفضة ينصح RNA عدة الاستخراج الجزئي. - إعداد 96-جيدا لوحة تجميد اسهم:
- عندما لوحة للأسهم الخلايا المجمدة (الأسهم-1) تصل 70-85٪ التقاء، نضح في وسائل الإعلام، إضافة 30 ميكرولتر من التربسين واحتضان عند 37 درجة مئوية لمدة 5 دقائق.
- تحييد التربسين بإضافة 100 ميكرولتر من وسائل الاعلام الخلية ES (الجدول 1) رس كل بئر وماصة صعودا وهبوطا لالتفكك الكامل إلى الخلايا وحيدة.
- نقل 15 ميكرولتر من خلايا معلقة من كل بئر لاثنين من الجيلاتين المغلفة لوحات 96-جيدا، وتحتوي كل منها على 185 ميكرولتر من وسائل الاعلام الخلية ES (الجدول 1)، والسماح لينمو بمعدل 37 درجة مئوية / 5٪ CO 2.
ملاحظة: هذا هو لمخزون 2 و -3 اللوحات التي هي إضافية استنساخ احتياطية في حالة إحياء الخلايا من الأوراق المالية-1 ليست ناجحة.
- وفي الوقت نفسه، إلى 100 ميكرولتر من الخلايا المتبقية في لوحة 96-جيدا (الأسهم-1)، إضافة 100 ميكرولتر من 2X تجميد وسائل الإعلام (الجدول 6). ختم لوحة مع الشريط اللاصق وسرعان ما عكس لوحة 4-5 مرات لخلط الصحيح. تخزين لوحة عند -80 درجة مئوية حتى يتم مرمزة الحيوانات المستنسخة.
- عندما لوحات الأسهم-2 والأوراق المالية 3 على استعداد لتجميد نضح في وسائل الإعلام، إضافة 30 ميكرولتر من التربسين واحتضان عند 37 درجة مئوية لمدة 5 دقائق. تحييد التربسين بإضافة 70 ميكرولتر من وسائل الاعلام الخلية ES (Tقادرة 1) إلى كل بئر وماصة صعودا وهبوطا لالتفكك الكامل إلى الخلايا وحيدة.
- إضافة 100 ميكرولتر من 2X وسائل الإعلام التجمد، وختم لوحة مع ختم الشريط، وسرعان ما عكس لوحة 4-5 مرات لخلط الصحيح. تخزين لوحة عند -80 درجة مئوية حتى هناك حاجة لهذه اللوحات.
التصميم التمهيدي 5. أليل محددة
- تصميم 4 مجموعات من الاشعال (الشكل 1C) لفحص الحيوانات المستنسخة لحذف المطلوب: داخل الاشعال، الاشعال الخارجية، وgRNA المرافقة الاشعال (لكل 5 "و 3" المواقع المستهدفة gRNA) كما هو موضح أدناه.
- الحصول على المسار SNP المقابلة ل129 و المصبوب المورثات في http://labs.csb.utoronto.ca/mitchell/crispr.html. يظهر مسار معين بدائل قاعدة بين 129 و المصبوب في الإحداثيات في التجمع الجينوم MM9 الماوس.
ملاحظة: الرابط في الموقع المذكور أعلاه إعادة توجيه إلى متصفح الجينوم UCSC وإضافة مسار مخصص يحتوي على تعدد الأشكال بين 129 و المصبوب جنرال الكتريكnomes. - أدخل احداثيات المنطقة المراد حذفه. التكبير في منطقة حوالي 500 شركة بريتيش بتروليوم في منتصف حذف المرجوة التي تحتوي على> 3 تعدد الأشكال.
- الذهاب لمشاهدة> الحمض النووي في شريط الخيار وانقر على الحصول على الحمض النووي لتحميل تسلسل الهدف في كل شكل حالة العلوي.
- إنشاء اثنين من سلاسل FASTA. واحدة ل 129 واحد ليلقي بالتعويض قاعدة في موقف الحزب الوطني الاسكتلندي. بمناسبة تعدد الأشكال التي كتبها بأحرف صغيرة.
- الذهاب إلى Primer3 الجمع (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) ولصق SNP استبدال 129 تسلسل. استخدام الإعدادات الافتراضية لتصميم الاشعال.
- لتصميم الاشعال-أليل معين داخل حدد Primer_List في القائمة المنسدلة المهمة، وانقر فوق اختيار الاشعال. اختيار الأمام أو عكس التمهيدي لديه SNP إما في "نهاية أو خلال 4 قواعد من 3 '3 ويسقط داخل المنطقة المراد حذفه.
ملاحظة: الاشعال التي لديها SNP في عرض 3'end زادت أليل خصوصية في QPCR. - لتحديد عودة التمهيدي الثاني إلى الصفحة الرئيسية، حدد الكشف في القائمة المنسدلة مهمة ولصق أول تسلسل التمهيدي في المربع المناسب في أسفل الصفحة. في علامة التبويب إعدادات عامة تغيير الإعداد لمجموعة حجم المنتج إلى 80-200 الغليان وانقر فوق انتقاء الاشعال. اختار التمهيدي مجموعة من أزواج التمهيدي المدرجة. هذه ستكون 129 داخل الاشعال-أليل معين.
- كرر الخطوات من 5.1.5 إلى 5.1.7 لتصميم الاشعال لأليل المصبوب.
- العودة إلى متصفح الجينوم UCSC وأدخل احداثيات المنطقة المراد حذفه. الذهاب لمشاهدة> الحمض النووي في شريط الخيار، ضمن خيارات تسلسل استرجاع منطقة إضافة 1000 سنة مضت المنبع والمصب انقر فوق الحصول على الحمض النووي لتحميل تسلسل الهدف.
- بمناسبة تسلسل الهدف gRNA بين قوسين. حفظ هذا التسلسل بالكامل قبل المتابعة.
- لتصميم الاشعال خارج إزالة تسلسل بين اثنين من متواليات gRNA الهدف. كرر الخطوة 5.1.4-5.1.8 لتصميم الخارجي التمهيدي أليل معينالصورة ولكن تغيير حجم المنتج 400-800 نقطة أساس.
- تقسيم تسلسل حصلت عليه في الخطوة 5.1.10 إلى اثنين من سلاسل، مع كل 500 سنة مضت 5 'و 3' تسلسل الهدف gRNA. كرر الخطوات من 5.1.5 إلى 5.1.7 تصميم غير أليل الاشعال محددة للمناطق المحيطة gRNA لكن تغيير حجم المنتج 400-800 نقطة أساس.
ملاحظة: للحصول على بادئات محددة غير أليل، إما 129 أو تسلسل المصبوب يمكن استخدام وينبغي اختيار الاشعال التي لا تحتوي على الحزب الوطني الاسكتلندي. فإنه من المستحسن لتحديد حجم المنتج من 400-800 نقطة أساس لتصميم خارج وgRNA المرافقة الاشعال. وهذا يسمح لتضخيم حتى لو indels صغيرة موجودة.
- الحصول على المسار SNP المقابلة ل129 و المصبوب المورثات في http://labs.csb.utoronto.ca/mitchell/crispr.html. يظهر مسار معين بدائل قاعدة بين 129 و المصبوب في الإحداثيات في التجمع الجينوم MM9 الماوس.
- اختبار الاشعال داخل لخصوصية أليل من QPCR باستخدام نقية 129 و المصبوب سلالة الحمض النووي الجيني في 2 نانوغرام / ميكرولتر. اتبع الخطوة 6،2-6،4 لإعداد رد فعل QPCR.
ملاحظة: إذا كان النمط الجيني 129 في المنطقة المستهدفة هي نفسها C57BL / 6J، الحمض النووي من C57BL / 6J يمكن استخدامها في مكان من 129 الحمض النووي. يجب عرض الاشعال-أليل معين لا يقل عن 5 دوراتالفرق بين قيمة ط م (عتبة دورة) على الصحيح مقابل النمط الجيني غير صحيحة. ويمكن اختبار الاشعال الخارجية للتأكد من أنهم تضخيم الحذف باستخدام Cas9 / gRNA transfected خلايا ES، والضوابط F1 الحمض النووي الجيني على التوالي الإيجابية والسلبية. يمكن اختبار أليل خصوصية الاشعال خارج مرة واحدة وقد تم التعرف على الحيوانات المستنسخة monoallelic.
6. انماط والحذف
- استخراج الحمض النووي الجيني من التنميط الجيني لوحة 96-جيدا باستخدام لوحة من الخطوة 4.6 الذي تم إنشاؤه بعد التوسع الاستيطاني.
- تحضير مزيج استخراج الحمض النووي الجيني: 89 ميكرولتر من الماء، و 10 ميكرولتر من 10X العازلة و1 ميكرولتر من كاشف استخراج (المزودة من قبل الشركة المصنعة). إضافة 100 ميكرولتر من مزيج استخراج الحمض النووي الجيني إلى كل بئر وختم لوحة مع الشريط اللاصق.
- احتضان لوحة عند 75 درجة مئوية لمدة 5 دقائق تليها 95 درجة مئوية لمدة 5 دقائق.
- تسمح لوحة لتبريد التي يحتضنها على الجليد لعدد قليل من دقيقةد ثم لفترة وجيزة أجهزة الطرد المركزي لتسوية أي التكثيف إلى قاع البئر. وهذا بمثابة لوحة قالب الحمض النووي للكشف عن الحذف.
- إعداد ردود الفعل QPCR من نسختين لكل استنساخ على النحو التالي: 5 ميكرولتر من مزيج 2X SYBR QPCR، إلى الأمام وعكس التمهيدي (3 ميكرومتر) كل 1 ميكرولتر و 1 ميكرولتر من المياه. استخدام ماصة الأقنية إضافة 2 ميكرولتر من الحمض النووي القالب تليها 8 ميكرولتر من مزيج رد فعل على كل بئر من 384 لوحة جيدا.
- ختم لوحة مع ختم الشريط وتدور في 600 x ج لمدة 2 دقيقة لخلط المحتويات. وضع مجموعة 384 لوحة جيدا في cycler على الوقت الحقيقي.
- برنامج cycler على الوقت الحقيقي ل2-خطوة PCR تليها تحليل منحنى تذوب مع الكشف على النحو التالي: 1 دورة في درجة حرارة 95 درجة مئوية لمدة 10 دقيقة، 40 دورات من 95 درجة مئوية لمدة 15 ثانية، 62 درجة مئوية لمدة 30 ثانية مع لوحة قراءة و 95 درجة مئوية لمدة 10 ثانية، 65 درجة مئوية إلى 95 درجة مئوية مع زيادة 5 درجات مئوية لمدة 5 ثانية + لوحة القراءة.
ملاحظة: بالإضافة إلى التمهيدي التصميم، وQPCR ميلتسهم المعلمات x و دورة أيضا التمهيدي خصوصية. المعلمات المذكورة أعلاه والكواشف المدرجة في المواد على نحو أكثر تواترا تسفر عن التضخيم محددة أليل. - تحليل نتائج QPCR
- التحقق من كل أليل لتضخيم مع الاشعال-أليل معين في الداخل. لا التضخيم من أليل واحد أو الاختلافات قيمة ط عالية (> 5 دورات) بين الأليلات تشير هذه الحيوانات المستنسخة تحمل الحذف متخالف من الأليل مع ارتفاع قيمة ط م / غائبة. لا التضخيم من كل من الأليلات يوحي بأنها تحمل الحذف متماثل.
- التحقق من كل أليل لتضخيم مع الاشعال خارج-أليل معين. عندما حذف الهدف أكبر من 1 كيلوبايت تضخيم مع الاشعال خارج يحدث فقط عندما يكون الحذف الحالي. قيمة ط م من 22-28 تؤكد الحذف. لحذف الهدف أصغر من 1 كيلو بايت، تأكد من حجم amplicon من قبل الكهربائي.
ملاحظة: إذا تبين الاشعال الخارجية المعتدلة فقط أليل خصوصية (انظر فيقوإعادة 2)، ويمكن الحصول على amplicons مع كل من مجموعات التمهيدي أليلية في استنساخ monoallelic بسبب بعيدة عن التضخيم الهدف من الاشعال الخارجية. في هذه الحالة فرق قيمة ط م بين اثنين من الأليلات لا تقل عن خمس دورات يجب التأكد من أليل الصحيح يتم حذف (قيمة ط م أقل) على أساس النتائج التي تم الحصول عليها من الاشعال في الداخل. إذا كان على هدف مقابل خارج أليل الهدف ط م الفرق أقل من خمس دورات تصميم أليل جديدة الاشعال خارجية محددة. - في استنساخ حذف monoallelic تحقق من سلامة أليل غير حذف باستخدام فحص الثانوي، gRNA المرافقة الاشعال.
ملاحظة: Indels من> 25 حجم الغليان في جميع أنحاء الموقع المستهدف gRNA يمكن تحديدها من خلال مراقبة تحولا في منحنى تذوب في QPCR عن 400-800 amplicons مضت. بدلا من ذلك، amplicons من الاشعال المرافقة gRNA يمكن أن تكون متسلسلة للكشف عن indels صغيرة من <25 نقطة.- أداء QPCR مع 2 مجموعات من gRNA المرافقة الاشعال أي 5 "و 3" زRNA تستخدم في توليد كريسبر الحذف. لا التضخيم مع هذه المجموعات من الاشعال يشير indels أكبر من amplicon QPCR موجودة في الموقع المستهدف gRNA على أليل غير حذف من الحيوانات المستنسخة حذف monoallelic. استنساخ تجاهل تحتوي على هذه indels كبيرة من مزيد من التحليل إلى نتائج قد يكون من الصعب تفسيرها دون معرفة مدى الحذف.
- تنقية amplicons تم الحصول عليها من الخارج التمهيدي QPCR رد فعل باستخدام PCR تنظيف عدة التالية تعليمات الشركة الصانعة.
- تأكيد تسلسل أليل حذفها من قبل تسلسل الحمض النووي المنتج PCR تنقيته من الخطوة السابقة. استخدام الاشعال التضخيم QPCR لالأمام وعكس التسلسل.
ملاحظة: في هذه المرحلة تعدد الأشكال ضمن قانون amplicon تأكيدا الثانوية من النمط الجيني للأليل حذفها.
7. تحليل التعبير مع أليل الاشعال محددة
- ذوبان الجليد في 96-جيدا رر الأسهم خليةأكلت المخزنة في -80 درجة مئوية (الأسهم 1 من الخطوة 4.9) عن طريق وضعه في حمام دافئ حبة. عندما أكثر من نصف الآبار في لوحة وإذابة، وتدور في 300 x ج لمدة 5 دقائق.
- بعناية، وإزالة الشريط اللاصق وبسرعة نقل الخلايا من حذف الآبار الإيجابية في، لوحات 12-جيدا الجيلاتين المغلفة التي تحتوي على 1 مل من وسائل الاعلام الخلية ES (الجدول 1)، واحتضان عند 37 درجة مئوية / 5٪ CO 2.
- عندما تصل إلى لوحة 70-85٪ التقاء، مرور الخلايا وتقسيمها إلى ثلاثة آبار ل، لوحة 6 جيدا الجيلاتين المغلفة، كل منها يحتوي على 2 مل من وسائل الاعلام الخلية ES (الجدول 1). استخدام اثنين من الآبار لإعداد 2 قارورة من الأسهم خلية المجمدة للتخزين على المدى الطويل في النيتروجين السائل (كما هو موضح في الخطوة 8) والبئر الثالث لاستخراج الحمض النووي الريبي.
- استخراج الحمض النووي الريبي باستخدام طقم استخراج الحمض النووي الريبي.
- تحويل 100-500 نانوغرام من الحمض النووي الريبي إلى [كدنا من النسخ العكسي (RT) الحمض النووي الريبي باستخدام عدة توليف [كدنا التالية بروتوكول الشركة المصنعة. تشمل ن RTرد فعل egative لكل عينة الحمض النووي الريبي لمراقبة كمية تلويث الحمض النووي في عينات الحمض النووي الريبي.
- تمييع [كدنا قبل QPCR في نسبة تتراوح بين 1: 2 و 1: 4؛ تبعا لمستوى التعبير عن الجينات المستهدفة في الخلايا ES.
- تعيين QPCR كما هو موضح أعلاه بما في ذلك F1 الحمض النووي الجيني كما المنحنى القياسي (5 التخفيفات أضعاف 250-،08 نانوغرام / ميكرولتر) لتقدير المطلق من مستويات النص. مقارنة التعبير عن كل أليل من الجينات في المصالح في كل استنساخ حذف أكدت لجين السيطرة مناسبة، على سبيل المثال GAPDH (الاشعال المدرجة في الجدول 7).
ملاحظة: الاشعال الجينات تحكم لا تحتاج إلى أن تكون محددة أليل. ، أليل معين التصميم التمهيدي هو نفسه بالنسبة لالاشعال RT-QPCR كما هو موضح لالاشعال التنميط الجيني باستثناء المنطقة المستهدفة لتضخيم. يجب استخدام التسلسل الجيني. إذا باستخدام بادئات لاكسون واحد أو الحدود اكسون-إنترون (لمراقبة النسخة الأولية) الحمض النووي الجيني F1 يمكن استخدامها لمنحنى القياسية. لمزيد من التفاصيل حول RT-QPCR يرجى الرجوع إلى Forlenza وآخرون. 2012 33.
8. إعداد الأوراق المالية تجميد للتخزين طويل الأجل للخلايا ES
- إضافة 300 ميكرولتر من التربسين إلى كل جيدا 6 (من الخطوة 7.3)، واحتضان لمدة 5 دقائق عند 37 درجة مئوية. إضافة 2 مل من وسائل الاعلام تدور (الجدول 2) لتحييد التربسين وماصة صعودا وهبوطا عدة مرات لفصل إلى الخلايا وحيدة.
- نقل الخلايا إلى أنبوب 15 مل وتدور في 300 x ج لمدة 5 دقائق.
- نضح طاف وإضافة 500 ميكرولتر من وسائل الاعلام الخلية ES (الجدول 1). ماصة صعودا وهبوطا لresuspend الخلايا.
- نقل المحتوى إلى 1.5 مل أنبوب cryovial وإضافة 500 ميكرولتر من خلية تجميد 2X وفاق وسائل الإعلام (الجدول 3). مزيج جيد من قبل قلب الأنبوب ووضع أنبوب في وعاء خلية تجميد خالية من الكحول. ضع هذه الخلية تجميد حاويات في -80 درجة مئوية لمدة لا تقل عن 12 ساعة قبل ترانسفيرينز في خزان تخزين النتروجين السائل.
Access restricted. Please log in or start a trial to view this content.
النتائج
بروتوكول الموصوفة هنا يستخدم خلايا F1 ES لدراسة -regulation رابطة الدول المستقلة في التعبير الجيني في الخلايا محسن حذف monoallelic تم إنشاؤها باستخدام كريسبر / Cas9 تحرير الجينوم (الشكل 1). وgRNA وأليل معين التصميم التمهيدي لالتنميط الجيني والتعبير ال?...
Access restricted. Please log in or start a trial to view this content.
Discussion
تقدم كريسبر / Cas9 تكنولوجيا التحرير الجينوم بوساطة طريقة واضحة وسريعة وغير مكلفة لتعديل الجينوم. طريقة مفصلة هنا لتوليد وتحليل حذف محسن monoallelic لتوصيف وظيفي محسن يستفيد من تعدد الأشكال في الخلايا F1 الماوس. من مزايا هذا النوع من النهج هي: 1) الحذف محسن monoallelic لا تنتج آثار ?...
Access restricted. Please log in or start a trial to view this content.
Disclosures
قرأت الكتاب السياسات إن الرب على تضارب المصالح وليس لديهم الصراعات في الكشف عنها.
Acknowledgements
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
References
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152(2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187(2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485(2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159(2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved