
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
יצירת CRISPR / Cas9 מתווכת Monoallelic מחיקות ללמוד לתפקד Enhancer בתאי עכבר גזע עובריים
In This Article
Summary
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Abstract
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introduction
גורמי רגולטוריים תעתיק הם קריטיים עבור כוונון במרחב ובזמן קנס של ביטוי גנים במהלך התפתחות 1 ושינוי של אלמנטים אלו יכולים לגרום למחלות בשל ביטוי 2 גנים סוטים. אזורים רבים הקשורים מחלה שזוהו על ידי מחקרי עמותה רחבים הגנום נמצאים באזורים ללא קידוד ויש להם תכונות של משפרי תעתיק 3-4. זיהוי משפר והתאמתי עם הגנים שהם מווסתים מסובכים כפי שהם בדרך כלל ממוקמים כמה kilobases מן הגנים שהם מווסתים ועשויים להיות מופעלים באופן רקמות ספציפיות 5-6. תחזיות Enhancer מבוססים בדרך כלל על סימני שינוי היסטון, מתחמי-cohesin מתווך ומחייבת של שעתוק סוג תא ספציפי גורמי 7-10. אימות של משפר חזוי לרוב נעשתה באמצעות assay וקטוריות שבו משפרים מפעיל ביטוי של גן כתב 11-12. נתונים אלה מספקים נמידע aluable מהפוטנציאל רגולטוריות של רצפים משפרים משוערים אבל לא חושף את תפקידם בהקשר הגנומי אנדוגני שלהם או לזהות את הגנים שהם מווסתים. הגנום עריכה משמש ככלי רב עוצמה כדי לחקור את הפונקציה של גורמים רגולטוריים תעתיק בהקשר אנדוגני שלהם על ידי ניתוח הפסד של פונקציה.
התקדמות עריכת הגנום, כלומר מערכת עריכת הגנום CRISPR / Cas9, לסייע בחקירת פונקציה הגנום. מערכת CRISPR / Cas9 קלה לשימוש להתאמה למערכות ביולוגיות רבות. החלבון Cas9 הוא ממוקד לאתר מסוים בגנום ידי RNA מדריך (gRNA) 13. המתחם SpCas9 / gRNA סורק את הגנום רצף גנומי היעד שלה אשר חייב להיות 5 'כדי מוטיב הסמוך protospacer (PAM) רצף, NGG 14-15. זיווג מאגר של gRNA למטרתה, 20 נוקלאוטידים (NT) רצף משלים gRNA, יפעיל פעילות nuclease SpCas9 וכתוצאה מכך doublדואר גדיל Break (DSB) 3 נ"ב במעלה הזרם של רצף PAM. סגולי מושג באמצעות זיווג בסיס שלם באזור הזרע gRNA, 6-12 nt סמוך PAM; לעומת זאת, חוסר התאמה 5 'של הזרע בדרך כלל נסבלים 16-17. הציג DSB ניתן לתיקון או עד הסוף שאינו הומולוגי שהצטרף (NHEJ) תיקון דנ"א או תיקון מכוון הומולוגיה (HDR) תיקון דנ"א mechanisms.NHEJ יוצר לעתים קרובות כניסה / מחיקה (indels) של כמה נ"ב באתר היעד שיכול לשבש מסגרת הקריאה הפתוחה (ORF) של גן. כדי ליצור מחיקות גדולות בגנום שני gRNAs, אשר לאגף את האזור של עניין, ניתן להשתמש 18-19. גישה זו שימושית במיוחד עבור המחקר של משפרי תעתיק התקבצו לאזורים מלאי לוקוס או-משפרי סופר אשר שגודלם עולים על משפר קונבנציונלי 9,18,20-22.
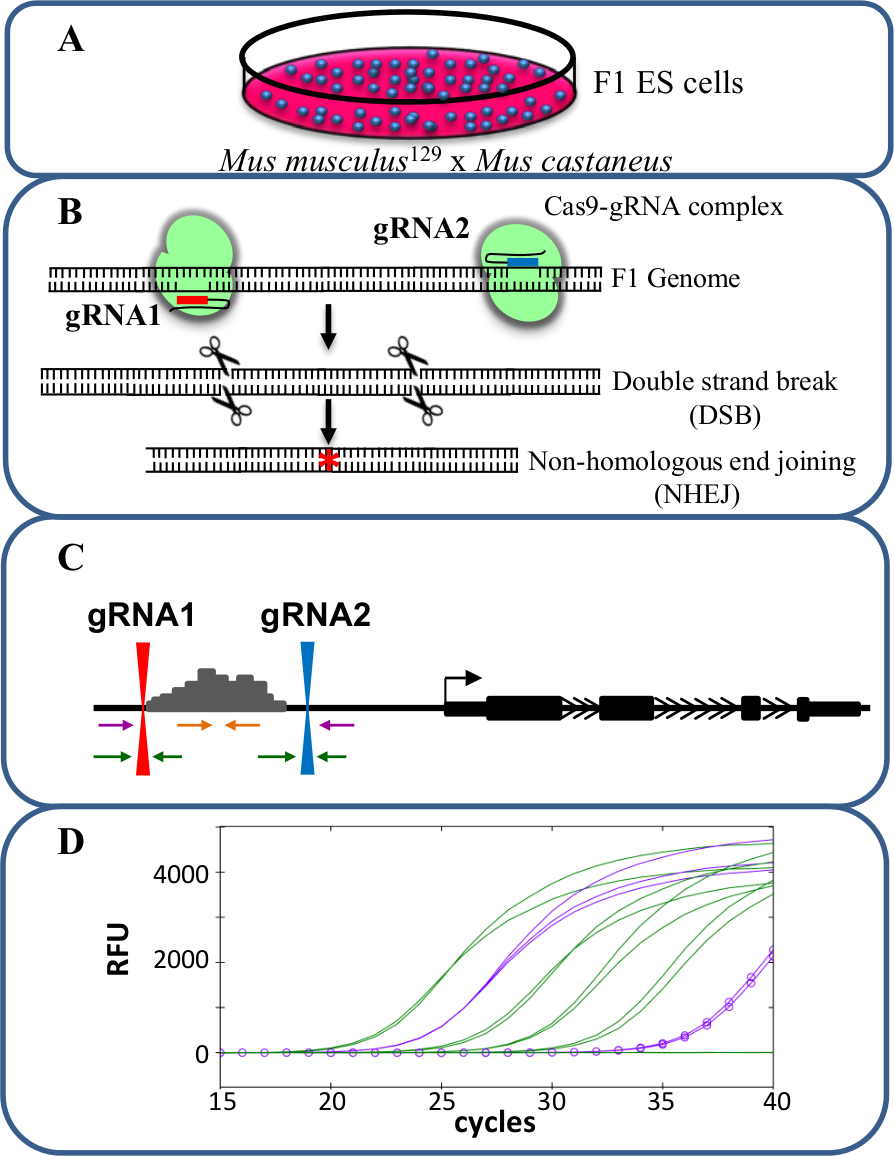
מחיקות Monoallelic הם מודל ערך ללימוד -regulation cis של שעתוק. הנצפה צ'אנגדואר ברמת תמליל לאחר מחיקת monoallelic של משפר וקושר לתפקיד משפר כי ויסות גנים ללא תופעות הבלבול שעלול להתרחש כאשר שעתוק של שני אללים מושפע ההשפיע כושר הסלולר פוטנציאלי. הערכת ביטוי מופחת קשה אולם ללא היכולת להבחין בין נמחק מן אלל הסוג הבר. יתר על כן, genotyping מחיקות בכל אלל ללא היכולת להבחין בין שני אללים הוא מאתגר, במיוחד עבור מחיקות גדולות של> 10 kb עד 1 Mb 23 שבו קשה להגביר את האזור סוג בר כולו על ידי PCR. השימוש בתאי גזע עובריים F1 שנוצר על ידי מעבר Mus musculus 129 עם Mus castaneus מאפשר שני אללים כדי להיות מובחן על ידי אלל ספציפי PCR 18,24. הגנום ההיברידי בתאים אלו, מסייע הקרנה מחיקה ספציפית אלל וניתוח ביטוי. בממוצע יש SNP כל נ"ב 125 בין שני אלה הגנוםמתן, גמישות בעיצוב פריימר לביטוי גנוטיפ מנתח. הנוכחות של SNP אחד יכול להשפיע על טמפרטורת ההתכה פריימר (מ T), ולמקד סגוליות PCR כמותי בזמן אמת (qPCR) הגברה המאפשר אפליה של שני אללים 25. יתר על כן חוסר התאמה בתוך הסוף '3 של פריימר משפיעה מאוד על היכולת של DNA פולימרז להאריך מן פריימר למנוע הגברה של יעד אלל הרצוי 26. כמתואר בפרוטוקול הבא הוא השימוש בתאי גזע עוברי F1 עבור מחיקות משפרות ספציפית אלל של יותר מ 1 kb וניתוח ביטוי שלאחר מכן באמצעות מערכת עריכת הגנום CRISPR / Cas9 (איור 1).

איור 1. מחיקת Enhancer באמצעות CRISPR / Cas9 ללמוד ציס -regulation של ביטוי גנים. (א) בתאי גזע עובריים F1 שנוצר על ידי הכלאה בין Mus musculus 129 Mus castaneus משמשים כדי לאפשר למחיקה ספציפית אלל. (ב) שני RNAs המדריך (gRNA) משמש כדי לעורר מחיקת Cas9 בתיווך גדולה של האזור משפר. (ג) קובע פריימר ומשמשים לזיהוי מונו גדול ומחיקות דו-אללים. פריימרים הכתומים הם פריימרים בפנים, פריימרים הסגול הם פריימרים מחוץ ואת פריימרים הירוקים הם פריימרים איגוף gRNA. (ד) שינויים בביטוי הגנים מנוטרים באמצעות qPCR אלל ספציפי. RFU מציין יחידות קרינה יחסיות. אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocol
1. תכנון והקמת gRNA
- להסיר אזורים משפר תעתיק להשתמש בשני gRNAs, אחד 5 'ואחד 3' של האזור של עניין. השתמש מסלול דפדפן גנום העכבר UCSC שנוצר על ידי מעבדת ג'אנג לזהות רצפי gRNA ייחודיים (http://www.genome-engineering.org 15). הבא לבדוק gRNAs אלה PAM הסמוך אליהם עבור SNPs ו indels באמצעות כלים מקוונים הניתנים על ידי מכון סנגר (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. כדי למקד הן אללים עם יעילות שווה, למנוע רצפי gRNA / PAM המכילים SNP או indel.
- בעת בחירת gRNA, לבדוק את הכדאיות של עיצוב פריימרים ספציפיים אלל עבור genotyping את המחיקה. ראה גם סעיף 5 עבור עיצוב פריימר אלל ספציפי.
- הרכב את שני פלסמידים gRNA מבוססים על הפרוטוקול המתואר מאלי et al. 2013 15. לשלב את int רצף 20 נ"ב היעד הייחודי שנבחרO את oligonucleotides 61mer כפי שמוצג בטבלה 7 (רצפים מוצגים 5 'ל 3' כיוון, ואת הבסיסים מודגשים הם רצף היעד 20 נ"ב כי הם משלים הפוכים זה מזה).
- מערבבים 10 μl של 10 מיקרומטר gRNA Primer_F ו -10 μl של 10 מיקרומטר משלימים Primer_R בתוך שפופרת.
- לחשל פריימרים על ידי דוגרי תמהיל פריימר ב 100 מעלות צלזיוס למשך 5 דקות ולאחר מכן לצנן 1 ° C / sec 25 ° C. לצעד זה, להשתמש במכונת PCR או במקום הצינור במים רותחים ולאפשר לו להתקרר RT.
- לתערובת פריימר annealed, מוסיפים את תערובת התגובה הבאה לדגור על 72 מעלות צלזיוס למשך 30 דקות כדי להאריך כל צבע יסוד: 18.5 μl של מים, 10 μl של חיץ 5x HF, 1 μl של 10 מ"מ dNTP לערבב 0.5 μl של גבוהה פולימראז נאמנות DNA.
- הפעל 10 μl של שבר היעד על ג'ל agarose 2% כדי לאשר את שברי מדריך 100 נ"ב הופקו.
- Linearize וקטור gRNA (מתנה George הכנסייה; Addgene פלסמיד # 41,824) 15 עם AFL השנייה באמצעות התגובה הבאה להגדיר: 5 μl של עמוד השדרה וקטור gRNA (2-4 מיקרוגרם), 5 μl של חיץ 10x, 3 μl של AFL השנייה (20 יחידות / μl) ו -32 μl של מים. דגירת תערובת התגובה במשך 3 שעות ב 37 מעלות צלזיוס.
- הפעל את המוצר מתעכל על ג'ל 1% agarose ולטהר את הלהקה DNA המתאים 3.5 kb לינארית וקטור gRNA באמצעות ערכת חילוץ ג'ל על פי ההוראות של היצרן.
- הגדרת התגובות הרכבה גיבסון 29-30 באמצעות וקטור gRNA לינארית ולכוון שבר משלב 1.2.3 כדלקמן: 1 μl של וקטור ליניארי gRNA (50 ng / μl), 1 μl של שבר היעד, 10 μl של תמהיל מאסטר הרכבה 2x גיבסון ו -8 μl של מים. דגירת תגובות על 50 מעלות צלזיוס למשך 60 דקות.
- טרנספורמציה של תאים E.coli עם מורכבים gRNA וקטור.
- מערבבים 1 μl של וקטור gRNA המורכב מ 1.2.7ו 50 μl של DH5α (זן E.coli) תאים צינור. להפוך את תאי DH5α בשיטת הלם חום על ידי חשיפת התאים ל -42 מעלות צלזיוס למשך 45 שניות.
- צמד לצנן את הצינורות על קרח דק 5; ולאחר מכן להוסיף 400 μl של מדיום SOC ו לדגור על 37 מעלות צלזיוס במשך 45 דקות בתוך חממה רועדת.
- מורחים 100 μl של תאים DH5α על LB-kanamycin (50 מיקרוגרם / מ"ל) צלחת סלקציה חיובית של תאים טרנספורמציה דגירה O / N ב 37 מעלות צלזיוס.
- הקרנת מושבות חיוביות E.coli עבור gRNA הכנס.
- פיק מושבה ו resuspend kanamycin עמיד ב 3 מ"ל LB המכיל 50 מיקרוגרם / מ"ל של kanamycin. חזור על אותו במשך 6-8 מושבות דגירה כל הצינורות ב 37 מעלות CO / N בתוך חממה רועדת.
- חלץ פלסמידים מתרבה מבוגר O / N באמצעות פלסמיד ערכת הכנת מיני על ידי ביצוע הוראות היצרן.
- הכן תערובת תגובת עיכול Ecor לי לבדוק כנס רצף gRNAפלסמיד. עבור כל דגימה, להכין את תערובת התגובה כדלקמן: 2 μl של חיץ האנזים, 1 μl של לי Ecor, 15 μl של מים. Aliquot את תערובת התגובה לתוך צינורות 1.5 מ"ל ולהוסיף 2 μl של פלסמיד. דגירת הצינורות ב 37 מעלות צלזיוס למשך 2 שעות.
- הפעל את המוצר מתעכל על ג'ל 1.5% agarose.
הערה: דגימות עם הכנס יציג גודל הלהקה 475 נ"ב כי הוא 100 נ"ב גבוה יותר מאשר שיבוטים ללא מוסיף.
הערה: לחלופין, השיבוטים החיוביים יכולים להיות מוקרנים על ידי מושבה PCR באמצעות SP6 (קדימה) ו T7 (הפוך) פריימרים (לוח 7) אשר נקלטים על ידי רצף הווקטור לתת שבר גודל 642 נ"ב בנוכחות של gRNA הכנס. גישת מושבת PCR יש יתרון כאשר שיש אתר הגבלת Ecor שאני בתוך רצף gRNA.
- אשר את הרצף של gRNA הכנס ידי DNA רצף שימוש פריימר T7.
2. Transfection
הערה:Electroporation היא שיטה יעילה של transfecting פלסמידים לתוך בתאי גזע עובריים. השיטה המתוארת כאן משתמשת בטכנולוגית transfection microporator.
- לגדל תאים ES F1 בצלחת 10 ס"מ מצופה ג'לטין המכיל 10 מ"ל של התקשורת תא גזע עובריים (טבלה 1) על 37 מעלות צלזיוס / 5% CO 2. כאשר התאים מגיעים 85% confluency להסיר את המדיה ומוסיפים 2 מ"ל של טריפסין. לדגור על 37 מעלות צלזיוס CO 2 באינקובטור במשך 5 דקות.
הערה: תאי F1 ES התקבלו ברברה Panning 24 והם זמינים על פי בקשה. - לנטרל את טריפסין על ידי הוספת 10 מ"ל של התקשורת ספין (טבלה 2). פיפטה מספר פעמים כדי לנתק את התאים לחלוטין.
- לאסוף את כל התאים בתוך שפופרת 15 מ"ל ו ספין על XG 300 במשך 5 דקות. Resuspend ב 3 מיליליטר PBS ולספור את התאים באמצעות hemocytometer או נגד תא אוטומטי.
- גלולה 1 x 10 6 ES תאים צינור 1.5 מ"ל על ידי צנטריפוגה XG ב 300 במשך 5 דקות ו resuspend ב 100 μl של R (resuspension) חיץ כפי שסופק על ידי יצרן הערכה.
- הוסף 5 מיקרוגרם כל אחת pCas9_GFP (מתנה קיראן Musunuru; Addgene פלסמיד # 44,719) 31, 5 'ו 3' פלסמידים gRNA למחיקה של אזור היעד ומערבבים בעדינות עם טפטפת, כדי למנוע את כניסתה של בועות.
- השתמש קצה פיפטה האלקטרוני לשאוב 100 μl של תמהיל electroporation, להיות זהיר, כדי למנוע בועת הקצה.
- תכנת את וולט, רוחב וקטניות עבור electroporation. עבור תאים F1 ES, השתמש 1,400 V, 10 msec 3 פולסים.
- בעוד electroporation פועל להתבונן הקצה לצפות לכל ניצוצות בתמיסה. ניצוץ מעיד על קיומו של בועת אוויר יפריע transfection.
- הוצא את בתאי גזע עובריים transfected לתוך צלחת 10 ס"מ מצופה ג'לטין המכיל 10 מ"ל ES תא התקשורת (טבלה 1) ו לדגור על 37 ° C / 5% CO 2.
3. FACS מיון תאים transfected
- לאחר 48 שעות, לנתק את התאים על ידי הוספת 2 מ"ל של טריפסין ו לדגור על 37 מעלות צלזיוס CO 2 באינקובטור במשך 5 דקות.
- לנטרל את הצלחת על ידי הוספת 10 מ"ל של חיץ אוסף (לוח 3). אוספים את התאים בתוך שפופרת 15 מ"ל ו ספין על XG 300 במשך 5 דקות.
- בטל supernatant ו resuspend התאים 1 מ"ל של חיץ מיון (לוח 4). ספירת התאים לדלל המבוסס על פלטפורמת המיון. לדלל את התאים 0.5-1 x 10 6 תאים / מ"ל למיון ל -15 מ"ל צינורות למיון תאים בודדים ישירות לתוך צלחות 96-היטב, לדלל את התאים 2-5 x 10 6 תאים / מ"ל.
- מיין תאים Cas9-GFP + ES באמצעות זרימת FACS cytometer 32. איסוף תאים בתפזורת צינורות עם 2 מיליליטר תקשורת התאוששות (לוח 5) וצלחת כמתואר 3.5 לקטיף מושבה, או מעין תאים בודדים ישירות לתוך צלחות ג'לטין מצופה 96-היטב המכילות 100 μl תקשורת תא ES / טוב ( טבלה 1).
- זרע 1-1.5 x 10 4 GFP + ES תאים בצלחת 10 ס"מ מצופה ג'לטין המכיל 10 התקשורת תא מ"ל ES (טבלה 1). ציפוי בצפיפות נמוכה זה יקל לקטוף מושבות תאי ES פרט.
4. Culturing המשובטים עבור Genotyping, ניתוח ביטוי מניות תא הקפאה
- ביום 4-5 לאחר המיון, להבקיע היטב בכל 96-גם צלחות מסודרות ישירות לנוכחות של מושבות תאי ES.
- לנתק מושבות תאים ES על ידי הסרת התקשורת והוספת 30 μl של טריפסין. לדגור על 37 מעלות צלזיוס למשך 5 דקות. לנטרל את טריפסין על ידי הוספת 170 μl של התקשורת תא ES (טבלה 1), ו פיפטה למעלה ולמטה עבור ניתוק מוחלט של המושבה לתוך תאים בודדים. לגדל את התאים ב 37 ° C / 5% CO 2 עד שרוב בארות למעלה מ -70% ומחוברות (בדרך כלל 2-3 ימים).
- לחלופין לבחור מושבות תאי ES בודדות 10 מנות סנטימטר באמצעותמיקרוסקופ הפוכה. לאחר aspirating המושבה לתוך צעד מעקב פיפטה טיפ 4.1.1 הצבת כל מושבה אחד טוב של צלחת 96-היטב, pretreated עם ג'לטין המכיל 30 μl של טריפסין.
הערה: מושבות יכולות לשבת טריפסין ב RT בעוד שורה שלמה אחת המושבות נקלטת. - לאחר כל המושבות כבר הרים ו ניתקו לתוך תקשורת לגדל את התאים ב 37 מעלות צלזיוס CO 2 באינקובטור עד שרוב הבארות למעלה מ -70% ומחוברות (בדרך כלל 2 ימים).
- כאשר 96-גם הצלחות מוכנות עבור פיצול, להסיר את המדיה, להוסיף 30 μl של טריפסין ו לדגור על 37 מעלות צלזיוס למשך 5 דקות. לנטרל את טריפסין על ידי הוספת 180 μl של התקשורת תא ES (טבלה 1) לכל היטב פיפטה ולמטה עבור ניתוק מוחלט לתוך תאים בודדים.
- מתוך 210 μl וכתוצאה מכך, זרע 70 μl לשלוש מצופה ג'לטין 96-גם צלחות שכל אחת מהן מכילה 130 μl של התקשורת תא ES / טוב (טבלה 1). להשתמש בצלחות אלה עבור GEnotyping, ניתוח ביטוי והקפאת מניות תא עבור כל שיבוט כמתואר להלן.
- כשהצליח גנוטיפ מגיע 70-85 מפגש%, לטפל הצלחת כמתואר בסעיף 6 "גנוטיפ את המחיקה".
- כשהצליח ניתוח הביטוי מגיע 70-85% מפגש, להסיר את המדיה, לאטום את הצלחת עם איטום קלטת ולאחסן ב -80 ° C עד השיבוטים כבר genotyped.
הערה: צלחת ניתוח הביטוי שימושית לנתח שינויים בביטוי גנים ב הקטעים הראשונים של השיבוטים. ניתוח התבטאות גנים מהצלחת 96-היטב אפשרי אבל כמו מספרי התא נמוכים ערכת חילוץ מיקרו RNA מומלץ. - הכנת מאגר להקפיא 96-היטב פלייט:
- כשהצליח למניות תא קפוא (מלאה-1) מגיע 70-85% מפגש, לשאוב את התקשורת, להוסיף 30 μl של טריפסין ו לדגור על 37 מעלות צלזיוס למשך 5 דקות.
- לנטרל את טריפסין על ידי הוספת 100 μl של תקשורת תא ES (טבלה 1) to כל טוב פיפטה מעלה ומטה עבור ניתוק מוחלט לתוך תאים בודדים.
- העברת 15 μl של תאים מושעה מכל טוב לשתי צלחות ג'לטין מצופה 96-היטב, כל 185 μl המכיל של התקשורת תא ES (טבלה 1) ולאפשר לגדול ב 37 ° C / 5% CO 2.
הערה: אפשרות זו מיועדת מנייה-2 ו -3 צלחות שהן שיבוטי גיבוי נוספים במקרה החייאת התאים ממלאי-1 הוא לא מוצלחת.
- בינתיים, אל 100 μl של תאים הנותרים בצלחת 96-היטב (מלאה-1), להוסיף 100 μl של 2x הקפאת תקשורת (לוח 6). חותם את הצלחת עם סרט איטום ובמהירות להפוך את הצלחת 4-5 פעמים עבור ערבוב נאות. אחסן את הצלחת ב -80 מעלות צלזיוס עד שיבוטים הם genotyped.
- כאשר המנייה-2 ומלאים-3 הצלחות מוכנות להקפאה לשאוב התקשורת, להוסיף 30 μl של טריפסין ו לדגור על 37 מעלות צלזיוס למשך 5 דקות. לנטרל את טריפסין על ידי הוספת 70 μl של תקשורת תא ES (Tמסוגל 1) לכל היטב פיפטה ולמטה עבור ניתוק מוחלט לתוך תאים בודדים.
- הוספת 100 μl של תקשורת הקפאת 2x, לאטום את הצלחת עם איטום קלטת ובמהירות להפוך את הצלחת 4-5 פעמים עבור ערבוב נאות. אחסן את הצלחת ב -80 מעלות צלזיוס עד הצלחות אלה נדרשים.
עיצוב 5. אלל ספציפי פריימר
- עיצוב 4 סטים של פריימרים (תרשים 1C) להקרין את שיבוטים למחיקה הרצוי: בתוך Primers, בחוץ, gRNA איגוף פריימרים (עבור 5 שני 'ו 3' gRNA היעד אתרים) כמפורט להלן.
- השג את המסלול SNP המתאים גנוטיפים 129 ו יצוקה ב http://labs.csb.utoronto.ca/mitchell/crispr.html. המסלול הנתון תערוכות החלפות בסיס בין 129 ו יצוק ב קואורדינטות באסיפת גנום עכבר mm9.
הערה: הקישור באתר מעל יפנה אל דפדפן הגנום UCSC ולהוסיף מסלול מותאם אישית המכיל את SNPs בין 129 ו משתתף geמחוזות. - הזן את הקואורדינטות של האזור למחיקה. זום על אזור של כ -500 נ"ב באמצע המחיקה הרצויה המכילה> 3 SNPs.
- הקלק על תצוגה> DNA בבר אפשרות ולחץ לקחת דנ"א להוריד רצף היעד בכל פורמט רישית.
- צור שני רצפים FASTA; אחד עבור 129 והאחד יצוק ידי החלפת בסיס במיקום SNP. סמן את SNPs ידי אותיות קטנות.
- עבור אל Primer3 פלוס (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) ולהדביק את SNP להחליף 129 ברצף. השתמש בהגדרות ברירת המחדל לתכנן פריימרים.
- כדי לעצב את פריימרים בתוך ספציפי אלל לבחור Primer_List בתפריט ירידת משימה ולחץ הפיק פריימרים. בחר פריימר קדימה או אחורה כי יש SNP או ב 'בסוף או בתוך 4 בסיסים מן 3' 3 ונופל שבתחומי האזור למחיקה.
הערה: Primers שיש להם SNP בתצוגת 3'end גדל סגולי אלל ב qPCR. - כדי לבחור את החזר פריימר השני לדף הראשי, בחר איתור בתפריט ירידת המשימה ולהדביק את רצף פריימר הראשון בתיבה המתאימה בתחתית הדף. בלשונית ההגדרות הכלליות שנה את ההגדרה עבור טווח גודל מוצר 80-200 נ"ב ולחץ פיק פריימרים. בחר פריימר שתחילתו הזוגות פריימר המפורטים; אלה יהיו פריימרים אלל ספציפי בתוך 129.
- חזור על שלבים 5.1.5 5.1.7 לתכנן פריימרים עבור האלל יצוקה.
- חזור אל דפדפן הגנום UCSC והזן את הקואורדינטות-שיתוף של האזור למחיקה. הקלק על תצוגה> DNA בבר האפשרות, תחת אפשרויות אזור יחזור רצף להוסיף 1,000 נ"ב במורד זרם לחץ לקבל DNA להוריד רצף היעד.
- סמן את רצף יעד gRNA בסוגריים. שמור רצף כולו המעשה לפני שאתם ממשיכים.
- כדי לעצב את פריימרים מחוץ להסיר את הרצף בין רצפי יעד שני gRNA. חזור על שלב 5.1.4-5.1.8 לעצב את פריימר אלל ספציפי מחוץים אך לשנות את גודל המוצר 400-800 נ"ב.
- פיצול הרצף שהושג בשלב 5.1.10 לשני רצפים, כל אחד עם 500 נ"ב 5 'ו 3' של רצף יעד gRNA. חזור על שלבים 5.1.5 5.1.7 בעיצוב פריימרים ספציפיים שאינם אלל עבור אזורים gRNA איגוף אבל לשנות את גודל המוצר 400-800 נ"ב.
הערה: פריימרים ספציפיים שאינם אלל, או 129 או רצף עופרת ניתן להשתמש צריך להבחר פריימרים שאינם מכילים SNP. מומלץ להגדיר גודל המוצר של 400-800 נקודות בסיס לעיצוב החוצה gRNA איגוף פריימרים. זה מאפשר הגברה גם אם indels הקטן נוכח.
- השג את המסלול SNP המתאים גנוטיפים 129 ו יצוקה ב http://labs.csb.utoronto.ca/mitchell/crispr.html. המסלול הנתון תערוכות החלפות בסיס בין 129 ו יצוק ב קואורדינטות באסיפת גנום עכבר mm9.
- בדוק את פריימרים בתוך עבור סגוליות אלל ידי qPCR באמצעות DNA גנומי זן 129 ו יצוקה טהור 2 ng / μl. בצע צעד 6.2-6.4 להקים התגובה qPCR.
הערה: אם הגנוטיפ 129 האזור זהה C57BL / 6J, דנ"א C57BL / 6J יכול לשמש במקום 129 DNA. פריימרים אלל ספציפי צריכים להציג לפחות 5 מחזוריםההפרש בין השווי CT (סף מחזור) על הנכונות לעומת הגנוטיפ שגוי. פריימרים מחוץ יכול להיבדק על מנת להבטיח שהם להגביר את המחיקה באמצעות Cas9 / gRNA transfected לתאי גזע עובריים, ו- DNA F1 גנומי בהתאמה כמו בקרות חיוביות ושליליות. אלל-הסגולי של פריימרים מחוץ ניתן לבדוק פעם שיבוטי monoallelic זוהו.
6. Genotyping את המחיקה
- חלץ הדנ"א הגנומי מהצלחת 96-היטב genotyping באמצעות צלחת משלב 4.6 שנוצר לאחר הרחבת המושבה.
- מכינים את תערובת מיצוי DNA גנומי: 89 μl של מים, 10 μl של 10x חיץ 1 μl של מגיב מיצוי (המסופקים על ידי היצרן). הוספת 100 μl של תערובת מיצוי DNA גנומי זה טוב לאטום את הצלחת עם סרט האיטום.
- דגירה את הצלחת על 75 מעלות צלזיוס למשך 5 דקות ואחריו 95 מעלות צלזיוס למשך 5 דקות.
- אפשר הצליח להתקרר על ידי דוגר על קרח למשך דקות ספורותד אז צנטריפוגות בקצרה ליישוב מי עיבוי לתחתית הבאר. זה משמש את הצלחת ה- DNA תבנית להקרנה המחיקה.
- הגדר את התגובות qPCR בשני עותקים עבור כל שיבוט כדלקמן: 5 μl של תמהיל 2x SYBR qPCR, קדימה לאחור תחל (3 מיקרומטר) כל 1 μl ו 1 μl של מים. השתמש פיפטה רב להוסיף 2 μl של תבנית ה- DNA ואחריו 8 μl של תערובת התגובה היטב בכל צלחת 384 גם.
- חותם את הצלחת עם איטום קלטת ספין ב 600 XG למשך 2 דקות לערבב את תוכנו. מניחים את מערך צלחת 384 גם ב Cycler בזמן אמת.
- תכנית את Cycler בזמן האמת עבור PCR 2-צעד ואחריו ניתוח עקום להמס עם זיהוי כדלקמן: 1 מחזור ב 95 מעלות צלזיוס למשך 10 דקות, 40 מחזורים של 95 מעלות צלזיוס למשך 15 שניות, 62 ° C למשך 30 שניות עם קריאת צלחת ו -95 מעלות צלזיוס למשך 10 שניות, 65 ° C עד 95 ° C עם תוספת של 5 מעלות צלזיוס למשך 5 שניות + צלחת לקרוא.
הערה: בנוסף פריימר העיצוב, qPCR מילx ופרמטרי מחזור גם לתרום פריימר סגולי. הפרמטרים שתוארו לעיל ריאגנטים המפורטים חומרים יותר להניב לעתים קרובות הגברת אלל ספציפית. - ניתוח תוצאות qPCR
- בדוק כל אלל הגברה עם פריימרים אלל ספציפי בפנים. אין הגברה של אלל אחד או הבדלי ערך Ct גבוה (> 5 מחזורים) בין אללים מציעים שיבוטים אלה לשאת מחיקה הטרוזיגוטיים של האלל עם ערך Ct הגבוה / נעדרים. אין הגברה של שני אללים עולה כי הם נושאי מחיקה הומוזיגוטים.
- בדוק כל אלל הגברה עם פריימרים אלל ספציפי בחוץ. כאשר מחיקת היעד היא גדולה יותר מאשר הגברת 1 kb עם פריימרים מחוץ מתרחש רק כאשר מחיקה היא הווה. ערך ה- CT של 22-28 מאשר את המחיקה. לקבלת מחיקות היעד שלהן קטנה מ- 1 kb, לאשר את גודל amplicon ידי אלקטרופורזה.
הערה: אם פריימרים מחוץ להראות אלל-סגוליות מתונה בלבד (ראה figuבתשובה לשאלה 2), ניתן לקבל amplicons עם שתי קבוצות פריימר אללים שיבוטי monoallelic עקב הגברת יעד הנחה של פריימרים בחוץ. במקרה זה בדל ערך Ct בין שני אללים של מחזורים חמישה לפחות אמור לאשר את אלל הנכון (ערך ה- CT נמוך) נמחק על בסיס התוצאות שהתקבלו פריימרים בפנים. אם על היעד לעומת הבדל Ct אלל יעד ההנחה הוא פחות מחמישה מחזורים לעצב אלל חדש פריימרים מחוץ ספציפי. - שיבוטים מחיקים monoallelic לבדוק את תקינות האלל הלא שנמחק באמצעות ההקרנה משני, gRNA איגוף פריימרים.
הערה: indels של> גודל 25 נ"ב סביב האתר היעד gRNA ניתן לזהות על ידי התבוננות שינוי בעקום להמיס את qPCR עבור 400-800 amplicons נ"ב. לחלופין, amplicons מן פריימרים איגוף gRNA יכול להיות רצף לזהות indels קטן של <25 נ"ב.- בצע qPCR עם 2 סטים של פריימרים gRNA איגוף כלומר, 5 'ו 3' זRNA שבשימוש בתהליך יצירת מחיקת CRISPR. אין הגברה עם קבוצות אלה של פריימרים מציינת indels הגדול יותר amplicon qPCR נוכח באתר יעד gRNA על האלל הלא-deleted של שיבוטים מחיקים monoallelic. שיבוטי מחק המכילים indels הגדול אלה בניתוחים נוספים כמו התוצאות עשויים להיות קשים לפרש בלי לדעת את ההיקף המחיק.
- לטהר את amplicons המתקבל התגובה מחוץ פריימר qPCR באמצעות PCR לנקות ערכה ממלא את ההוראות של היצרן.
- אשר את הרצף של אלל נמחק על ידי ה- DNA רצף מוצר PCR מטוהר מהשלב הקודם. השתמש פריימרים הגברת qPCR עבור קדימה הפוך רצף.
הערה: בשעה SNPs בשלב זה בתוך מעשה amplicon כאישור משני של הגנוטיפ של אלל נמחק.
7. ביטוי ניתוח עם פריימרים ספציפיים אלל
- הפשרת pl מניות תא 96-היטבאכול מאוחסן ב -80 ° C (מלאה-1 משלב 4.9) על ידי הצבתו על אמבט חרוז חם. כאשר יותר ממחצית הבארות בצלחת הם הפשירו, ספין ב XG 300 במשך 5 דקות.
- בזהירות, הסר את סרט האיטום ולהעביר את התאים במהירות מבארות חיובית המחיקה לתוך מצופה ג'לטין, צלחות 12-היטב המכיל 1 מ"ל של התקשורת תא גזע עובריים (טבלה 1) ו לדגור על 37 ° C / 5% CO 2.
- כשהצלחת מגיעה 70-85% המפגש, מעבר התאים ולפצל אותם לשלוש בארות צלחת ג'לטין מצופה, 6-היטב, שכל אחת מהן מכילה 2 מ"ל של התקשורת תא גזע עובריים (טבלה 1). השתמש שתי בארות להכין 2 בקבוקונים של מניות תא קפוא עבור אחסון לטווח ארוך בחנקן נוזלי (כמתואר בשלב 8) ואת הבאר השלישית להפקת RNA.
- חלץ RNA באמצעות ערכת חילוץ RNA.
- המרת 100-500 ננוגרם של רנ"א cDNA ידי בהעתקה הפוכה (RT) רנ"א באמצעות ערכת סינתזת cDNA בעקבות הפרוטוקול של היצרן. כלול n RTתגובת egative עבור כל דגימת RNA כדי לפקח על כמות זיהום DNA בדגימות RNA.
- לדלל את cDNA לפני qPCR על יחס של בין 1: 2 ו -1: 4; בהתאם לרמת הביטוי של גן היעד בתאי גזע עוברי.
- הגדר את qPCR כמתואר לעיל כולל DNA F1 גנומי כמו עקומת סטנדרט (5 דילולים לקפל 250-.08 ng / μl) כימות מוחלטת של רמות תמליל. השווה את הביטוי של כל אלל של הגן של הריבית בכל שיבוט נמחק אישר גן בקרה מתאימה, למשל GAPDH (פריימרים המפורטים בטבלה 7).
הערה: פריימרים גן הבקרה לא צריך להיות אלל ספציפי. עיצוב פריימר אלל ספציפי זהה עבור פריימרים RT-qPCR כמתואר עבור פריימרים גנוטיפ למעט לאזור המיקוד של הגברה. רצף הגן אמור לשמש; אם באמצעות פריימרים עבור אקסון בודד או גבול אקסון-אינטרון (לפקח תמליל העיקרי) DNA F1 הגנומי יכול לשמשעקומת סטנדרט. לפרטים נוספים על RT-qPCR עיין לפורלנזה et al. 2012 33.
8. הכנת מאגר הקפאה עבור אחסון לטווח ארוך של בתאי גזע עובריים
- הוספת 300 μl של טריפסין לכל היטב 6 (משלב 7.3) ו דגירה במשך 5 דקות ב 37 מעלות צלזיוס. הוסף 2 מ"ל של ספין תקשורתי (טבלה 2) כדי לנטרל טריפסין ו פיפטה מעלה ומטה מספר פעמים כדי לנתק לתוך תאים בודדים.
- העברת התאים לתוך צינור ספין 15 מ"ל ב 300 XG במשך 5 דקות.
- לשאוב supernatant ולהוסיף 500 μl של התקשורת תא ES (טבלה 1). פיפטה מעלה ומטה כדי resuspend התאים.
- מעביר את התוכן לצינור 1.5 מיליליטר cryovial ולהוסיף 500 μl של תקשורת הקפאת תא 2x ES (לוח 3). מערבבים היטב על ידי צינור היפוך ובמקום הצינור לתוך מיכל הקפאה תא נטול אלכוהול. מניחים את התא הזה הקפאת מכולות ב -80 מעלות צלזיוס למשך 12 שעות לפחות לפני transferring לתוך מיכל אחסון חנקן נוזלי.
Access restricted. Please log in or start a trial to view this content.
תוצאות
הפרוטוקול המתואר כאן משתמש בתאי F1 ES ללמוד -regulation cis של ביטוי גנים בתאים נמחקו משפרים monoallelic שנוצרו באמצעות CRISPR / עריכת הגנום Cas9 (איור 1). GRNA ועיצוב פריימר ספציפי אלל עבור genotyping ביטוי גנים הם גורמי המפתח בגישה זו. כל קבוצת פריימר אלל ספציפי ...
Access restricted. Please log in or start a trial to view this content.
Discussion
עריכת טכנולוגיה הגנום בתיווך CRISPR / Cas9 מספק שיטה פשוטה, מהירה וזולה עבור שינוי הגנום. השיטה המפורטת כאן כדי ליצור ולנתח מחיקה משפר monoallelic לאפיון משפר תפקודי מנצלת SNPs בתאי עכבר F1. היתרונות של סוג זה של גישה הם: 1) מחיקות משפרים monoallelic אינן מייצרות תופעות בלבול המתרחשות כאש...
Access restricted. Please log in or start a trial to view this content.
Disclosures
המחברים קראו מדיניות של יופיטר על ניגוד העניינים ואין להם קונפליקטים לחשוף.
Acknowledgements
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
References
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152(2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187(2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485(2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159(2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved