
Bu içeriği görüntülemek için JoVE aboneliği gereklidir. Oturum açın veya ücretsiz deneme sürümünü başlatın.
Method Article
Yaratma CRISPR / Cas9 aracılığı monoallelik Delesyonlar Fare Embriyonik Kök Hücreleri Artırıcı Fonksiyonu Eğitim için
Bu Makalede
Özet
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Özet
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Giriş
Kopyalama düzenleyici elemanlar nedeniyle anormal gen ifadesinin 2 hastalığın neden olabilir, bu elemanların geliştirilmesi 1 ve modifikasyonu esnasında gen sentezlenmesinin yerleşim-zaman ince ayar için kritik öneme sahiptir. Genom ilişki çalışmaları ile tanımlanan birçok hastalıkla ilişkili bölgeler kodlayıcı olmayan bölgelerinde ve transkripsiyon arttırıcı 3-4 özelliklerine sahiptir. Güçlendiricileri belirlenmesi ve genellikle birkaç kilobazlık uzaklıkta da düzenleyen genler arasında yer almaktadır ve dokuya özel bir tarzda 5-6 aktive edilebilir karmaşık düzenleyen genler ile bulundu. Artırıcı tahminleri yaygın histon modifikasyonu işaretleri, arabulucu-kohezinin kompleksleri dayalı ve hücre tipine spesifik transkripsiyon bağlayıcı 7-10 faktörler vardır. Tahmin edilen arttırıcı validasyonu çoğunlukla arttırıcı bir raportör gen 11-12 ekspresyonunu aktive olan bir vektör bazlı deney yapılır. Bu veriler, v sağlarvarsayılan arttırıcı dizilerinin düzenleyici potansiyeli hakkında aluable bilgi ama onların endojen genomik bağlamda kendi işlevini açığa ya da düzenleyen genleri tanımlamak yok. Genom düzenleme kaybı fonksiyon-analizi ile kendi endojen bağlamda transkripsiyonel düzenleyici elemanlarının fonksiyonunu incelemek için güçlü bir araç olarak hizmet vermektedir.
genom düzenleme, yani CRISPR / Cas9 genom düzenleme sisteminde son gelişmeler, genom fonksiyonunun soruşturma kolaylaştırmak. CRISPR / Cas9 sistemi birçok biyolojik sistemler için kullanımı kolay ve uyarlanabilir. Cas9 proteini bir kılavuz RNA (gRNA) 13 tarafından genomunda belirli bir site hedeflenmektedir. SpCas9 / gRNA kompleksi protospacer bitişik motifi (PAM) dizisi, NGG 14-15 5 'olmalıdır hedef genomik dizisi için genom tarar. hedefine gRNA, gRNA tamamlayıcı olan bir 20 nükleotid (nt) dizisi baz çiftleşmesi, doubl elde SpCas9 nükleaz aktivitesi aktivee iplikli sonu (DSB) PAM dizisinin yukarı 3 bp. Özgünlük gRNA tohum bölgede tam baz eşleşmesi ile elde edilir, 6-12 PAM bitişik nt; Bunun aksine, tohum, genellikle 16-17 tolere edilir '5 uymuyordur. katılmadan homolog olmayan sonunda (NHEJ) DNA onarım veya homoloji yönettiği onarım (HDR) mechanisms.NHEJ DNA onarım sık sık bozabilir hedef bölgeye bir kaç bp ekleme / silme (indeller) oluşturur ya tanıttı DSB tamir edilebilir bir genin açık okuma çerçevesinin (ORF). Ilgili bölgeyi kuşatan genom iki gRNAs, daha büyük silme oluşturmak için 18-19 kullanılabilir. Bu yaklaşım, lokus kontrol bölgeleri ya da geleneksel arttırıcılar 9,18,20-22 daha büyük olan süper arttırıcı olarak kümelenmiş transkripsiyonel arttırıcılar çalışma için özellikle yararlıdır.
Monoallelik silmeler transkripsiyon sis -Yönetmeliği çalışmak için değerli bir modeldir. gözlenen changBir artırıcı madde monoallelik silindikten sonra transkript düzeyinde e hem alel transkripsiyon hücresel uygunluğu etkileyen potansiyel etkilenir oluşabilir karıştırıcı etkileri olmadan gen regülasyonu bu güçlendirici rolü ilişkilidir. azaltılmış ifade değerlendirilmeden ancak vahşi tip allel silinmiş ayırt yeteneği olmadan zordur. Ayrıca, iki allel ayırt yeteneği olmadan her alel de silmeleri genotipleme özellikle PCR ile tüm yabani tip bölgeyi yükseltmek için zor olduğu 1 Mb 23> 10 kb büyük delesyonlar için, zordur. Mus castaneus ile aradaki Mus musculus 129 tarafından üretilen F1 ES hücrelerinin kullanımı iki aleli alel-spesifik PCR 18,24 ayırt edilmesini sağlar. Bu hücrelerde melez genom alel spesifik silme tarama ve ifade analizi kolaylaştırır. Ortalama olarak bu iki genomları arasındaki her 125 bp bir SNP varİfade ve genotip için astar tasarım esnekliği sağlayan analiz eder. Bir SNP varlığı astar erime sıcaklığı (Tm) etkilemek ve iki allel 25 ayrımcılık sağlayan gerçek zamanlı kantitatif PCR (qPCR) amplifikasyon özgüllük hedefleyebilir. Ayrıca primerin 3 'ucu olan bir uyumsuzluk büyük ölçüde istenmeyen bir alel hedef 26 amplifikasyonunu önleyici astar uzanacak şekilde DNA polimerazın yeteneğini etkiler. CRISPR / Cas9 genom düzenleme sistemi (Şekil 1) kullanılarak daha büyük 1 kb alel spesifik güçlendirici kromozom anormallikleri ve daha sonra ekspresyon analizi için F1 ES hücrelerinin kullanımı, aşağıdaki protokolde tarif edilmektedir.

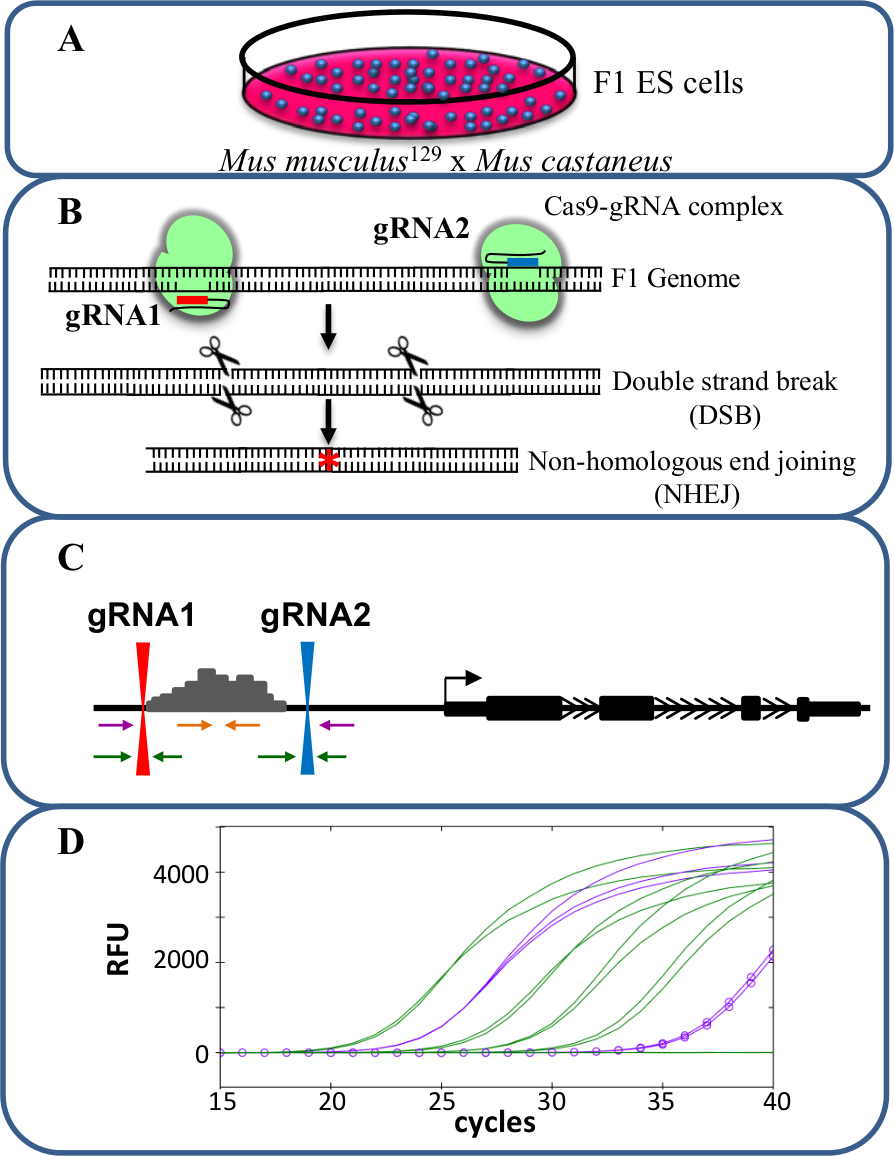
Sis -reg incelemek için CRISPR / Cas9 kullanarak Şekil 1. Artırıcı silmeGen ifadesinin ulation. Mus musculus 129 ve Mus castaneus arasında çapraz tarafından üretilen (A) F1 ES hücreleri alel spesifik silinmesi için izin vermek için kullanılır. (B) iki kılavuz RNA'lar (gRNA) geliştirici bölgesinin büyük Cas9 aracılı silme indüklemek için kullanılır. (C) Primer setleri büyük mono- ve bi-alelik silme tanımlamak için kullanılır. Turuncu primerler içinde primerler olarak mor primerler dış primerleri ve yeşil primerler gRNA kuşatan primerlerdir bulunmaktadır. Gen ekspresyonunda (D) değişiklikleri alel spesifik qPCR kullanılarak izlenmektedir. RFU nispi floresan üniteleri gösterir. Bu rakamın büyük halini görmek için lütfen buraya tıklayınız.
{kind=link}
Protokol
1. Tasarımı ve gRNA oluşturulması
- transkripsiyonel arttırıcı bölgeleri iki gRNAs, ilgilenilen bölgenin bir 5 've bir 3' kullanmak silmek için. (Http://www.genome-engineering.org 15) eşsiz gRNA sekansları tanımlamak için Zhang laboratuvar tarafından üretilen fare UCSC genom tarayıcı parça kullanın. Sonraki bu gRNAs ve Sanger Enstitüsü (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28 tarafından sağlanan çevrimiçi araçlar kullanarak SNP ve indellerin onların bitişik PAM kontrol edin. Eşit verimliliği ile hem allel hedeflemek için, bir SNP veya Indel ihtiva gRNA / PAM dizileri kaçının.
- gRNA seçerken, silme genotipleme için alel-spesifik primerler tasarlama fizibilite kontrol edin. alel-spesifik primer tasarımı için bölüm 5'e bakın.
- Mali ve ark., 2013 15 tarif edilen protokole göre iki gRNA plazmidler birleştirin. Seçilen benzersiz 20 bp hedef sekans int dahilTablo 7'de gösterildiği gibi 61mer oligonükleotidlerin O (diziler yönlenme 'ila 3' 5'te gösterildi ve kalın bazlar birbirinin ters tamamlayıcı olan 20 bp'lik hedef sekans olarak).
- 10 uM gRNA Primer_F 10 ul bir tüp içinde 10 uM, tamamlayıcı Primer_R 10 ul karıştırın.
- 5 dakika boyunca 100 ° C 'de primer karışımı inkübe edilerek primerler tavlanması ve daha sonra 25 ° C'ye kadar 1 ° C / saniye soğutulur. Bu aşama için, bir PCR makinesi kullanmak ve kaynar su içinde tüp yerleştirmek ve oda sıcaklığına soğumaya bırakın.
- su 18.5 ul 5x HF tamponu 10 ul 10 mM dNTP, 1 ul karıştırmak ve yüksek- 0.5 ul: tavlanmış primer karışımı için her primeri uzatmak için 30 dakika boyunca 72 ° C 'de, aşağıdaki reaksiyon karışımı ekleyin ve inkübe doğrulukta bir DNA polimerazı ile birleştirildi.
- 100 bp'lik bir kılavuz parçalan üretilmiştir teyit etmek için bir% 2 agaroz jeli üzerinde hedef fragmanın 10 ul çalıştırın.
- gRNA vektörü (Geo bir hediye Linearizerge Kilisesi; Aşağıdaki reaksiyonu kullanarak Afi II ile Addgene plazmid # 41824) 15 kurmak: gRNA vektör omurgası (2-4 ug), 10x tampon 5 ul, Afi II 3 ul 5 ul (20 adet / ul) ve 32 ul suyun. 37 ° C'de 3 saat boyunca reaksiyon karışımı inkübe edin.
- % 1 agaroz jel üzerinde sindirilmiş ürünü çalıştırın ve 3.5 kb tekabül eden DNA bant üreticinin yönergeleri izleyerek bir jel ekstraksiyon kiti kullanılarak gRNA vektör linearize arındırmak.
- Ayarlama Gibson düzeneği reaksiyonları 29-30 doğrusallaştırılmış gRNA vektörü kullanılarak aşağıdaki şekilde aşama 1.2.3 fragmanı hedef lineer gRNA vektörü (50 ng / | il), hedef fragmanın 1 ul, 2x Gibson düzeneği ana karışımı 10 ul 1 ul su ve 8 ul. 60 dakika boyunca 50 ° C 'de reaksiyonlar inkübe edin.
- Montajlı gRNA Vector ile E.coli hücrelerinin dönüştürülmesi.
- 1.2.7 monte gRNA vektörü 1 ul karıştırınve DH5α (E.coli) bir tüp hücre 50 ul. 45 saniye boyunca 42 ° C'ye kadar hücrelerin maruz bırakarak ısı şoku yöntemi ile DH5α hücrelerini dönüştürmek.
- Ek bileşen 5 dakika boyunca buz üzerinde tüpler soğuk; SOC ortamı içinde 400 ul ve bir çalkalama inkübatöründe 45 dakika boyunca 37 ° C'de inkübe edin.
- LB-kanamisin (50 ug / ml), transforme edilmiş hücrelerin pozitif seçim için plaka DH5α 100 ul hücre yayıldı ve 37 ° C 'de O / N inkübe edin.
- GRNA Ekle Pozitif E.coli kolonileri Eleme.
- kanamisin 50 ug / ml ihtiva eden 3 mi LB bir kanamisin dirençli koloni ve tekrar süspansiyon al. 6-8 koloniler için tekrarlayın aynı ve bir çalkalama inkübatörde 37 ° CO / N tüm tüpler kuluçkaya yatmaktadır.
- Üreticinin kılavuzuna takip ederek plazmid, mini hazırlık kiti kullanılarak O / N yetiştirilen kültür plazmidler ayıklayın.
- GRNA dizisi insert kontrol etmek için bir EcoR I sindirim reaksiyon karışımı hazırlayınplasmidde. Enzim tampon maddesi, 2 ul, EcoR I 1 ul su, 15 ul aşağıdaki gibidir: Her bir örnek için, reaksiyon karışımı hazırlayın. 1.5 ml tüpler içinde, reaksiyon karışımı kısım ve plazmidin 2 ul ekle. 2 saat boyunca 37 ° C'de inkübe edin.
- % 1.5 agaroz jeli üzerinde sindirilmiş ürün çalıştırın.
Not: ekleme ile numuneler ekler olmadan klonlar 100'den bp yüksek olan bir 475 bp bandı boyutunu gösterecektir.
Not: Alternatif olarak, pozitif klonlar uç bir gRNA mevcudiyetinde bir 642 bp boyutlu fragmanı elde SP6 (ileri) ve T7 (geri) primerleri (Tablo 7) vektörü sekansına bağlanan kullanılarak koloni PCR ile taranabilir. GRNA dizisi içinde bir EcoR I sınırlandırma bölgesi olduğu zaman, koloni PCR yaklaşımı avantajlıdır.
- T7 kapsülü kullanılarak DNA dizilemesi ile gRNA ucun dizisini doğrulamak.
2. Transfeksiyon
Not:Elektroporasyon ES hücrelerine plazmidler transfekte etkili bir yöntemdir. Burada anlatılan yöntemi microporator transfeksiyon teknolojisini kullanır.
- 37 ° C /% 5 CO2 de ES hücre ortamı (Tablo 1) 10 ml içeren 10 cm'lik bir jelatin kaplı tabak F1 ES hücrelerinin büyütün. Hücreler% 85 ulaştığınızda confluency ortamını çıkarın ve tripsin 2 ml ekleyin. 5 dakika boyunca CO2 inkübatöründe 37 ° C'de inkübe edin.
Not: F1 ES hücreleri Barbara Panning 24 elde edilmiş ve istek üzerine mevcuttur bulundu. - Spin medya (Tablo 2) 10 ml ekleyerek tripsin nötralize. Pipet defalarca tamamen hücreleri ayırmak için.
- 5 dakika boyunca 300 x g'de 15 ml'lik bir tüp ve bir dönüş tüm hücreleri toplamak. 3 ml PBS içinde süspanse ve bir hemasitometre veya otomatik hücre sayacını kullanarak hücreleri saymak.
- (R 100 ul, 5 dakika tekrar süspansiyon 300 x g santrifüj ile 1.5 ml bir tüp içinde 1 x 10 6 ES hücrelerinin PeletKit, üretici tarafından tedarik edilen yeniden süspansiyon) tampon maddesi.
- (Kiran Musunuru bir hediye; Addgene plazmid # 44719) 5 mikrogram pCas9_GFP her ekleyin 31, 5 've 3' hedef bölgenin silinmesi için gRNA plazmidler ve kabarcıkların giriş önlemek için bir pipet ile hafifçe karıştırın.
- ucunda bir balon önlemek için dikkatli olmak, elektroporasyon karışımı 100 ul aspire elektronik pipet kullanın.
- Program volt, genişlik ve elektroporasyon için bakliyat. F1 ES hücreleri için, 3 bakliyat 1.400 V, 10 msn kullanın.
- elektroporasyon çalışırken çözelti içinde herhangi bir kıvılcım izlemek için ucu gözlemlemek. Bir kıvılcım bir hava kabarcığı varlığını gösterir ve transfeksiyon engel olacaktır.
- 10 mi ES hücre ortamı (Tablo 1) ihtiva eden bir 10 cm jelatin kaplı tabak içine transfekte ES hücrelerinin çıkarın ve 37 ° C /% 5 CO2 inkübe edilir.
Transfekte edilmiş hücreler Sıralama 3. FACS
- 48 saat sonra, tripsin 2 ml ekleyerek hücreleri ayırmak ve 5 dakika CO2 inkübatöründe 37 ° C'de inkübe edin.
- Toplama tamponu (Tablo 3), 10 ml ilave edilerek plaka nötralize. 5 dakika boyunca 300 x g'de 15 ml'lik bir tüp ve bir dönüş hücreleri toplamak.
- Süpernatantı atın ve sıralama tamponu (Tablo 4) 1 ml hücreleri tekrar süspansiyon. sıralama platformu dayalı hücreleri ve seyreltik güvenin. 15 ml tüpler içine sıralama için 96 oyuklu plakalar doğrudan tek tek hücrelerin sıralamak için 0.5-1 x 10 6 hücre / ml hücre seyreltilir 2-5 x 10 6 hücre / ml hücre seyreltin.
- 32 sitometresi FACS akış kullanarak sırala Cas9-GFP + ES hücreleri. Şirketinden / göz 100 ul ES hücre ortamı içeren jelatin kaplı 96-çukurlu plakalara Hücre çıkarma ya da sıralama tek tek hücreler için 3.5 de tarif edildiği gibi (2 mi kurtarma ortamı (Tablo 5) ve plaka tüplerine toplu hücreleri toplamak Tablo 1).
- Tohum 1-1.5 x 10 4 GFP 10 ml ES hücre ortamı (Tablo 1) ihtiva eden bir 10 cm jelatin kaplı çanak + ES hücreleri. Bu düşük yoğunlukta Kaplama bireysel ES hücre kolonileri toplama kolaylaştıracaktır.
Genotiplendirme, İfade Analizi ve Donma Hücre Stoklar 4. Kültürleme Klonlar
- sıralama gün sonra 4-5, ES hücre kolonilerinin mevcudiyetinde doğrudan kriteri 96 oyuklu plakaların her bir puan.
- medya kaldırılması ve tripsin 30 ul ekleyerek ES hücre kolonileri ayırmak. 5 dakika boyunca 37 ° C'de inkübe edin. ES hücre medya (Tablo 1) 170 ul ekleyerek tripsin nötralize ve tek hücre içine koloninin tam ayrışma için aşağı yukarı pipet ve. En kuyular% 70 konfluent üzerinde (genellikle 2-3 gün) kadar 37 ° C /% 5 CO 2 hücreleri büyütün.
- Alternatif kullanarak 10 cm yemekleri tek tek ES hücre kolonileri almakters mikroskop. jelatin ile ön-muamele, 96 oyuklu bir plakanın bir oyuk her koloni yerleştirilmesi pipet takip aşama 4.1.1 içine koloni aspirasyon ve tripsin 30 ul ihtiva eden sonra.
Not: kolonilerin biri tüm satır aldı ise Kolonileri oda sıcaklığında tripsin oturabilir. - Bir kez tüm koloniler toplanmış ve en kuyu% 70 konfluent (genellikle 2 gün) bitene kadar ortam CO2 inkübatöründe 37 ° C'de hücrelerin büyümesine ayrışması edilmiştir.
- 96 oyuklu plakalar, yarma için hazır olduğunda, ortamını çıkarın tripsin 30 ul eklenir ve 5 dakika boyunca 37 ° C'de inkübe edin. Tek hücre içine tam bir ayrılma için aşağı doğru her bir ve pipet up ES hücre ortamı 180 ul (Tablo 1) ilave edilmesi ve tripsin nötralize.
- Elde edilen 210 ul kaynaktan üç jelatin kaplı 96 oyuklu plakalar ES hücre ortamı / oyuk (Tablo 1) 130 ul ihtiva eden her birine 70 uL tohum. ge için bu tabak kullanınnotyping ekspresyon analizi ve aşağıda tarif edildiği gibi, her klon için hücre stokları dondurulması.
- genotiplendirme plakası% 70-85 izdiham ulaştığında "silme genotiplenmesi" bölümünde 6 açıklandığı gibi, plaka davranın.
- söylem analizi plakası% 70-85 izdiham ulaştığında, ortamı çıkarın klonlar genotip edilene kadar -80 ° C'de bandı ve mağaza sızdırmazlık plakasını mühür.
Not: ifade analiz plaka klonları erken pasajlar gen ekspresyonu değişiklikleri analiz etmek için kullanışlıdır. 96 gözlü plaka gen ekspresyonu analizi mümkündür, ancak hücre sayıları düşük bir RNA mikro ekstraksiyon kiti önerilir. - 96 gözlü levhalar Donma Stokta hazırlanması:
- Dondurulmuş hücre stokları (stok-1) plakası 70-85% konfluansa ulaştığında, ortam aspire tripsin 30 ul eklenir ve 5 dakika boyunca 37 ° C'de inkübe edin.
- ES hücre ortamı 100 ul eklenmesiyle tripsin nötralize (Tablo 1) tHer iyi ve pipet yukarı ve aşağı tek hücre içine tam ayrılma için o.
- Aktarım her bir oyuğa, iki jelatin kaplı 96 oyuklu plakalar asılı hücre 15 ul, ES hücre ortamı (Tablo 1), her biri 185 ul ve 37 ° C /% 5 CO2 büyümeye izin verir.
Not: Bu stokta-2 için ve hisse senedi-1 hücreler canlandırıcı durumunda ek yedekleme klonlar -3 tabak başarılı değildir.
- Bu arada, 96 oyuklu bir plaka (stok-1) 'de kalan hücreler, 100 ul, 2x 100 ul ortam (Tablo 6) dondurma ekleyin. bir sızdırmazlık bandı ile plaka Seal ve hızlı bir şekilde uygun karıştırma için plaka 4-5 kez ters çevirin. klonlar genotip kadar -80 ° C 'de plaka saklayın.
- Hazır-2 ve hazır-3 plakalar medya aspire dondurma için hazır olduğunda, tripsin 30 ul eklenir ve 5 dakika boyunca 37 ° C'de inkübe edin. (ES hücre medya 70 ul ekleyerek T tripsin nötralize) 1 mümkün her kuyuya ve pipet yukarı ve aşağı komple ayrışma için tek hücre içine.
- 2x donma medya 100 ul ekleyin sızdırmazlık bandı ile plaka mühür ve hızlı bir şekilde plaka düzgün karıştırma için 4-5 kez ters çevirin. Bu plakalar, gerekli olana kadar -80 ° C'de plaka saklayın.
5. Allel özel Primer Tasarım
- Tasarım primerler (Şekil 1C) 4 adet, istenen silme için klonları taramak üzere: primerler dış primerler, aşağıda tarif edildiği gibi gRNA (hem 5 've 3' gRNA hedef sahaları için), flanking primerler içinde.
- http://labs.csb.utoronto.ca/mitchell/crispr.html az 129 ve Cast genotipleri karşılık gelen SNP parçayı alın. Verilen parça mm9 fare genomu montaj koordinatlarında 129 ve Cast arasındaki baz değiştirmelerin gösterir.
Not: Yukarıdaki yerinde bağlantı ge UCSC genom tarayıcı yönlendirme arasındaki 129 SNP içeren özel bir parça eklemek ve Cast olacaknomes. - Bölgenin koordinatları girin silinir. > 3 SNP içeren istenen silme ortasına yaklaşık 500 bp'lik bir bölge büyüt.
- seçenek çubuğunda> DNA görüntüleyebilir ve tüm büyük harf biçiminde hedef dizisini indirmek için olsun DNA tıklayın gidin.
- İki FAŞTA dizileri oluşturun; 129 için bir ve SNP konumundaki baz ikamesi Cast için. bir küçük harf ile SNP işaretleyin.
- artı (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) Primer3 gidin ve SNP 129 dizisi ikame yapıştırın. primerler tasarlamak için varsayılan ayarları kullanın.
- iç alel-spesifik primerler görev açılan menüde Primer_List seçin ve Astarlar seçin tıklayın tasarlanması. 3 'ucu ya da 3 ila 4 baz içinde "ya bir SNP ile bölgenin silinmesi tam içine ileri ya da ters primeri seçin.
Not: 3 'ucuna ekrana bir SNP sahip Astarlar qPCR alleli özgüllük arttı. - ana sayfaya ikinci primer dönüşünü seçmek için, görev açılan menüden Algılama seçin ve sayfanın altındaki uygun kutuya ilk primer dizisi yapıştırın. Genel Ayarlar sekmesinde 80-200 bp ürün boyut aralığı ayarını değiştirmek ve Astarlar seçin tıklayın. Listelenen primer çiftleri erkene bir primer seçti; Bu 129 içindeki alel-özgül primerler olacaktır.
- Tekrarlayın Cast alel için primerler tasarlamak için 5.1.5 5.1.7 için yineleyin.
- UCSC genom tarayıcıya dönmek ve bölgenin koordinatları silinecek girin. görüntülemek için Git> Dizi Alma Bölge Seçenekleri altında çubuğunda, DNA yukarı 1000 bp eklemek ve alt hedef dizisini indirmek için DNA elde tıklayın.
- Parantez içindeki gRNA hedef dizisini işaretleyin. Devam etmeden önce tüm bu diziyi kaydedin.
- dış primerler iki gRNA hedef diziler arasında dizisini kaldırmak tasarlanması. dış alel-spesifik primer tasarım adımı yineleyin 5.1.4-5.1.8s ancak ürün boyutunu 400-800 bp değiştirin.
- Iki dizilerine gRNA hedef dizinin 500 bp 5 've 3' ile birlikte, her adımda 5.1.10 elde edilen sekansı bölün. Yineleyin gRNA yan bölgeler için 5.1.5 5.1.7 tasarlayarak olmayan alel spesifik primerler adımları ancak ürün boyutunu 400-800 bp değiştirin.
olmayan alel spesifik primerler için, ya da 129 ya da dökme dizisi kullanılabilir ve primerleri, bir SNP içermeyen seçilmelidir: edin. Bunun dışında tasarımı ve gRNA primerler yan için 400-800 bp ürün boyutunu ayarlamak için tavsiye edilir. Bu küçük indeller mevcut olsa bile amplifikasyonu için izin verir.
- http://labs.csb.utoronto.ca/mitchell/crispr.html az 129 ve Cast genotipleri karşılık gelen SNP parçayı alın. Verilen parça mm9 fare genomu montaj koordinatlarında 129 ve Cast arasındaki baz değiştirmelerin gösterir.
- 2 ng / ul saf 129 ve Döküm suşu genomik DNA kullanılarak qPCR alel özgüllük iç primerler test edin. qPCR reaksiyonu kurmak için adım 6,2-6,4 izleyin.
Not: hedef bölgede 129 genotipi 129 DNA yerine kullanılabilir C57BL / 6J, C57BL / 6J DNA ile aynı ise. Alel-özgül primerler, en az 5 döngü göstermelidirYanlış genotip vs doğru üzerinde Ct (döngüsü eşik) değeri arasındaki farktır. dış primerleri Cas9 / gRNA ES hücreleri transfekte kullanarak silinmesi yükseltmek sağlamak için test ve F1 genomik DNA sırasıyla pozitif ve negatif kontroller yapılabilir. monoallelik klon tanımlanmış olan dış primerlerin alel-özgüllük test edilebilir.
6. Genotiplemesi Silme
- Koloni genişlemesinden sonra oluşturulan aşama 4,6 plaka kullanılarak genotipleme 96 oyuklu plaka genomik DNA ekstrakte edin.
- genomik DNA ekstraksiyon karışım hazırlayın: su 89 ul 10x tampon 10 ul (üretici tarafından sağlanan) ekstraksiyon reaktifi 1 ul. her bir genomik DNA ekstraksiyon karışımı 100 ul ilave edin ve bir sızdırmaz bant plaka mühür.
- 5 dakika boyunca 95 ° C'de ve ardından 5 dakika süreyle 75 ° C'de inkübe plakası.
- Plaka birkaç dakika An buz üzerinde inkübe edilerek soğumasını bekleyind sonra santrifüj kısaca kuyunun dibine herhangi yoğunlaşmayı yerleşmek için. Bu silme taraması için model DNA plakası olarak işlev görür.
- aşağıdaki gibi her klon için iki nüsha halinde qPCR reaksiyonlar ayarlayın: 2x SYBR qPCR karışımı 5 ul, ileri ve primer (3 uM) her 1 ul ve su 1 ul ters. 384 oyuklu plakanın her bir reaksiyon karışımı 8 ul ve ardından şablon DNA, 2 ul eklemek için bir çok kanallı pipet kullanın.
- 2 dk içeriğini karıştırmak için 600 x g'de bandı ve spin sızdırmazlık plakasını mühür. Gerçek zamanlı cycler 384-plaka dizi yerleştirin.
- Program algılama erime eğrisi analizine ve ardından 2-aşamalı bir PCR için gerçek zamanlı Cycler aşağıdaki gibi 10 dakika boyunca 95 ° C 'de 1 döngü, 15 saniye 95 ° C 40 döngü, 62 ° C plaka okuma 30 sn 10 sn için 95 ° C, 5 saniye + plaka boyunca 5 ° C'lik artış ile 95 ° C ila 65 ° C oku.
Not: Ayrıca tasarım primer için, qPCR mix ve döngü parametreleri de özgüllüğü primer katkıda bulunur. malzeme listelenen yukarıda tarif edilen parametreler ve reaktifler daha sık alel spesifik amplifikasyon verir. - qPCR Sonuçları çözümleme
- içindeki alel-özgül primerler ile amplifikasyonu için, her alel edin. Hiç kimse alel amplifikasyonu ya da alel arasındaki yüksek Ct değer farkları (> 5 siklus) Bu klonlar yüksek / yok Ct değeri ile allelinin heterozigot silinmesini taşımak öneririz. Her iki alel hiçbir amplifikasyon bir homozigot delesyonu taşıyan önerir.
- dış alel-özgül primerler ile amplifikasyonu için, her alel edin. Hedef silinmesi dış primerleri ile 1 kb amplifikasyon daha büyük olduğunda, bir silinmesi, bu olduğu zaman meydana gelir. 22-28 A Ct değeri silme onaylar. 1 kb daha küçük hedef silmeleri için elektroforez ile amplikon boyutunu onaylayın.
Not: Dış primerler sadece ılımlı alel-özgüllük gösterirse (figu bkz) 2 Yeniden, amplikonları dışında nedeniyle primerlerin hedef dışı amplifikasyon monoallelik klonlar alel astar setleri hem de elde edilebilir. Bu durumda en az beş döngüleri iki allel arasındaki Ct değeri farkı (alt Ct değeri) iç primerler elde edilen sonuçlara dayalı olarak silinir doğru alel teyit etmelidir. kapalı hedef alel Ct fark karşı hedef ise en az beş döngüleri yeni alel spesifik dış primerler dizayn. - monoallelik silme klonlar içinde gRNA primerler kuşatan, ikincil tarama kullanılarak silinmemiş alel bütünlüğü için kontrol edin.
gRNA hedef bölgede yaklaşık> 25 bp büyüklüğü indeller 400-800 bp'lik amplikonlarının için qPCR olarak erime eğrisi bir kayma gözlemlenerek belirlenebilir: edin. Seçenek olarak ise, gRNA komşu tetikleyiciler amplikonlarının <25 bp küçük indeller tespit etmek için sekanslanabilir.- 2 gRNA komşu tetikleyiciler örneğin 5 setleri 've 3' g qPCR gerçekleştirmekRNA CRISPR silme üretilmesinde kullanılan. primerleri bu setleri ile hiçbir amplifikasyon qPCR amplikon daha büyük indeller monoallelik silme klonlarının olmayan silinen alleli gRNA hedef yerinde mevcut olduğunu gösterir. sonuçlar olarak daha fazla analiz gelen bu büyük indellerin içeren ıskarta klonlar silme kapsamını bilmeden yorumlamak zor olabilir.
- PCR üreticinin talimat aşağıdaki kiti temizlemek kullanarak dış astar qPCR reaksiyon elde edilen amplikonları arındırın.
- önceki adımda elde edilen saflaştırılmış PCR ürünü DNA dizilimi tarafından silinen alel dizisi onaylayın. İleri için qPCR amplifikasyon primerleri kullanın ve sıralama ters.
Not: Silinen alel genotipi ikincil onay olarak amplikon hareket içinde bu sahne SNP anda.
Allel Spesifik astar 7. Analiz Anlatım
- 96-iyi hücre hazır pl çözülmeSıcak bir boncuk banyosu üzerine yerleştirerek -80 ° C (stok-1 adım 4,9) saklanan yedik. plakasındaki kuyuların yarısından fazlası 5 dakika boyunca 300 xg'de, sıkma çözülmüş zaman.
- Dikkatle, sızdırmazlık bantı çıkarın ve hızlı bir şekilde ES hücre ortamı (Tablo 1), 1 ml ihtiva eden jelatin kaplı 12-çukurlu plakalara silinmesi pozitif olan yuvalardan hücreleri transferi ve 37 ° C /% 5 CO2 inkübe edilir.
- Plakası 70-85% konfluansa, geçiş hücreleri ulaşır ve jelatin kaplı, 6-çukurlu plaka üç kuyu, ES hücre ortamı (Tablo 1), her biri 2 ml içine split. Uzun süreli (aşama 8'de tarif) Sıvı azot içinde saklama ve RNA ekstraksiyonu için, üçüncü bir kuyu için donmuş hücre stokları 2 şişe hazırlamak için iki kuyu kullanın.
- bir RNA özütleme kiti kullanılarak RNA ekstrakte edin.
- transkripsiyonunu (RT), üretici protokolüne göre bir cDNA sentez kiti kullanılarak RNA cDNA'ya RNA'nın 100-500 ng dönüştürün. bir RT N,Her RNA örnek için egative reaksiyon RNA örnekleri DNA kirletici miktarını izlemek için.
- 2 ve 1: 1 arasında bir oranda qPCR önce cDNA seyreltilir 4; ES hücreleri, hedef genin ekspresyonu seviyesine bağlı olarak.
- transkript seviyeleri mutlak ölçümü için standart bir eğri (0.08 ng / ul, 250 ila 5 kat seyreltmeleri) halinde F1 genomik DNA da dahil olmak üzere yukarıda tarif edildiği gibi qPCR olarak ayarlayın. Örnek GAPDH (Tablo 7'de listelenen primerler), uygun bir kontrol geni için her teyit silinmiş klonun ilgi konusu genin her alel ekspresyonunu karşılaştır.
Not: Kontrol gen primerler alel spesifik olması gerekmez. amplifikasyonu için hedef bölgesinin dışında genotipleme primerler için tarif edildiği gibi alel-spesifik primer tasarımı RT-qPCR primerleri için de aynıdır. Gen sekansı kullanılmalıdır; Tek bir ekson veya bir ekson-intron sınırı için primerler kullanılarak, eğer F1 genomik DNA, için kullanılabilir (primer transkriptin izlemek için)standart eğri. RT-qPCR ilgili daha fazla bilgi için Forlenza ve arkadaşlarına bakınız. 2012 33.
ES Hücrelerinin Uzun vadeli Depolama 8. Freeze Stok Hazırlık
- (Aşama 7.3) her biri 6-oyuğuna tripsin 300 ul ilave edin ve 37 ° C'de 5 dakika inkübe edilir. Tek hücre içine ayrılma yukarı ve aşağı birkaç kez tripsin ve pipet nötralize etmek için eğirme ortamı (Tablo 2) 2 ml ilave edilir.
- 5 dakika boyunca 300 x g'de 15 ml'lik bir tüp ve döndürme hücreleri aktarın.
- Süpernatant aspire ve ES hücre ortamı (Tablo 1) 500 ul ilave edin. hücrelerin tekrar süspansiyon aşağı yukarı pipet ve.
- 1.5 ml cryovial tüp içerikleri transferi ve 2x ES hücre dondurma ortamı (Tablo 3) 500 ul ilave edin. Tersine tüp İyice karıştırın ve alkol içermeyen hücre dondurma kabına tüp yerleştirin. transferrin önce en az 12 saat boyunca -80 ° C 'de konteyner dondurma bu hücre yerleştirinSıvı azot depolama tankı içine örn.
Sonuçlar
Burada açıklanan protokol CRISPR / Cas9 genom düzenleme (Şekil 1) kullanılarak oluşturulan monoallelik güçlendirici silinen hücrelerin gen ekspresyonunun cis Ziraî çalışma F1 ES hücreleri kullanır. genotipleme ve gen ifadesi için gRNA ve alel-spesifik primer tasarımı bu yaklaşımda önemli faktörlerdir. Her alel-spesifik primer seti alel özgünlüğünü teyit etmek qPCR tarafından onaylanmalıdır. Sadece, ilgili genomik DNA hedef amplifiy...
Tartışmalar
CRISPR / Cas9 aracılı genom düzenleme teknolojisi genom değişiklik için, basit, hızlı ve ucuz bir yöntem sağlar. Fonksiyonel güçlendirici karakterizasyonu için monoallelik güçlendirici iptali gerçekleştirilebilir ve analiz etmek için burada açıklanan yöntem F1 fare hücrelerinde SNP yararlanır. Bu tip bir yaklaşımın avantajları şunlardır: 1) monoallelik güçlendirici silme kritik güçlendirici örneğin, her iki allellerinin ölümcül hücre gelen düzenlenmiş genin protein düze...
Açıklamalar
Yazarlar çıkar çatışması Jüpiter politikalarını okumak ve ifşa çatışmaları hiçbir sahibiz.
Teşekkürler
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Malzemeler
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Referanslar
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiThis article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır