
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Создание CRISPR / cas9 опосредованного моноаллельной Пропуски для изучения Enhancer Функция в эмбриональных стволовых клетках мышей
В этой статье
Резюме
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Аннотация
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Введение
Транскрипциональные регуляторные элементы имеют решающее значение для пространственно-временной тонкой настройки экспрессии генов в процессе развития 1 и модификации этих элементов может привести к болезни из - за аберрантной экспрессии генов 2. Многие болезни ассоциированные регионы , выявленные генома широких исследований ассоциации в некодирующих регионах и имеют особенности транскрипционных энхансеров 3-4. Идентификация и улучшающие сопоставления их с генами , которые они регулируют затруднено , поскольку они часто расположены несколько т.п.н. от генов , которые они регулируют и могут быть активированы в порядке 5-6 тканесецифического. Прогнозы Enhancer обычно основаны на модификации гистонов марок, медиаторов-Cohesin комплексов и связывание транскрипции типа специфических клеток факторов 7-10. Проверка предсказанных усилителями чаще всего делается с помощью основанного анализа вектора , в котором энхансер активирует экспрессию гена - репортера , 11-12. Эти данные обеспечивают Valuable информация о регуляторного потенциала предполагаемых последовательностей энхансера, но не раскрывают их функции в их эндогенного геномного контекста или идентифицировать гены, которые они регулируют. редактирования Геном служит мощным инструментом для изучения функции транскрипционных регуляторных элементов в их эндогенного контекст с потерей функции анализа.
Последние достижения в области редактирования генома, а именно / cas9 системы редактирования генома CRISPR, облегчают исследование функции генома. Система / cas9 CRISPR проста в использовании и предназначен для многих биологических систем. Белок cas9 нацелен на определенный сайт в геноме с помощью РНК направляющей (gRNA) 13. Комплекс SpCas9 / gRNA сканирует геном для своей целевой геномной последовательности , которая должна быть не менее 5 'к последовательности protospacer смежно мотив (РАМ), NGG 14-15. База спаривание gRNA к своей цели, то 20 нуклеотидов (нт) последовательность, комплементарную gRNA, активирует нуклеазы активность SpCas9 приводит в DOUBLе нить брейк (DSB) 3 б.п. выше последовательности PAM. Специфичность достигается за счет полного спаривания оснований в запальной зоне gRNA, 6-12 нт рядом с ПАС; И наоборот, не соответствует 5 'от семени, как правило , переносится 16-17. Введенный DSB можно отремонтировать либо негомологичные конец соединительной (NHEJ) репарации ДНК или гомологии направлены ремонт репарации ДНК (HDR) mechanisms.NHEJ часто создает вставки / удаления (вставкам) из нескольких пар оснований на целевом сайте, что может привести к нарушению открытая рамка считывания (ORF), гена. Для того, чтобы генерировать более крупные делеции в геноме двух gRNAs, которые фланкируют область интереса, может быть использован 18-19. Этот подход особенно полезен для изучения транскрипционных энхансеров , сгруппированных в локус контроля областей или супер-усилителями , которые больше , чем обычные усилители 9,18,20-22.
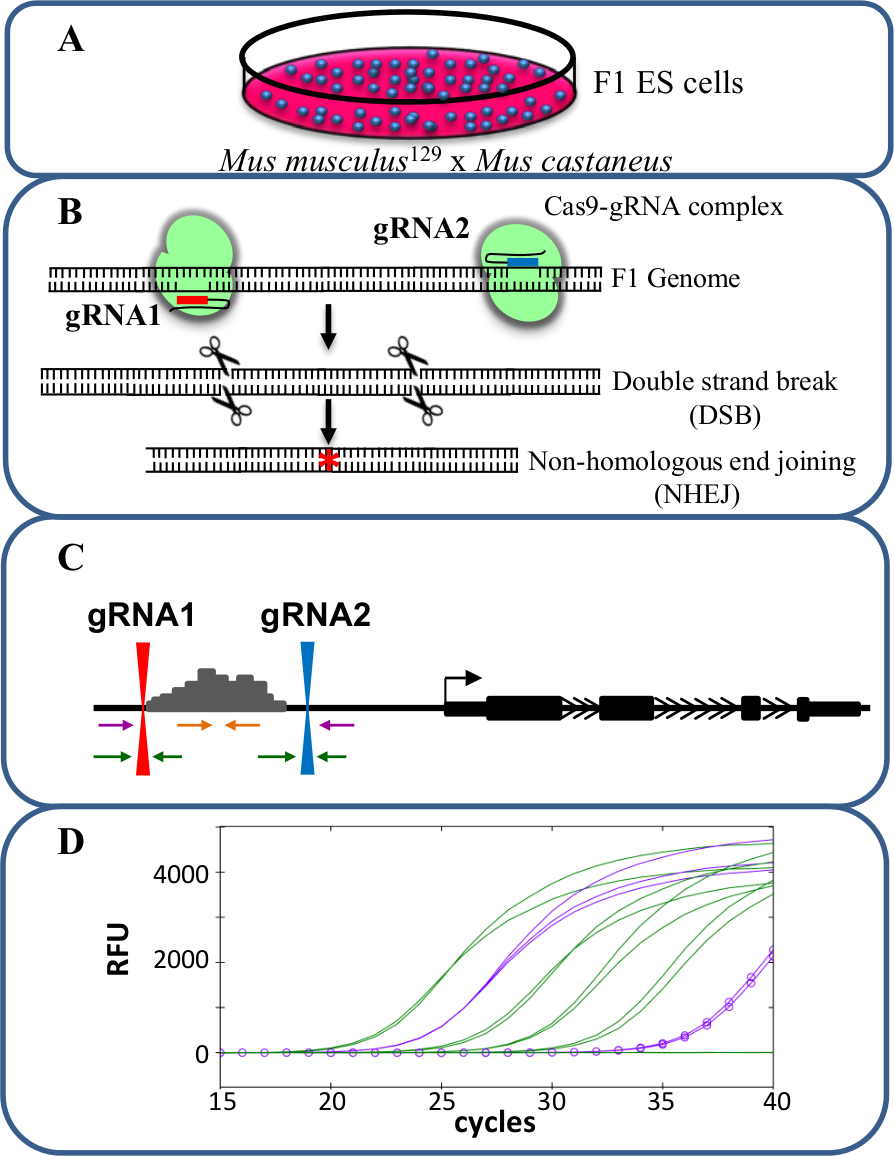
Моноаллельной делеции являются ценным моделью для изучения цис -regulation транскрипции. Наблюдаемое чане в уровне транскрипта после моноаллельной удаления энхансер коррелирует с ролью этого усиливающего в регуляции генов без искажающих эффектов, которые могут возникнуть при транскрипции обоих аллелей влияет, потенциально влияющих на клеточную пригодность. Оценивая приведенное выражение трудно, однако без способности различать удаленные от дикого типа аллеля. Кроме того, генотипирование делеции в каждом аллеля без способности различать два аллеля является сложной задачей, особенно для больших удалений> 10 кб до 1 Мб 23 , в котором оно трудно усилить весь дикий регион типа с помощью ПЦР. Использование клеток F1 ES , генерируемых путем скрещивания Mus Musculus 129 с Mus castaneus позволяет двум аллели быть дифференцированы по аллель-специфической ПЦР 18,24. Гибридный генома в этих клетках способствует аллель специфический скрининг удаления и анализа экспрессии. В среднем является SNP каждые 125 пар оснований между этими двумя геномами, Обеспечивая гибкость при проектировании праймера для экспрессии и генотипирование анализов. Наличие одного SNP может влиять на температуру плавления праймера (Т м) и целевой специфичность в реальном масштабе времени количественной ПЦР (КПЦР) амплификации , допускающие дискриминацию двух аллелей 25. Кроме того несоответствие в пределах 3' - конца праймера в значительной степени влияет на способность ДНК - полимеразы , чтобы простираться от праймера амплификации , предотвращая нежелательную мишени аллеля 26. Описанная в следующем протоколе является использование клеток F1 ES для аллельных специфических усиливающих делеции более 1 кб и последующего анализа экспрессии с помощью / cas9 системы редактирования генома CRISPR (Рисунок 1).

Рисунок 1. Enhancer удаление с помощью CRISPR / cas9 для изучения цис -regавляет экспрессии генов. (A) F1 ES - клетки , генерируемые помесь Mus Musculus 129 и Mus castaneus используются для обеспечения аллель специфического удаления. (B) , два направляющих РНК (gRNA) используются , чтобы вызвать большой cas9-опосредованное удаление области энхансера. (C) наборов праймеров используются для идентификации больших моно- и би-аллельные делеции. Оранжевые праймеры внутренние праймеры, пурпурные праймеры являются внешними праймеры и зеленые праймеры фланкирующие праймеры gRNA. (D) Изменения в экспрессии генов контролируются с помощью аллель-специфической КПЦР. РФС обозначает относительные единицы флуоресценции. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
протокол
1. Проектирование и Построение gRNA
- Для удаления транскрипционных энхансеров регионы используют два gRNAs, один 5 'и 3' один из интересующей области. Используйте дорожку генома мыши браузера УСК , порожденную лаборатории Чжан , чтобы идентифицировать уникальные последовательности gRNA (http://www.genome-engineering.org 15). Затем проверить эти gRNAs и прилегающие к ним РАМ для ОНП и вставкам с помощью онлайн - инструментов , предоставляемых Sanger Institute (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211~~HEAD=pobj) 27-28. Чтобы настроить таргетинг на оба аллеля с одинаковой эффективностью, избегать / ПАМ последовательностей gRNA, которые содержат SNP или INDEL.
- При выборе gRNA, проверьте возможности проектирования аллель-специфических праймеров для генотипирования удаления. Обратитесь к разделу 5 для разработки праймера аллель-специфической.
- Соберите два плазмид gRNA на основе протокола , описанного в Мали и др. 2013 15. Включать выбранный уникальный 20 н.п. целевой последовательности IntO олигонуклеотиды 61mer , как показано в таблице 7 (последовательности отображаются в 5' - 3 'ориентации, а полужирный основаниями являются 20 пар оснований последовательности - мишени , которые являются обратные комплементы друг друга).
- Смешайте 10 мкл 10 мкМ gRNA Primer_F и 10 мкл 10 мкМ комплементарной Primer_R в трубке.
- Отжигу праймеров путем инкубирования смеси праймера при 100 ° С в течение 5 мин, а затем охлаждают на 1 ° C / сек до 25 ° C. Для этого шага, используйте машину PCR или поместите трубку в кипящую воду и дайте ему остыть до комнатной температуры.
- Для отожженного праймера смеси, добавьте следующую реакционную смесь и инкубировать при 72 ° С в течение 30 мин, чтобы расширить каждого праймера: 18,5 мкл воды, 10 мкл 5х буфера HF, 1 мкл 10 мМ дНТФ Перемешать и 0,5 мкл высоко- точность ДНК-полимеразы.
- Выполнить 10 мкл целевого фрагмента на агарозном геле 2%, чтобы подтвердить 100 фрагменты руководства BP были произведены.
- Линеаризовать вектор gRNA (подарок от GeoRGE Церковь; Addgene плазмида # 41824) 15 с AFL II с помощью следующей реакции установить: 5 мкл gRNA вектора позвоночника (2-4 мкг), 5 мкл 10х буфера, 3 мкл AFL II (20 ед / мкл) и 32 мкл воды. Инкубируют реакционную смесь в течение 3 ч при 37 ° С.
- Запуск расщеплении на агарозном геле 1% и очищают полоса ДНК, соответствующая 3,5 кб линеаризуется gRNA вектор с использованием набора для экстракции из геля, следуя инструкциям производителя.
- Установить монтажные реакции Gibson с использованием 29-30 линеаризованного вектора gRNA и целевой фрагмент из шага 1.2.3 следующим образом : 1 мкл линейного gRNA вектора (50 нг / мкл), 1 мкл целевого фрагмента, 10 мкл 2x Gibson сборочного мастер - микс и 8 мкл воды. Инкубируйте реакции при 50 ° С в течение 60 мин.
- Трансформация клеток E.coli с Собранный gRNA Вектор.
- Смешайте 1 мкл собранного вектора gRNA из 1.2.7и 50 мкл DH5 & alpha ; (Е. coli штамма) клеток в трубке. Трансформации клеток DH5 & alpha; методом теплового шока путем экспозиции клеток до 42 ° С в течение 45 сек.
- Привязка охладить трубы на льду в течение 5 мин; Затем добавляют 400 мкл SOC среды и инкубируют при температуре 37 ° С в течение 45 мин в качалке инкубаторе.
- Спред по 100 мкл клеток DH5 & alpha на пластине для позитивной селекции трансформированных клеток в LB-канамицин (50 мкг / мл) и инкубировать O / N при 37 ° С.
- Скрининг положительный E.coli Колонии для gRNA вставки.
- Выберите устойчивую колонию канамицину и ресуспендируют в 3 мл LB, содержащей 50 мкг / мл канамицина. Повторите то же самое для 6-8 колоний и инкубировать все пробирки при 37 ° CO / N в шейкере инкубаторе.
- Извлечение плазмид из O / N выращенной культуры с использованием плазмиды мини-набор подготовительную, следуя инструкцией производителя.
- Подготовить реакционную смесь переваривание EcoR I для проверки последовательности вставки gRNAв плазмиде. Для каждого образца готовят реакционную смесь следующим образом : 2 мкл буфера фермента, 1 мкл EcoR I, 15 мкл воды. Алиготе реакционную смесь в 1,5 мл пробирки и добавляют 2 мкл плазмиды. Инкубировать пробирки при 37 ° С в течение 2 часов.
- Запуск расщеплении на 1,5% -ном агарозном геле.
Примечание: Образцы с вставкой будет отображаться размер полосы на 475 б.п., что на 100 б.п. выше клонов без вставок.
Примечание: В качестве альтернативы, положительные клоны могут быть подвергнуты скринингу с помощью ПЦР колоний с использованием SP6 (вперед) и Т7 (обратный) праймеров (таблица 7) , которые связываются с векторной последовательности , чтобы дать фрагмент размером в 642 пар оснований в присутствии gRNA вставки. Колонию ПЦР подход выгоден , когда имеется сайт рестрикции EcoR I в пределах последовательности gRNA.
- Подтвердите последовательность gRNA вставку Секвенирование ДНК с помощью Т7 праймера.
2. Трансфекция
Заметка:Электропорация является эффективным методом трансфекции плазмид в клетки ES. Описанный здесь метод использует технологию microporator трансфекции.
- Рост клеток F1 ES в 10 см желатин покрытием блюдо, содержащую 10 мл клеточных сред ES (таблица 1) при 37 ° C / 5% CO 2. Когда клетки достигают 85% конфлуэнтности удалить медиа и добавьте 2 мл трипсина. Инкубируют при 37 ° С в инкубаторе СО 2 в течение 5 мин.
Примечание: F1 ES - клетки были получены из Барбаре панорамирование 24 и доступны по запросу. - Нейтрализовать трипсина путем добавления 10 мл спин носителя (таблица 2). Пипетка несколько раз, чтобы полностью отделить клетки.
- Соберите все клетки в 15 мл трубки и спина при 300 мкг в течение 5 мин. Ресуспендируют в 3 мл PBS и подсчета клеток с помощью гемоцитометра или автоматического счетчика клеток.
- Гранул 1 × 10 6 клеток ES в 1,5 мл трубки центрифугированием при 300 х г в течение 5 мин и ресуспендируют в 100 мкл R (ресуспендирования) буфер, поставляемый изготовителем комплекта.
- Добавьте 5 мкг каждого из pCas9_GFP (подарок от Kiran Musunuru; Addgene плазмида # 44719) 31, 5 'и 3' плазмид gRNA для удаления целевого региона и аккуратно перемешать с помощью пипетки , чтобы избежать введения пузырьков.
- Используйте электронный наконечник пипетки отсосать 100 мкл электропорации смесь, соблюдая осторожность, чтобы избежать пузыря на кончике.
- Программирование вольт, ширина и импульсы для электропорации. Для клеток F1 ES, используют 1,400 В, 10 мс в течение 3 импульсов.
- В то время как электропорация работает наблюдать за кончик, чтобы наблюдать за искр в растворе. Искра указывает на наличие воздушных пузырьков и будет мешать трансфекцией.
- Выброс трансфектированных ES клеток в 10 см желатин покрытием блюдо, содержащую 10 мл эмбриональных стволовых клеток к среде (таблица 1) и инкубировать при 37 ° C / 5% CO 2.
3. FACS Сортировка трансфицированных клеток
- Через 48 ч открепления клеток путем добавления 2 мл трипсина и инкубировать при 37 ° С в СО 2 инкубаторе в течение 5 мин.
- Нейтрализовать пластины путем добавления 10 мл сбора буфера (таблица 3). Сбор клеток в 15 мл трубки и спина при 300 мкг в течение 5 мин.
- Жидкость над осадком сливают и клетки вновь суспендируют в 1 мл буфера сортировочный (таблица 4). Граф клеток и разбавить на платформе сортировочный. Развести клеток 0.5-1 х 10 6 клеток / мл для сортировки в 15 мл пробирки и при сортировке отдельных клеток непосредственно в 96-луночные планшеты, разбавленных клетки до 2-5 х 10 6 клеток / мл.
- Сортировать клетки cas9-GFP + ES с использованием проточного цитометра FACS 32. Сбор клеток навалом в трубках с помощью 2 мл носителя восстановления (таблица 5) и пластины , как описано в 3.5 для захвата колоний, или сортировать отдельные клетки непосредственно в покрытые желатином 96-луночные планшеты , содержащие 100 мкл эмбриональных стволовых клеток носитель / лунку ( Таблица 1).
- Семенной 1-1.5 х 10 4 + GFP ES - клетки в 10 см желатин покрытием блюдо, содержащую 10 мл ES клеток среды (таблица 1). Покрытие при этой низкой плотности будет способствовать собирание отдельных колоний ES клеток.
4. Культивирование Клоны для генотипирования, анализа экспрессии и морозильное клеточных запасов
- В день 4-5 после сортировки, забить каждую лунку прямых отсортированных 96-луночных планшетах на наличие колоний ES-клеток.
- Диссоциируют колоний ES клеток путем удаления носителя и добавление 30 мкл трипсина. Инкубируют при 37 ° С в течение 5 мин. Нейтрализовать трипсина путем добавления 170 мкл клеточных сред ES (таблица 1), и пипетку вверх и вниз для полной диссоциации колонии на отдельные клетки. Расти клетки при 37 ° C / 5% CO 2 , пока большинство скважин не более 70% сплошности (обычно 2-3 дня).
- В качестве альтернативы выбрать отдельные колонии клеток ES от 10 см чашки с помощьюинвертированный микроскоп. После того, как аспирационные колонии в пипетку В соответствии с шагом 4.1.1 помещая каждую колонию в одну лунку 96-луночного планшета, предварительно обработанную с желатином и содержащим 30 мкл трипсина.
Примечание: Колонии могут сидеть в трипсин при комнатной температуре, а один весь ряд колоний выбрали. - После того, как все колонии были собраны и диссоциируют на медиа растут клетки при 37 ° С в СО 2 инкубаторе до тех пор , пока большинство скважин более 70% сплошности (обычно 2 дня).
- Когда 96-луночные планшеты готовы к расщеплению, носитель извлечь, добавить 30 мкл трипсина и инкубируют при температуре 37 ° С в течение 5 мин. Нейтрализовать трипсина путем добавления 180 мкл клеточных сред ES (таблица 1) в каждую лунку и пипетку вверх и вниз для полной диссоциации на отдельные клетки.
- Из полученного в результате 210 мкл, 70 мкл семян на три покрытые желатином 96-луночные планшеты , каждый из которых содержит 130 мкл ES клеточных сред / лунку (таблица 1). Используйте эти пластины для геnotyping, анализ экспрессии и замораживание запасов клеток для каждого клона, как описано ниже.
- Когда генотипирование пластина достигает 70-85% слияния, относиться к плашку, как описано в разделе 6 "генотипирования удаление".
- Когда анализ экспрессии пластина достигает 70-85% сплошности, носитель извлечь, запечатать пластину с уплотнительной лентой и хранят при -80 ° С до тех пор, пока клоны были генотипирование.
Примечание: Анализ экспрессии пластины полезен для анализа изменений в экспрессии генов на ранних пассажах из клонов. Экспрессия гена анализа из 96-луночного планшета возможна, но поскольку количество клеток с низким набор микро Выделение РНК рекомендуется. - Получение 96-луночного планшета Зафиксированные складе:
- Когда пластина для запасов замороженных клеток (акций-1) достигает 70-85% сплошности, аспирация СМИ, добавить 30 мкл трипсина и инкубируют при температуре 37 ° С в течение 5 мин.
- Нейтрализовать трипсина путем добавления 100 мкл клеточных сред ES (таблица 1) то каждую лунку и пипетку вверх и вниз для полной диссоциации на отдельные клетки.
- Передача 15 мкл суспендированных клеток из каждой лунки на два покрытые желатином 96-луночных планшетов, каждый из которых содержит 185 мкл клеточных сред ES (таблица 1) и позволяют расти при 37 ° C / 5% CO 2.
Примечание: Это для штока-2 и -3 пластин, дополнительные резервные клоны в случае возрождая клетки складе-1 из не увенчались успехом.
- В то же время, к 100 мкл остальных клеток в 96-луночный планшет (сток-1), добавляют 100 мкл 2x морозильными носителя (таблица 6). Уплотнение пластины с помощью герметизирующей ленты и быстро пластину перевернули 4-5 раз для правильного смешивания. Хранить планшет при температуре -80 ° С до тех пор, клоны не генотипирование.
- Когда шток-2 и шток-3 плиты готовы к замораживанию аспирация СМИ, добавляют 30 мкл трипсина и инкубировать при температуре 37 ° С в течение 5 мин. Нейтрализовать трипсина путем добавления 70 мкл клеточных сред ES (Tв состоянии 1) в каждую лунку и пипетку вверх и вниз для полной диссоциации на отдельные клетки.
- Добавьте 100 мкл 2x замораживания средств массовой информации, запечатать пластину с уплотнительной лентой и быстро пластину перевернули в 4-5 раз для правильного смешивания. Хранить планшет при температуре -80 ° С до тех пор, пока необходимы эти пластины.
5. аллель-специфического праймера Дизайн
- Дизайн 4 набора праймеров (фиг.1С) для скрининга клонов для желаемого удаления: внутри праймеров, внешних праймеров и gRNA фланкирующих праймеров (для обоих 5'- и 3' - сайтов - мишеней gRNA) , как описано ниже.
- Получить дорожку SNP, соответствующую 129 и Cast генотипов в http://labs.csb.utoronto.ca/mitchell/crispr.html. Данный трек показывает базовые замещения между 129 и брось в точке с координатами в геноме сборки MM9 мыши.
Примечание: Ссылка на указанном выше сайте будет перенаправлять генома браузера УСК и добавить пользовательский трек, содержащий SNPs между 129 и Cast GEномы. - Введите координаты области, которые будут удалены. Нажмите на область около 500 б.п. в середине требуемого удаления, содержащей> 3 ОНП.
- Перейти для просмотра> ДНК в панели опций и нажмите получить ДНК для загрузки целевой последовательности во всех верхнем формат регистра букв.
- Создание двух последовательностей FASTA; один для 129 и один для произнесения на базовой замещения в положении SNP. Отметьте ОНП на нижнем регистре.
- Перейти к Primer3 плюс (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) и вставьте SNP 129 замещенные последовательности. Используйте настройки по умолчанию для разработки праймеров.
- Для того, чтобы разработать внутренние аллель-специфических праймеров выберите Primer_List в раскрывающемся меню задачи и нажмите Pick Грунтовки. Выберите прямой или обратный праймер, который имеет SNP или в 'конце или в течение 4-х оснований из 3' 3 и попадает внутрь области, чтобы быть удалены.
Примечание: Грунтовки, имеющие SNP на дисплее 3'-конца увеличилась аллель-специфичность в кПЦР. - Для выбора второго праймера возврата на главную страницу, выберите Обнаружение в раскрывающемся меню задач и вставить первую последовательность праймера в соответствующем поле в нижней части страницы. На вкладке Общие настройки изменить настройку диапазона размеров продукта 80-200 б.п. и нажмите Pick Грунтовки. Выбрал набор праймеров из перечисленных пар праймеров; это будут 129 внутри аллель-специфических праймеров.
- Повторите шаги 5.1.5 5.1.7 для разработки праймеров для Cast аллеля.
- Возвращение в геном браузера УСК и введите координаты региона, которые будут удалены. Перейти для просмотра> ДНК в панели опций, в разделе Параметры последовательности Retrieval область добавить 1000 п.о. вверх и вниз нажмите получить ДНК для загрузки целевой последовательности.
- Отметьте целевую последовательность gRNA в скобках. Сохранить всю эту последовательность, прежде чем продолжить.
- Для того, чтобы проектировать внешние праймеры удаления последовательности между двумя целевыми gRNA последовательностями. Повторите шаг 5.1.4-5.1.8 для разработки внешнего аллель-специфического праймераs, но изменить размер продукта 400-800 пар оснований.
- Разделить последовательность, полученную на стадии 5.1.10 на две последовательности, каждый из которых имеет 500 п.о. 5 'и 3'-последовательности-мишени gRNA. Повторите шаги 5.1.5 5.1.7 проектирование без аллель-специфических праймеров для gRNA фланкирующих но изменить размер продукта 400-800 пар оснований.
Примечание: Для не аллельных специфических праймеров, либо 129 или Cast последовательность может быть использована и праймеры должны быть выбраны, которые не содержат SNP. Желательно, чтобы установить размер продукта 400-800 пар оснований для проектирования снаружи и gRNA фланговые праймеров. Это позволяет амплификации, даже если небольшие вставки подсчитывали присутствуют.
- Получить дорожку SNP, соответствующую 129 и Cast генотипов в http://labs.csb.utoronto.ca/mitchell/crispr.html. Данный трек показывает базовые замещения между 129 и брось в точке с координатами в геноме сборки MM9 мыши.
- Проверьте внутренние праймеры для аллеля специфичности КПЦР с использованием чистого 129 и Cast штамм геномной ДНК на 2 нг / мкл. Выполните шаг 6.2-6.4, чтобы настроить реакцию КПЦР.
Примечание: Если 129 генотип в целевом регионе является такой же, как C57BL / 6J, ДНК из C57BL / 6J можно использовать вместо 129 ДНК. Аллель-специфических праймеров следует отображать по меньшей мере, 5 цикловразность между значением Ct (пороговый цикл) на правильный против неверного генотипа. Внешние праймеры могут быть проверены, чтобы гарантировать, что они усиливают удаление с помощью cas9 / gRNA трансфецированных клеток ES, и F1 геномную ДНК, соответственно, как положительные, так и отрицательные контроли. Аллель-специфичность внешних праймеров могут быть проверены один раз были определены моноаллельной клоны.
6. генотипирование делеции
- Извлечение геномной ДНК из генотипирования 96-луночного планшета с использованием пластины из шага 4.6, который генерируется после расширения колонии.
- Подготовить геномную смесь экстракции ДНК: 89 мкл воды, 10 мкл 10х буфера и 1 мкл экстракционного реагента (поставляется производителем). Добавьте 100 мкл геномной ДНК смеси для экстракции в каждую лунку и запечатать пластину с уплотнительной лентой.
- Инкубируйте планшет при 75 ° С в течение 5 мин, затем 95 ° С в течение 5 мин.
- Дайте пластины остыть инкубированием на льду в течение нескольких минут апd затем Центрифуга кратко урегулировать конденсат на дне колодца. Это служит ДНК пластины шаблона для удаления скрининга.
- Настройка реакции КПЦР в двух экземплярах для каждого клона следующим образом: 5 мкл 2x SYBR КПЦР смеси, прямого и обратного праймера (3 мкМ) каждый 1 мкл и 1 мкл воды. С помощью многоканальной пипетки для добавления 2 мкл матричной ДНК с последующим 8 мкл реакционной смеси в каждую лунку 384-луночного планшета.
- Уплотнение пластины с уплотнительной лентой и спина при 600 мкг в течение 2 мин, чтобы перемешать содержимое. Поместите блок плиты 384-а в реальном времени термоячейке.
- Программа в реальном времени циклователь для 2-х ступенчатый ПЦР с последующим анализом кривой плавления с детектированием следующим образом: 1 цикл при 95 ° С в течение 10 мин, 40 циклов при 95 ° С в течение 15 с, 62 ° С в течение 30 сек с пластиной чтения и 95 ° с в течение 10 с, 65 ° с до 95 ° с с шагом 5 ° с в течение 5 сек + пластины чтения.
Примечание: В дополнение к праймер дизайн, КПЦР милиПараметры х и цикл также способствуют затравки специфичности. Описанные выше параметры и реагенты, перечисленные в материалах более часто дают аллель специфической амплификации. - Анализ результатов КПЦР
- Проверьте каждый аллель для амплификации с внутренней аллель-специфических праймеров. Нет усиления одного аллеля или высокое значение Ct различия (> 5 циклов) между аллелями не предлагают, чтобы эти клоны несут гетерозиготную удаление аллеля с высокой / отсутствующего значения Ct. Нет усиления обоих аллелей не говорит о том, что они несут гомозиготная делеция.
- Проверьте каждый аллель для амплификации с внешними аллель-специфических праймеров. Когда цель удаления больше, чем 1 кб амплификации с внешними праймеров происходит только тогда, когда удаление присутствует. Значение Ct 22-28 подтверждает удаление. Для целевых удалений меньше 1 кб, подтвердить размер ампликона с помощью электрофореза.
Примечание: Если внешние праймеры показывают только умеренное аллель-специфичность (см FIGUповторно 2), ампликонов могут быть получены с обоими наборами праймеров аллельных в моноаллельной клонов из - за слишком поторопился усиления внешних праймеров. В этом случае разница Ct значение между двумя аллелями по крайней мере, пять циклов должен подтвердить правильность аллель (нижнее значение Ct) удаляется на основе результатов, полученных из внутренних праймеров. Если на цель по сравнению с поторопился с аллель разницы Ct составляет менее пяти циклов разработки новых аллеля специфических внешних праймеров. - В моноаллельной клонов делеции проверки целостности Неудаленные аллеля с использованием вторичного скрининга, gRNA фланговые праймеров.
Примечание: вставкам из> 25 размер п.н. вокруг целевого участка gRNA могут быть идентифицированы путем наблюдения сдвиг кривой расплава в кПЦР для 400-800 п.н. ампликонов. В качестве альтернативы, ампликонов из фланкирующих праймеров gRNA можно секвенировать обнаружить небольшие вставкам <25 пар оснований.- Выполните КПЦР с 2 - мя наборами gRNA фланкирующих праймеров т.е., 5 'и 3' гРНК используется в генерации CRISPR удаления. Нет амплификации с этими наборами праймеров указывает на более крупные, чем вставки подсчитывали ампликону КПЦР присутствуют на целевом сайте gRNA на Неудаленные аллеля моноаллельной клонов удаления. Отбросить клоны, содержащие эти большие вставкам из дальнейшего анализа как результаты могут быть трудно интерпретировать, не зная степень удаления.
- Очищают ампликонов, полученных от внешнего праймера реакции КПЦР с использованием ПЦР очистить комплект следуя инструкции производителя.
- Повторите последовательность удаляемого аллеля путем секвенирования ДНК очищенного продукта ПЦР из предыдущей стадии. С помощью праймеров для амплификации КПЦР для прямого и обратного секвенирования.
Примечание: На этом этапе ОНП в пределах ампликона акта в качестве вторичного подтверждения генотипа удаляемого аллеля.
7. Анализ экспрессии с аллель специфичными праймерами
- Оттепель 96-луночный запасов плели хранили при -80 ° C (1-складе с шага 4.9), поместив его на теплый шарик ванны. Когда более половины скважин в пластине оттаивают, спина при 300 мкг в течение 5 мин.
- Осторожно снимите упаковочную ленту и быстро передавать клетки из делеций положительных лунок в покрытые желатином, 12-луночные планшеты , содержащие 1 мл клеточных сред ES (таблица 1) и инкубируют при 37 ° C / 5% CO 2.
- Когда пластина достигает 70-85% сплошности, прохождение клеток и разделить их на три лунки желатин покрытием, 6-луночного планшета, каждый из которых содержит 2 мл клеточных сред ES (таблица 1). Используйте две скважины для подготовки 2 ампул запасов замороженных клеток для длительного хранения в жидком азоте (описано в шаге 8) и третьей скважины для экстракции РНК.
- Извлечение РНК с использованием набора для экстракции РНК.
- Преобразование 100-500 нг РНК в кДНК с помощью обратной транскрипции (RT) РНК с использованием набора для синтеза кДНК в соответствии с протоколом производителя. Включите п RTegative реакции для каждого образца РНК для мониторинга количества примесной ДНК в образцах РНК.
- Развести кДНК перед кПЦР в соотношении от 1: 2 и 1: 4; в зависимости от уровня экспрессии гена-мишени в клетках ES.
- Установите КПЦР, как описано выше, включая F1 геномную ДНК в качестве стандартной кривой (5 разведений сгиба от 250 до 0,08 нг / мкл) для абсолютного количественного определения уровней транскриптов. Сравнение экспрессии каждого аллеля представляющего интерес гена в каждом подтвержденного удален клон подходящего контрольного гена, например , GAPDH (праймеры , перечисленные в таблице 7).
Примечание: контрольного гена праймеры не должны быть аллель конкретными. Конструкция праймера аллельспецифической одинакова для RT-КПЦР праймеров, как описано для генотипирования праймеров за исключением целевой области для усиления. Последовательность гена следует использовать; при использовании праймеров для одного экзона или границы экзон-интрон (для мониторинга первичных транскриптов) F1 геномной ДНК может быть использована длястандартной кривой. Более подробную информацию о RT-КПЦР пожалуйста , обратитесь к Forlenza и др. 2012 33.
8. Стоп-кадр изображения Подготовка к длительному хранению ЭС клеток
- Добавить 300 мкл трипсина в каждую лунку 6-(со стадии 7.3) и инкубировали в течение 5 мин при 37 ° С. Добавляют 2 мл спин носителя (таблица 2) , чтобы нейтрализовать трипсина и пипетку вверх и вниз несколько раз , чтобы диссоциировать на отдельные клетки.
- Передача клеток в 15 мл трубки и спина при 300 х г в течение 5 мин.
- Аспирируйте супернатант и добавить 500 мкл клеточных сред ES (таблица 1). Пипетировать вверх и вниз для ресуспендирования клеток.
- Передача содержимого в 1,5 мл криопробирку пробирку и добавляют 500 мкл 2x ES клеток замораживании сред (таблица 3). Хорошо перемешайте переворачивания пробирки и поместите пробирку в безалкогольного контейнер ячейки замерзания. Поместите эту ячейку замораживания контейнеры при температуре -80 ° С в течение по меньшей мере 12 ч до трансферринаг в жидкий резервуар для хранения азота.
Access restricted. Please log in or start a trial to view this content.
Результаты
Протокол , описанный здесь использует клетки F1 ES для изучения цис -regulation экспрессии генов в моноаллельной усиливающих удалены клеток , полученных с помощью CRISPR / редактирования генома cas9 (Рисунок 1). GRNA и дизайн праймера аллель-специфической для генотипир?...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
CRISPR / cas9 технологии редактирования опосредованный геном обеспечивает простой, быстрый и недорогой способ для модификации генома. Подробно здесь, чтобы генерировать и анализировать моноаллельную энхансер для удаления функционального энхансер характеризации метод использует ОНП в кл...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторы читали политику Jove о конфликте интересов и не имеют никаких конфликтов раскрывать.
Благодарности
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Ссылки
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152(2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187(2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485(2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159(2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены