
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
생성 CRISPR은 / Cas9 중재 Monoallelic 삭제할 경우는 마우스 배아 줄기 세포의 증강 기능을 연구하는
요약
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
초록
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
서문
전사 조절 요소에 의한 비정상적인 유전자 발현이 질병을 초래할 수있다 이러한 요소 개발 1 변형시 유전자 발현 시공간 미세 조정이 중요하다. 게놈 넓은 협회 연구에 의해 확인 된 많은 질병 관련 지역 비 코딩 지역에 있고 전사 증강 3-4의 기능을 가지고 있습니다. 강화제를 파악하고 그들이 자주 몇 킬로베이스 거리가 조절하는 유전자의 위치와 조직 - 특이 적 방식으로 5-6으로 활성화 될 수 있으므로 복잡 조절하는 유전자로 일치. 증강 예측은 일반적으로 히스톤 수정 마크, 중재자-cohesin 단지를 기반으로 세포 유형 특정 전사의 결합은 7-10 요인 있습니다. 예측 증강의 유효성 검사는 가장 자주 인핸서는 리포터 유전자 11 ~ 12의 발현을 활성화하는 벡터 기반의 분석을 통해 이루어집니다. 이러한 데이터는 V를 제공추정 인핸서 서열의 규제 가능성에 대한 aluable 정보는하지만 자신의 내생 게놈 맥락에서 그 기능을 공개하거나 조절하는 유전자를 식별하지 않습니다. 게놈 편집 기능 상실 분석에 의해 자신의 내생 적 맥락에서 전사 조절 인자의 기능을 연구하는 강력한 도구 역할을한다.
게놈 편집, 즉 CRISPR / Cas9 게놈 편집 시스템의 최근의 진보는, 게놈 기능의 조사를 용이하게한다. CRISPR / Cas9 시스템은 많은 생물학적 시스템에 사용하기 쉽고 적용 할 수 있습니다. Cas9 단백질 가이드 RNA (gRNA) (13)에 의해 게놈의 특정 사이트를 대상으로합니다. SpCas9 / gRNA 단지는 protospacer 인접 모티브 (PAM) 시퀀스, NGG 14 ~ 15에 '5이어야 목표 게놈 시퀀스의 게놈을 스캔합니다. 목표에 gRNA의 gRNA에 상보적인 20 뉴클레오티드 (NT) 시퀀스의 염기쌍는 doubl 결과 SpCas9 클레아 활동을 활성화전자 가닥 브레이크 (DSB)가 PAM 시퀀스의 상류 3 염기쌍. 특이도가 gRNA 종자 지역의 전체 염기 쌍을 통해 달성되면, 6-12은 PAM에 인접한 NT; 반대로, 종자의 보통 16-17을 허용하는 '5 불일치. 가입 비 동종 말 (NHEJ)의 DNA 수리 또는 상동 감독 수리 (HDR) mechanisms.NHEJ의 DNA 수리가 종종 방해 할 수있는 대상 사이트에서 몇 bp의 삽입 / 삭제 (삽입과 삭제)을 작성 중 하나를 소개 DSB는 복구 할 수 있습니다 유전자의 오픈 리딩 프레임 (ORF). 관심 영역의 측면 게놈 두 gRNAs에서 큰 결실을 생성하기 위해, 18 ~ 19을 사용할 수있다. 이 접근법은 궤적 제어 영역 또는 종래 증진제 9,18,20-22보다 큰 수퍼 증강제로 클러스터 전사 인핸서의 연구에 유용하다.
Monoallelic 삭제는 전사의 시스 -regulation 연구를위한 귀중한 모델입니다. 관찰 장증강의 monoallelic 삭제 후 성적 수준에서 전자는 두 대립 유전자의 전사가 세포 적합성에 영향을 미치는 잠재적 영향을 때 발생할 수있는 혼란 효과없이 유전자 조절에서 해당 증강의 역할에 상관 관계. 감소 된 발현을 평가하기 그러나, 야생형 대립 유전자에서 삭제를 구별하는 능력없이 어렵다. 또한, 두 개의 대립 유전자를 구별하는 능력없이 각 대립 유전자의 결실을 유전형 특히이 PCR에 의해 전체 야생형 영역을 증폭하기 어렵다되는 1 메가 23> 10킬로바이트 큰 결실에 대한 도전이다. 뮤스 castaneus으로 건너 뮤스의 musculus (129)에 의해 생성 된 F1 ES 세포의 사용은 두 가지 대립 유전자가 대립 유전자 특이 PCR 18,24에 의해 구별 할 수 있습니다. 이러한 세포의 하이브리드 유전자는 대립 유전자 특정 삭제 검사 및 발현 분석을 용이하게한다. 평균적으로이 두 게놈 사이의 모든 125 BP는 SNP에게있다, 표현과 유전자형에 대한 프라이머 설계 유연성을 제공하는 분석한다. 하나의 SNP의 존재는 프라이머 융해 온도 (Tm)에 영향을 미치는 두 대립 (25)의 식별을 허용 실시간 정량 PCR (qPCR에) 증폭 특이성을 타겟팅 할 수있다. 또한, 프라이머의 3 '말단 내의 불일치 크게 원하지 대립 타겟 (26)의 증폭을 방지 프라이머로부터 연장하는 DNA 폴리머 라 아제의 능력에 영향을 미친다. CRISPR / Cas9 게놈 편집 시스템 (도 1)를 사용하여보다 큰 1킬로바이트의 특정 대립 유전자 인핸서 삭제 이후 발현 분석 F1 ES 세포의 사용은 다음의 프로토콜에 설명한다.

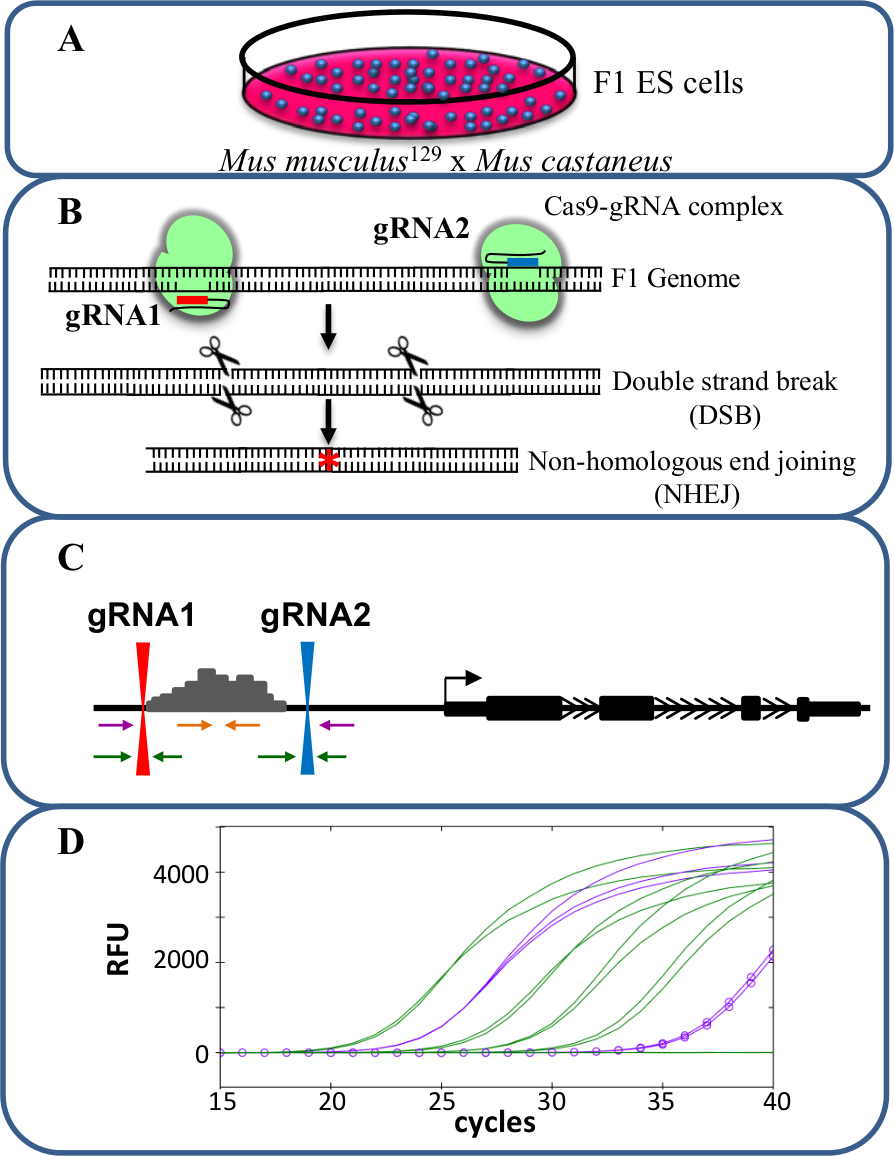
시스 -reg을 연구하는 CRISPR / Cas9를 사용하여 그림 1. 증강 삭제유전자 발현의 위험률. 뮤스의 musculus 129 뮤스 castaneus 사이의 교차에 의해 생성 (A) F1 ES 세포는 대립 유전자 특정 삭제를 허용하는 데 사용됩니다. (B)는 두 개의 RNA를 가이드 (gRNA)는 인핸서 영역의 큰 Cas9 매개 결실을 유도하기 위해 사용된다. (C) 프라이머 세트가 많은 모노 - 및 이중 대립 유전자 결실을 식별하는 데 사용된다. 오렌지 프라이머 내부 프라이머이며, 보라색 프라이머는 외부 프라이머 및 녹색 프라이머는 gRNA 측면 프라이머되어 있습니다. 유전자 발현의 (D)의 변화는 대립 유전자 특정 qPCR에를 사용하여 모니터링됩니다. RFU 상대 형광 단위를 의미한다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
프로토콜
1. 설계 및 gRNA를 구축
- 전사 인핸서 영역이 gRNAs, 관심 영역 중 하나 5 '하나 3'을 사용하여 삭제합니다. (http://www.genome-engineering.org 15) 고유 gRNA 시퀀스를 식별하기 위해 장 기관에 의해 발생 된 마우스 UCSC 게놈 브라우저 트랙을 사용한다. 다음이 gRNAs와 생거 연구소 (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27 ~ 28에서 제공하는 온라인 도구를 사용하여 SNP를하고 삽입과 삭제에 대한 자신의 인접 PAM을 확인합니다. 동일한 효율로 두 대립 유전자를 대상으로하기 위해, SNP 또는 INDEL를 포함 gRNA / PAM 시퀀스를 피할 수 있습니다.
- gRNA을 선택하는 동안, 삭제를 유전형에 대한 대립 유전자 특이 적 프라이머를 설계의 타당성을 확인합니다. 대립 유전자 특이 프라이머 디자인 (5)을 참조하십시오.
- 말리 등. 2013 15에 기술 된 프로토콜에 따른 두 gRNA 플라스미드를 조립한다. 선택한 독특한 20 bp의 표적 서열 INT를 통합표 7에 나타낸 바와 같이 61mer 올리고 뉴클레오티드 O (서열 방향 '3'5에 표시하고, 굵은 염기는 서로 역방향 보완되어 20 염기쌍 표적 서열 임).
- 10 μM gRNA Primer_F 10 μL와 튜브에 10 μm의 보완 Primer_R의 10 μl를 섞는다.
- 5 분 동안 100 ° C에서 프라이머 믹스를 배양함으로써 프라이머를 어닐링 한 후 25 ° C로 1 ° C / sec의 냉각. 이 단계를 들어, PCR 기기를 사용하거나 끓는 물에 튜브를 배치하고 RT로 냉각 할 수있다.
- 물 18.5 μL, 5 배 HF 완충액 10 μL, 10 mM의 dNTP를 1 ㎕를 혼합하고 높은 0.5 μL : 어닐링 된 프라이머를 혼합하여, 각 프라이머 연장을 30 분 동안 72 ° C에서 다음 반응 혼합물을 추가 부화 충실도 DNA 폴리머 라제.
- 100 bp의 가이드 프래그먼트가 생성되고 확인하는 2 % 아가 로스 겔상에서 표적 단편의 10 μl를 실행.
- gRNA 벡터 (지오의 선물을 선형화RGE 교회; 다음과 같은 반응을 사용하여 AFL II와 Addgene 플라스미드 # 41824) (15)는 설정 : gRNA 벡터 백본 (2-4 μg의), 10 배 버퍼의 5 μL, AFL II의 3 μL의 5 μL (20 단위 / μL) 32 μl를 물. 37 ° C에서 3 시간 동안 반응 혼합물을 인큐베이션.
- 1 % 아가로 오스 겔에서 분해 생성물을 실행하여 3.5 kb의 DNA에 대응하는 대역은 제조자의 지시에 따라, 겔 추출 키트를 사용하여 gRNA 벡터를 선형화 정제.
- 설정 깁슨 조립 반응 29 ~ 30 선형화 gRNA 벡터를 사용하여 다음과 같이 단계 1.2.3에서 조각 대상 : 선형 gRNA 벡터 (50 NG / μL), 대상 조각의 1 μL, 배 깁슨 어셈블리 마스터 믹스 10 μl를 1 μl를 물 8 μL. 60 분 동안 50 ℃에서 반응을 인큐베이션.
- 조립 gRNA 벡터와 대장균 세포의 변형.
- 1.2.7에서 조립 gRNA 벡터의 1 μl를 혼합및 DH5α (대장균 균주) 튜브에 세포를 50 μL. 45 초 동안 42 ° C까지 세포를 노출시킴으로써 열 쇼크 법에 의해 DH5α 세포 변형.
- 스냅 5 분 동안 얼음에 튜브를 진정; 다음 SOC 배지 400 ㎕를 추가하고, 진탕 배양기에서 45 분 동안 37 ℃에서 배양한다.
- 하는 LB-카나마이신 (50 μg의 / ㎖) 형질 전환 된 세포의 긍정적 인 선택을위한 접시에 DH5α 세포 100 μl를 확산하고 37 ℃에서 O / N을 품어.
- gRNA 삽입에 대한 긍정적 인 대장균 콜로니를 선별.
- 카나마이신 50 ㎍ / ㎖의를 포함하는 3 ㎖ LB의 카나마이신 내성 식민지에 resuspend을 선택합니다. 6-8 식민지에 대해 동일한을 반복 진탕 배양기에서 37 ° CO / N의 모든 튜브를 품어.
- 제조업체의 매뉴얼에 따라 플라스미드에게 미니 준비 키트를 사용하여 O / N의 성장 문화에서 플라스미드의 압축을 풉니 다.
- gRNA 시퀀스 삽입을 확인하기를 EcoRI 소화 반응 믹스를 준비플라스미드있다. 효소 완충액 2 μL,를 EcoRI 1 μL, 물 15 μL를 다음과 같이 각각의 샘플의 경우, 반응 혼합물을 준비한다. 1.5 ml의 튜브에 반응 믹스를 나누어지는 플라스미드의 2 μl를 추가합니다. 2 시간 동안 37 ℃에서 인큐베이션 튜브.
- 1.5 % 아가 로스 겔에 소화 제품을 실행합니다.
참고 : 삽입과 함께 샘플을 삽입하지 않고 클론 100 bp의 높은 475 bp의 밴드 크기를 표시합니다.
참고 : 또는 포지티브 클론 삽입 gRNA의 존재하에 642 bp의 크기의 단편을 수득 SP6 (정방향) 및 T7 (역방향) 프라이머 (표 7) 벡터 서열에 해당 바인딩을 사용하여 콜로니 PCR로 스크리닝 될 수있다. gRNA 시퀀스 내를 EcoRI 제한 효소 부위가 있으면 콜로니 PCR 방법이 유리하다.
- T7 프라이머를 사용하여 DNA 시퀀싱에 의해 gRNA 삽입의 순서를 확인합니다.
2. 형질
노트 :전기는 ES 세포로의 플라스미드 형질 감염의 효과적인 방법이다. 여기에 기재된 방법은 microporator 형질 전환 기술을 사용한다.
- 37 ° C / 5 % CO 2에서 ES 세포 배지 (표 1) 10 ㎖를 포함하는 10cm 젤라틴 코팅 접시 F1 ES 세포 성장. 세포가 85 %에 도달하면 컨 플루 미디어를 제거하고 트립신 2 ㎖를 추가합니다. 5 분 동안 CO2 인큐베이터에서 37 ℃에서 인큐베이션.
참고 : F1 ES 세포는 바바라 패닝 (24)로부터 얻어진 요청에 사용할 수 있습니다되었다. - 스핀 배지 (표 2) 10 ㎖를 첨가함으로써 트립신을 중성화. 피펫을 반복해서 완전히 세포를 분리합니다.
- 5 분 300 XG에 15 ML 튜브와 스핀의 모든 세포를 수집합니다. 3 ml의 PBS에서 재현 탁하고 혈구 또는 자동 세포 계수기를 사용하여 세포를 계수.
- (R 100 ㎕에서 5 분,에 resuspend 300 XG에 원심 분리하여 1.5 ML 튜브에 1 × 10 6 ES 세포 펠렛키트 제조 업체가 제공하는 부유는) 버퍼.
- (키란 Musunuru의 선물, Addgene 플라스미드 # 44719) 5 μg의 pCas9_GFP 각각 추가 31, 5 '및 3'대상 영역의 삭제 gRNA 플라스미드를 거품의 도입을 방지하기 위해 피펫으로 가볍게 섞는다.
- 끝에서 거품 않도록주의하면서 전기 믹스 100 μl를 흡인하는 전자 피펫 팁을 사용합니다.
- 프로그램 볼트, 폭과 전기에 대한 펄스. F1 ES 세포의 경우, 3 펄스 1400 V, 10 밀리 초를 사용합니다.
- 일렉트로 실행 중에 용액 어떠한 스파크 시청할 팁을 관찰한다. 스파크는 기포의 존재를 나타내고 형질을 방해 할 것이다.
- 10 ML의 ES 세포 미디어 (표 1)를 포함하는 10cm 젤라틴 코팅 접시에 형질 ES 세포를 추출하고 37 ° C / 5 % CO 2 부화.
형질 감염된 세포를 정렬 3. FACS
- 48 시간 후, 트립신 2 ㎖를 첨가하여 세포를 분리하고 5 분 동안 CO2 인큐베이터에서 37 ℃에서 배양한다.
- 수집 버퍼 (표 3) 10 ㎖를 첨가하여 중화 판. 5 분 300 XG에 15 ML 튜브와 스핀에 세포를 수집합니다.
- 뜨는을 취소하고 정렬 버퍼 (표 4)의 1 ml의 세포를 재현 탁. 정렬 플랫폼을 기반으로 세포 및 희석을 계산합니다. 15 ㎖의 튜브에 정렬하고 96 웰 플레이트에 직접 개별 셀을 정렬에 0.5 × 106 세포 / ml로 희석하여 세포를 2-5 × 106 세포 / ml의 세포를 희석.
- (32) 사이토 FACS 흐름을 이용하여 정렬 Cas9-GFP + ES 세포. 직접 / 잘 100 μl의 ES 세포 미디어를 포함하는 젤라틴 코팅 된 96 웰 플레이트에 식민지 따기, 또는 종류의 개별 셀 3.5에 기술 된 바와 같이 (2 ml의 복구 미디어 (표 5) 접시와 튜브 대량으로 세포를 수집 표 1).
- 시드 1.5 × 104 GFP 10 ㎖ ES 세포 배지 (표 1)를 포함하는 10cm 젤라틴 코팅 접시 + ES 세포. 이 낮은 밀도로 도금하여 개별 ES 세포 식민지를 따기 촉진 할 것이다.
유전자형, 식 분석 및 냉동 세포 주식 4. 배양 클론
- 정렬 후 4-5 일에, ES 세포 콜로니의 존재를 직접적으로 정렬 된 96 웰 플레이트의 각 웰 점수.
- 미디어를 제거하고 트립신 30 μl를 추가하여 ES 세포 식민지를 떼어 놓다. 5 분 동안 37 ℃에서 인큐베이션. ES 세포 배지 (표 1) 170 μL를 첨가하여 트립신을 중화하고, 단일 세포로 콜로니의 완전한 분리를위한 상하 피펫. 대부분의 우물 70 %의 합류 이상 (보통 2~3일)가 될 때까지 37 ° C / 5 % CO 2에서 세포를 성장.
- 또는 사용하여 10cm 요리에서 개별 ES 세포 식민지를 선택거꾸로 현미경. 젤라틴으로 전처리 96 웰 플레이트 잘 하나에 각 식민지를 배치 피펫 팁의 후속 단계 4.1.1로 식민지를 흡입하고 트립신 30 μl를 포함하는 후.
참고 : 식민지의 한 행 전체가 선택됩니다 동안 식민지 실온에서 트립신에 앉아 있습니다. - 일단 모든 콜로니를 포착하고 대부분의 우물 70 %의 합류 (보통 2 일간) 이상 될 때까지 미디어가 CO 2 배양기에서 37 ° C에서 세포를 성장으로 해리되고있다.
- 96 웰 플레이트가 분할을위한 준비가되면, 미디어를 제거 트립신 30 μl를 추가하고 5 분 동안 37 ° C에서 품어. 하나의 세포로 완전한 분리를 위해 아래 각 웰 피펫 최대 ES 세포 미디어 180 μL (표 1)을 첨가하여 트립신을 중화.
- 얻어진 210 μL에서 세 젤라틴 코팅 된 96 웰 플레이트 ES 세포 배지 / 웰 (표 1) 130 μl를 함유 각각 70 μL를 시드. GE의 이러한 판을 사용하여notyping, 발현 분석 및 아래에 설명 된대로 각 복제에 대한 세포의 주식을 동결.
- 유전자형 판 70-85% 합류점에 도달하면 "삭제 유전자형"(6)에 기재된 바와 같이, 상기 플레이트를 처리한다.
- 발현 분석 플레이트 70~85% 합류점에 도달하면, 미디어를 분리 클론 유전자형 때까지 -80 ℃에서 저장 테이프 밀봉 접시를 밀봉.
주 : 발현 분석 플레이트 클론 초기 통로에서 유전자 발현의 변화를 분석하는 데 유용하다. 96 웰 플레이트에서 유전자 발현 분석이 가능하지만, 세포 수는 낮은 등의 RNA 마이크로 추출 키트를 권장합니다. - 96 웰 플레이트 고정 재고의 제조 :
- 냉동 세포 축적량 (주 1)에 플레이트 70-85% 합류점에 도달하면, 미디어 대기음 트립신의 30 μL를 추가로 5 분 동안 37 ℃에서 배양한다.
- ES 세포 배지 100 ㎕를 첨가함으로써 트립신을 중성화 (표 1) t각 웰과 피펫 위아래로 하나의 세포로 완전한 분리에 대한 오.
- 전송 각 웰에 두 젤라틴 코팅 된 96 웰 플레이트에서 정지 세포의 15 μL, ES 세포 미디어 (표 1)의 각 포함 185 ㎕를 37 ° C / 5 % CO 2 증가 할 수 있습니다.
참고 :이 주식 2이며, 주식 1에서 세포를 되살리는 경우 추가 백업 복제 -3 판은 성공하지 못합니다.
- 한편, 96 웰 플레이트 (주-1)의 나머지 셀의 100 μL에 2 배 100 ㎕ 미디어 (표 6) 동결을 추가합니다. 밀봉 테이프로 플레이트를 밀봉하고 신속하게 적절한 혼합을 위해 플레이트를 4 ~ 5 번 반전. 클론 유전자형 때까지 -80 ° C에서 접시를 저장합니다.
- 주식-2 및 주식 3 판 흡인에게 미디어를 동결 할 준비가되면, 트립신 30 μl를 추가하고 5 분 동안 37 ° C에서 품어. (ES 세포 배지의 70 μl를 첨가하여 T를 트립신 중화1) 수를 각 웰 피펫 최대 아래로 완전한 분리를위한 하나의 세포로.
- 2 배 동결 매체의 100 μl를 추가 테이프를 밀봉으로 접시를 밀봉하고 신속하게 판에게 적절한 혼합을위한 4 ~ 5 번 반전. 이 판은 필요할 때까지 -80 ° C에서 접시를 저장합니다.
5. 대립 유전자 특이 프라이머 디자인
- 디자인 프라이머 (그림 1C)의 4 세트는 원하는 삭제 클론을 선별하기 : 프라이머, 외부 프라이머, 아래에 설명 된대로 gRNA이 (모두 5 '및 3'gRNA 대상 사이트) 프라이머를 측면 내부.
- http://labs.csb.utoronto.ca/mitchell/crispr.html에서 129 캐스트 유전자형에 해당하는 SNP 트랙을 얻습니다. 주어진 트랙은 mm9 마우스 게놈 어셈블리의 좌표에 129 캐스트 사이베이스의 대체를 보여줍니다.
참고 : 위의 사이트에있는 링크가 GE를 UCSC 게놈 브라우저로 리디렉션 사이에 129을의 SNP를 포함하는 사용자 정의 트랙을 추가하고 주조한다nomes. - 영역의 좌표를 입력 삭제할. > 3 개의 SNP를 포함 원하는 삭제의 중간에 약 500 염기쌍의 영역에 확대합니다.
- 옵션 바에서> DNA를 확인하고 모든 대문자 형식으로 표적 서열을 다운로드받을 DNA를 클릭하여 이동합니다.
- 이 FASTA 시퀀스를 생성; 129 다른 하나는 SNP 위치의 염기 치환에 의해 캐스팅 하나. 소문자로의 SNP를 표시합니다.
- 플러스 (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) Primer3로 이동하여 SNP가 129 시퀀스를 대체 붙여 넣습니다. 프라이머를 설계하는 기본 설정을 사용합니다.
- 내부 대립 유전자 특이 프라이머 작업 드롭 메뉴에 Primer_List 선택하고 프라이머 선택을 클릭 설계합니다. 3 '말단 또는 3에서 4 기지 내'에서 중 하나 SNP를 가지고 영역이 삭제 될 내부에 해당하는 정방향 또는 역방향 프라이머를 선택합니다.
참고 : 말단 디스플레이에서 SNP가 프라이머는 qPCR에에서 대립 유전자 특이도를 증가했다. - 메인 페이지에 두 번째 프라이머 반환을 선택하려면 작업 드롭 메뉴에서 검색을 선택하고 페이지 하단의 해당 상자의 첫 번째 프라이머 시퀀스를 붙여 넣습니다. 일반 설정 탭에서 80-200 BP에 제품의 크기 범위에 대한 설정을 변경하고 프라이머 선택을 클릭합니다. 나열된 프라이머 쌍의 프라이머 세트를 선택; 이들은 129 내부 대립 유전자 특이 적 프라이머 될 것입니다.
- 반복 캐스트 대립 유전자에 대한 프라이머를 설계 5.1.5 5.1.7에 단계를 반복합니다.
- UCSC 게놈 브라우저로 돌아가서 지역의 좌표가 삭제 될 입력합니다. 보기로 이동> 시퀀스 검색 지역 옵션에서 옵션 바에서 DNA 상류 1000 bp의 추가 및 다운 스트림 대상 시퀀스를 다운로드 DNA를 얻을 클릭합니다.
- 괄호 안에 gRNA 대상 시퀀스를 표시합니다. 진행하기 전에이 전체 시퀀스를 저장합니다.
- 외부 프라이머는 두 gRNA 대상 서열 사이의 서열을 제거 설계합니다. 외부 대립 유전자 특이 프라이머를 설계하는 단계를 반복 5.1.4-5.1.8의하지만 제품의 크기를 400 ~ 800 bp의 변경합니다.
- 두 서열에 gRNA 표적 서열의 500 bp의 5 '및 3'각 단계 5.1.10에서 얻은 서열을 분할. 반복 gRNA 측면 지역에 대한 5.1.5 5.1.7에 디자인 비 대립 유전자 특이 적 프라이머 단계하지만 제품 크기를 400 ~ 800 bp의 변경합니다.
비 대립 유전자 특이 적 프라이머를 들어, 하나 (129) 또는 전송 시퀀스가 사용될 수 있고, 프라이머는 SNP를 포함하지 않도록 선택되어야한다 : 참고. 외부 설계 및 gRNA 프라이머를 측면에 대한 400-800 BP의 제품 크기를 설정하는 것이 좋습니다. 이 작은 삽입과 삭제가 존재하는 경우에도 증폭 할 수 있습니다.
- http://labs.csb.utoronto.ca/mitchell/crispr.html에서 129 캐스트 유전자형에 해당하는 SNP 트랙을 얻습니다. 주어진 트랙은 mm9 마우스 게놈 어셈블리의 좌표에 129 캐스트 사이베이스의 대체를 보여줍니다.
- 2 NG / μL에서 순수 129 캐스트 변형 게놈 DNA를 사용하여 qPCR에 의해 대립 유전자 특이성에 대한 내부 프라이머를 테스트합니다. qPCR에 반응을 설정하는 단계 6.2-6.4을 따르십시오.
주 : 대상 영역 129 유전자형 DNA (129) 대신에 사용될 수 C57BL / 6J, C57BL / 6J의 DNA와 동일한 경우. 대립 유전자 특이 적 프라이머는 적어도 5주기를 표시한다잘못된 유전자형 대 올바른에 코네티컷 (사이클 임계 값) 값의 차이. 외부 프라이머는 Cas9 / gRNA는 ES 세포를 형질 전환하여 삭제를 증폭하기 위해 테스트 및 F1 게놈 DNA를 각각 긍정적이고 부정적인 컨트롤 할 수 있습니다. monoallelic 클론이 확인 된 후 외부 프라이머의 대립 유전자 특이성을 테스트 할 수 있습니다.
6. 유전자형 삭제
- 식민지 확장 한 후 생성 단계 4.6에서 접시를 사용하여 유전자형 96 웰 플레이트에서 게놈 DNA를 추출합니다.
- 게놈 DNA 추출 혼합물을 준비 : 물 89 μL, 10 배 버퍼의 10 μL와 (제조업체에서 제공) 추출 시약 1 μl를. 각 웰에 게놈 DNA 추출 혼합 100 μl를 추가하고 밀봉 테이프로 번호판을 봉인.
- 5 분 동안 95 ° C이어서 5 분 동안 75 ℃에서 플레이트를 인큐베이션.
- 접시 몇 분 얼음 위에서 배양에 의해 냉각 허용D는 원심 분리기 짧게 잘 하단에 응축을 해결합니다. 이 삭제 검사의 템플릿 DNA 판 역할을한다.
- 다음과 같이 각 클론에 대해 중복으로 qPCR에 반응을 설정 : 배 SYBR qPCR에 믹스의 5 μL, 앞으로 프라이머 (3 μM) 각 1 μL 및 물 1 μl를 역. 384 웰 플레이트의 각 웰에 반응 혼합물의 8 μl를 다음에 주형 DNA의 2 μl를 추가하는 멀티 채널 피펫을 사용합니다.
- 2 분은 내용을 혼합 600 XG에 테이프와 스핀을 밀봉와 함께 접시를 밀봉합니다. 실시간 자전거 타는에서 384 웰 플레이트 배열을 놓습니다.
- 프로그램 검출을 용융 곡선의 분석에 이어 2 단계 PCR을위한 실시간 클러는 다음과 같이 10 분 동안 95 ° C에서 1 사이클을 15 초 동안 95 ° C, 40 사이클, 62 ° C 플레이트 읽기 30 초간 10 초 동안 95 ° C, 5 초 + 플레이트 5 ° C의 증가와 95 ° C에 65 ° C 읽기.
참고 : 또한 디자인 프라이머하려면 qPCR에 미x 및주기 매개 변수는 특이 프라이머에 기여. 자료에 나와있는 전술 매개 변수와 시약은 더 자주 대립 유전자 특이 적 증폭을 얻었다. - qPCR에 결과 분석
- 내부 대립 유전자 특이 적 프라이머와 증폭을위한 각각의 대립 유전자를 확인합니다. 아무도 대립 유전자의 증폭 또는 대립 유전자 사이의 높은 코네티컷 값의 차이 (> 5주기)이 클론은 높은 / 결석 코네티컷 값으로 대립 유전자의 이형 삭제를 수행 제안합니다. 두 대립 유전자의 어떤 증폭 그들이 동형 접합 삭제를 수행 할 것을 제안합니다.
- 외부 대립 유전자 특이 적 프라이머와 증폭을위한 각각의 대립 유전자를 확인합니다. 대상 삭제 외부 프라이머 1킬로바이트 증폭보다 큰 경우 삭제가 존재하는 경우에만 발생한다. 22-28의 코네티컷 값은 삭제를 확인합니다. 1킬로바이트보다 작은 대상 삭제를 들어, 전기 영동에 의한 앰플 리콘 크기를 확인합니다.
참고 : 외부 프라이머는 중간 대립 유전자 특이성을 표시하는 경우 (Figu 참조)이 재, 증폭 인해 외부 프라이머 오프 표적 증폭 monoallelic 클론 대립 프라이머 세트 모두를 얻을 수있다. 이 경우 적어도 다섯 사이클의 두 대립 유전자 사이 코네티컷 값 차이 (저급 코네티컷 값) 내부 프라이머로부터 얻어지는 결과들에 기초하여 삭제 올바른 대립 유전자를 확인한다. 오프 대상 대립 유전자 코네티컷 차이 비교 대상의 경우 5 개 미만의 사이클은 새로운 대립 유전자는 특정 외부 프라이머를 디자인합니다. - monoallelic 삭제 클론에서 gRNA 프라이머를 측면, 차 심사를 사용하여 삭제되지 않은 대립 유전자의 무결성을 확인합니다.
gRNA 대상 부위 주변> 25 bp의 크기의 삽입 - 삭제 400-800 염기쌍의 증폭 용 qPCR에의 용융 곡선의 변화를 관찰함으로써 확인 될 수있다 : 주. 대안 적으로, 플 랭킹 프라이머 gRNA의 증폭은 <25 bp의 작은 삽입과 삭제를 검출 서열화 될 수있다.- 2 gRNA 측면 프라이머 즉, 5 세트 '3'g과 qPCR에를 수행RNA는 CRISPR 삭제를 생성하는데 사용된다. 프라이머의이 세트와도 증폭은 qPCR에의 앰플 리콘보다 큰 삽입과 삭제가 monoallelic 삭제 클론의 삭제되지 않은 대립 유전자의 gRNA 대상 사이트에 존재 나타냅니다. 그 결과로 추가 분석에서 이러한 큰 삽입과 삭제를 포함 취소 클론 삭제의 범위를 알지 못하고 해석하기 어려울 수 있습니다.
- PCR을 제조 업체의 지시에 따라 키트를 정리하여 외부 프라이머 qPCR에 반응에서 얻은 증폭을 정화.
- 이전 단계의 정제 된 PCR 생성물을 DNA 시퀀싱에 의해 삭제 된 대립 유전자의 서열을 확인한다. 앞으로의 qPCR에 증폭 프라이머를 사용하여 순서를 반대로.
참고 : 삭제 된 대립 유전자의 유전자형의 차 확인과 앰플 리콘의 행위에서이 단계의 SNP에서.
대립 유전자 특정 프라이머 7. 분석 식
- 96 웰 세포 재고 와줘을 해동따뜻한 비드 욕조에 배치하여 -80 ° C (주식 1 단계 4.9에서)에 저장 먹었다. 접시에 우물의 절반 이상이 5 분 동안 300 XG에 스핀을 해동 할 때.
- 조심스럽게, 밀봉 테이프를 제거하고 신속하게 ES 세포 배지 (표 1) 1 ㎖를 함유하는 젤라틴 - 코팅 된 12 웰 플레이트로 삭제 양성 웰로부터 세포를 전송하고 37 ℃ / 5 % CO 2에서 배양한다.
- 플레이트를 70-85% 합류 통로 세포 도달 젤라틴 코팅 된 6- 웰 플레이트의 웰 세, ES 세포 배지 (표 1)의 각각 함유하는 2 ml의로 분할하는 경우. 장기 (8 단계에서 설명) 액체 질소 저장 및 RNA 추출을위한 세 번째 우물을위한 냉동 세포 주식의 2 병을 준비하는 두 개의 우물을 사용합니다.
- RNA를 추출 키트를 사용하여 RNA를 추출합니다.
- 역하게 스크립트 (RT) 제조 업체의 프로토콜 다음 cDNA 합성 키트를 사용하여 RNA에서 cDNA를에 RNA의 100 ~ 500 ng의 변환합니다. 는 RT n을 포함각 RNA 샘플 용 egative 반응은 RNA 샘플에서 DNA 오염의 양을 모니터링한다.
- 2 : 1 : 1 사이의 비율 qPCR의 앞에 cDNA를 희석 4; ES 세포에서 표적 유전자의 발현 수준에 따라.
- 성적 수준의 절대 정량 표준 곡선 (0.08 NG / μL 250에서 5 배 희석) 등 F1 게놈 DNA를 포함하여 상기 한 바와 같이 qPCR에 설정합니다. 예를 GAPDH (표 7에 나와 프라이머)에 대한 적절한 제어 유전자 각각 확인 삭제 복제에 대한 관심의 유전자의 각 대립 유전자의 발현을 비교합니다.
참고 : 제어 유전자 프라이머 대립 유전자 특정 할 필요가 없습니다. 증폭 대상 영역을 제외한 유전자형 프라이머에 상술 한 대립 유전자 특이 적 프라이머를 디자인 RT-qPCR에 프라이머 동일하다. 유전자 서열을 사용한다; 단일 엑손 또는 엑손 - 인트론 경계 용 프라이머를 사용하는 경우 F1 게놈 DNA가 사용될 수있다 (기본 증명서를 모니터링)표준 곡선. RT-qPCR에에 대한 자세한 내용은 Forlenza 등을 참조하시기 바랍니다. 2012 33.
ES 세포의 장기 저장 8. 동결 스톡의 제조
- (단계 7.3에서) 각 6 웰에 트립신의 300 μl를 추가하고 37 ℃에서 5 분 동안 품어. 하나의 세포로 해리 위아래로 여러 번 트립신과 피펫을 중화 스핀 미디어 (표 2) 2 ㎖를 추가합니다.
- 5 분 동안 300 XG에 15 ML 튜브와 스핀에 세포를 전송합니다.
- 뜨는을 기음과 ES 세포 미디어 (표 1)의 500 μl를 추가합니다. 세포를 재현 탁 아래로 피펫합니다.
- 1.5 ml의 cryovial 튜브에 콘텐츠를 전송하고 배 ES 세포 동결 매체 (표 3) 500 μl를 추가합니다. 튜브를 반전하여 잘 혼합 알코올이없는 세포 냉동 용기에 튜브를 배치합니다. 트랜스페린 전에 적어도 12 시간 동안 -80 ° C에서 냉동 컨테이너이 셀 배치액체 질소 저장 탱크로 g.
결과
여기에 설명 된 프로토콜 CRISPR / Cas9 게놈 편집 (도 1)를 이용하여 생성 monoallelic 인핸서 삭제 세포에서 유전자 발현의 시스 -regulation 공부 F1 ES 세포를 사용한다. 유전자형 유전자 발현의 gRNA 및 대립 유전자 특이 적 프라이머를 디자인이 방법에서 중요한 요소이다. 각 대립 유전자 특이 프라이머 세트는 대립 유전자 특이성을 확인하는 qPCR에 의해 검증되?...
토론
CRISPR / Cas9 매개 게놈 편집 기술은 게놈 수정에 대한 간단 빠르고 저렴한 방법을 제공한다. 기능 증강 특성에 대한 monoallelic 증강 삭제를 생성하고 분석하기 위해 여기에 설명 된 방법은 F1 마우스 세포에서 SNP를 활용합니다. 이러한 유형의 접근법의 이점은 : 1) monoallelic 인핸서 삭제가 중요한 증강제, 즉 두 대립 유전자에서 치사 셀에 이르는 조절 유전자의 단백질 수준에서 큰 감소를 삭제하...
공개
저자는 이해 충돌에 조브의 정책을 읽고 공개 충돌이 더이 없다.
감사의 말
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
자료
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
참고문헌
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152 (2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187 (2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485 (2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159 (2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유