
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Gerando CRISPR / Cas9 Mediated monoalélicos Eliminações para estudar a função Enhancer em células-tronco embrionárias de camundongos
Neste Artigo
Resumo
Experimental validation of enhancer activity is best approached by loss-of-function analysis. Presented here is an efficient protocol that uses CRISPR/Cas9 mediated deletion to study allele-specific regulation of gene transcription in F1 ES cells which contain a hybrid genome (Mus musculus129 x Mus castaneus).
Resumo
Enhancers control cell identity by regulating tissue-specific gene expression in a position and orientation independent manner. These enhancers are often located distally from the regulated gene in intergenic regions or even within the body of another gene. The position independent nature of enhancer activity makes it difficult to match enhancers with the genes they regulate. Deletion of an enhancer region provides direct evidence for enhancer activity and is the gold standard to reveal an enhancer's role in endogenous gene transcription. Conventional homologous recombination based deletion methods have been surpassed by recent advances in genome editing technology which enable rapid and precisely located changes to the genomes of numerous model organisms. CRISPR/Cas9 mediated genome editing can be used to manipulate the genome in many cell types and organisms rapidly and cost effectively, due to the ease with which Cas9 can be targeted to the genome by a guide RNA from a bespoke expression plasmid. Homozygous deletion of essential gene regulatory elements might lead to lethality or alter cellular phenotype whereas monoallelic deletion of transcriptional enhancers allows for the study of cis-regulation of gene expression without this confounding issue. Presented here is a protocol for CRISPR/Cas9 mediated deletion in F1 mouse embryonic stem (ES) cells (Mus musculus129 x Mus castaneus). Monoallelic deletion, screening and expression analysis is facilitated by single nucleotide polymorphisms (SNP) between the two alleles which occur on average every 125 bp in these cells.
Introdução
Elementos reguladores da transcrição são críticos para a sintonia fina espaço-temporais da expressão de genes durante o desenvolvimento e uma modificação destes elementos pode resultar em doença devido à expressão aberrante do gene 2. Muitas regiões associados à doença identificadas por estudos de associação de genoma de largura estão em regiões não codificantes e têm características de potenciadores da transcrição 3-4. Identificando potenciadores e combinando-os com os genes que regulam é complicado uma vez que são muitas vezes localizados vários quilobases de distância a partir dos genes que regulam e podem ser activados de um modo específico do tecido 5-6. Previsões Enhancer são comumente baseado em marcas de modificação das histonas, complexos mediador-cohesin e ligação de transcrição específicos do tipo de célula fatores 7-10. Validação dos intensificadores preditos é mais frequentemente feito por meio de um ensaio à base de vector em que o intensificador activa a expressão de um gene repórter 11-12. Estes dados fornecem vinformações aluable sobre o potencial de regulamentação de sequências potenciadoras putativos mas não revelam a sua função no seu contexto genómico endógeno ou identificar os genes que eles regulam. edição genoma serve como uma ferramenta poderosa para o estudo da função dos elementos reguladores da transcrição no seu contexto endógeno por análise por perda de função.
Os avanços recentes na edição genoma, ou seja, a / Cas9 sistema de edição genoma CRISPR, facilitar a investigação da função genoma. O sistema / Cas9 CRISPR é fácil de usar e adaptável para muitos sistemas biológicos. A proteína Cas9 é direcionado para um site específico no genoma por um RNA guia (gRNA) 13. O complexo SpCas9 / gRNA digitaliza o genoma para a sua sequência genómica alvo que deve ser 5 'para uma sequência adjacente protospacer motivo (PAM), NGG 14-15. o emparelhamento de bases do gRNA ao seu alvo, a 20 nucleótidos (nt) sequência complementar ao gRNA, activa a actividade de nuclease SpCas9 resultando numa DOUBLpausa e Strand (DSB) 3 pb a montante da sequência PAM. Especificidade é conseguido através de emparelhamento de base completa na região da semente do gRNA, a 6-12 nt ao lado do PAM; Por outro lado, não corresponde 5 'da semente são geralmente tolerada 16-17. O DSB introduzido podem ser reparados tanto por a extremidade não-homóloga de união (NHEJ) a reparação do ADN ou homologia reparação dirigida (HDR) mechanisms.NHEJ reparação do ADN, muitas vezes cria inserção / deleção (indels) de alguns pb no local alvo que pode perturbar a grelha de leitura aberta (ORF) de um gene. Para gerar delecções maiores no genoma dois gRNAs, que flanqueiam a região de interesse, podem ser usados 18-19. Esta abordagem é particularmente útil para o estudo de potenciadores da transcrição aglomeradas em regiões de controlo local ou super-potenciadores, que são maiores do que melhoradores convencionais 9,18,20-22.
Eliminações monoalélicos são um modelo valioso para estudar Regulamentação cis da transcrição. O chang observadoe no nível de transcrição após eliminação monoalélicos de um potenciador se correlaciona com o papel de que potenciador na regulação dos genes sem os efeitos de confusão que podem ocorrer quando a transcrição de ambos os alelos é afetado potencialmente influenciar a aptidão celular. Avaliando expressão reduzida é difícil no entanto, sem a capacidade de distinguir o excluído do alelo selvagem. Além disso, a genotipagem deleções em cada alelo sem a capacidade de distinguir os dois alelos é um desafio, em especial para grandes deleções de> 10 kb até 1 Mb 23 em que é difícil para amplificar toda a região de tipo selvagem por PCR. A utilização de células ES F1 gerados pelo cruzamento de Mus musculus 129 com Mus castaneus permite que os dois alelos de ser diferenciadas por PCR 18,24 específico de alelo. O genoma híbrido nestas células facilita a eliminação específica alelo rastreio e análise de expressão. Em média, existe um SNP a cada 125 pb entre estes dois genomas, Proporcionando flexibilidade no desenho de primers para a expressão e genotipagem analisa. A presença de um SNP pode influenciar a temperatura de fusão do iniciador (T m) e especificidade alvo em amplificação em tempo real quantitativa PCR (qPCR) permitindo a discriminação dos dois alelos 25. Além disso, um desfasamento dentro da extremidade 3 'do iniciador influencia grandemente a capacidade de ADN-polimerase para estender a partir do iniciador de amplificação de prevenir o alvo alelo indesejada 26. Descrito no seguinte protocolo é a utilização de células ES F1 para o alelo específico deleções potenciadoras de maior do que 1 kb e análise subsequente expressão que utilizam o sistema CRISPR / Cas9 edição genoma (Figura 1).

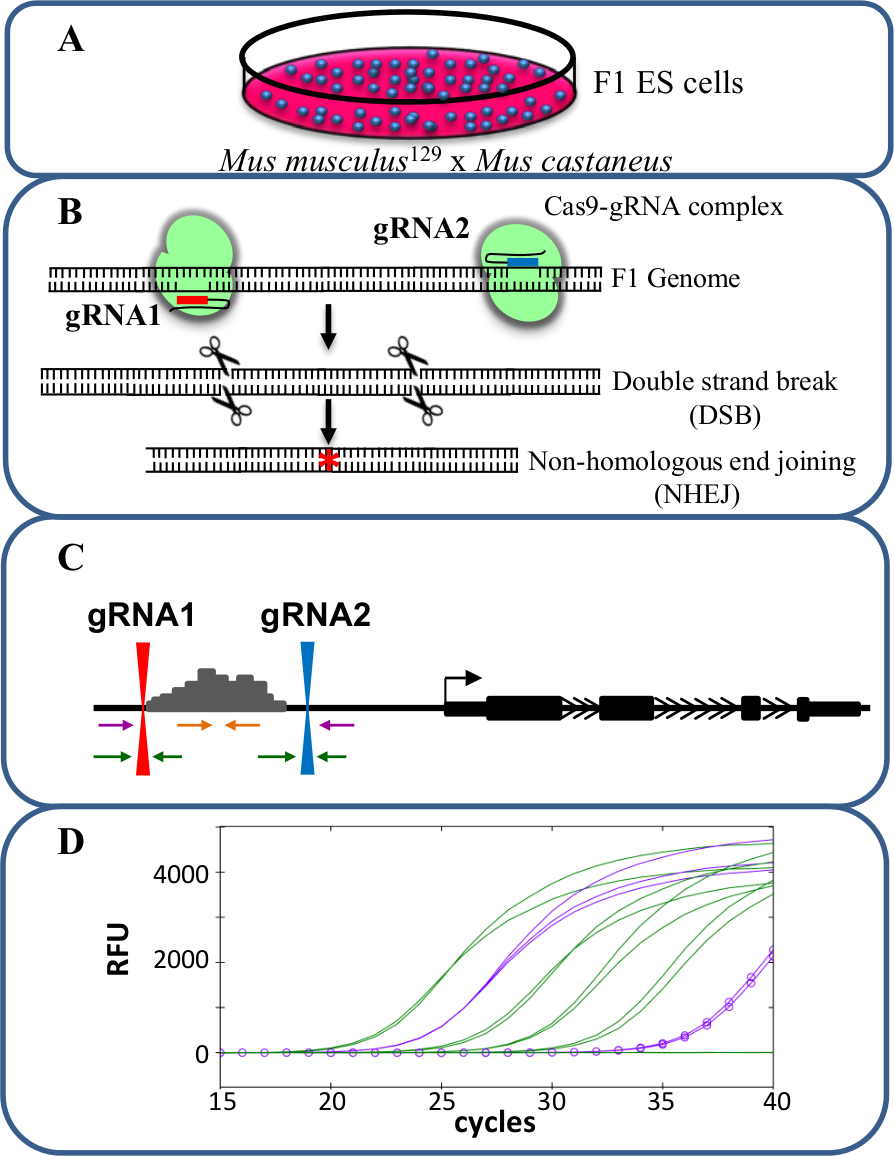
Figura 1. Enhancer eliminação utilizando CRISPR / Cas9 para estudar cis -reglação da expressão de genes (A) células F1 ES gerados por um cruzamento entre Mus musculus 129 e Mus castaneus. são usadas para permitir a eliminação específica dos alelos. (B) Dois RNAs de guia (gRNA) são utilizados para induzir um grande deleção Cas9 mediada da região do potenciador. (C) Os conjuntos de iniciadores são utilizados para identificar grande mono- e bi-supressões alélicas. Os iniciadores de laranja são os primers dentro, os primers roxo são os iniciadores externos e os primers verdes são os iniciadores flanqueando gRNA. (D) As mudanças na expressão genética são monitorados através de qPCR alelo-específico. RFU denota unidades de fluorescência relativas. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. projetar e construir o gRNA
- Para eliminar regiões potenciador da transcrição usar dois gRNAs, um 5 'e um 3' da região de interesse. Use o mouse UCSC faixa navegador genoma gerado pelo laboratório Zhang para identificar sequências únicas gRNA (http://www.genome-engineering.org 15). Em seguida verifique estes gRNAs e sua PAM adjacente, para SNPs e indels usando ferramentas on-line fornecidos pelo Instituto Sanger (www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1211) 27-28. Para direcionar ambos os alelos com igual eficiência, evitar sequências / PAM gRNA que contêm um SNP ou INDEL.
- Embora a escolha do gRNA, para verificar a viabilidade de projetar primers específicos de alelo para a genotipagem de eliminação. Consulte a seção 5 para desenho de primers específicos de alelo.
- Adaptar os dois plasmídeos gRNA com base no protocolo descrito no Mali et al., 2013 15. Incorporar o único 20 pb sequência alvo int seleccionadoO 61mer os oligonucleótidos, como mostrado na Tabela 7 (sequências são apresentadas na direcção 5 'para 3', e as bases em negrito são a sequência alvo de 20 pb, que são complementos reversos um do outro).
- Misturar 10 ul de 10 mM gRNA Primer_F e 10 ul de 10 mM Primer_R complementar em um tubo.
- Hibridar os iniciadores por incubação da mistura de iniciadores a 100 ° C durante 5 min e depois arrefecer 1 ° C / seg a 25 ° C. Para esta etapa, utilizar uma máquina de PCR ou colocar o tubo em água a ferver e deixá-lo arrefecer para a TA.
- Para a mistura de iniciadores emparelhados, adicionar a seguinte mistura de reacção e incuba-se a 72 ° C durante 30 min para estender cada um dos iniciadores: 18,5 ul de água, 10 ul de tampão 5x HF, 1 ul de dNTPs 10 mM e misturam 0,5 ul de alta a polimerase de ADN de fidelidade.
- Corra 10 uL de fragmento-alvo num gel de agarose a 2% para confirmar os 100 fragmentos de guia pb foram produzidos.
- Linearizar o vector gRNA (um presente de George Igreja; Addgene plasmídeo # 41824) com 15 Afl II, utilizando a seguinte reacção configurar: 5 ul de espinha dorsal gRNA vector (2-4 ng), 5 ul de tampão 10x, 3 jul de Afl II (20 unidades / ul) e 32 ul de água. Incubar a mistura de reacção durante 3 horas a 37 ° C.
- Executar o produto digerido num gel de agarose a 1% e purifica-se a banda de ADN correspondente aos linearizado de 3,5 kb gRNA vector utilizando um kit de extracção de gel, seguindo as instruções do fabricante.
- Defina-se reacções de montagem Gibson 29-30 usando linearizado gRNA vector e fragmento alvo a partir do passo 1.2.3 do seguinte: 1 uL de vector de gRNA linear (50 ng / mL), 1 uL de fragmento-alvo, 10 ul de 2x Gibson montagem master mix e 8 pi de água. Incubar as reacções a 50 ° C durante 60 min.
- Transformação de células de E. coli com montado gRNA Vector.
- Misturar 1 uL de vector de gRNA montado a partir de 1.2.7e 50 ul de DH5a (estirpe de E. coli) em células de um tubo. Transformar as células DH5a pelo método de choque de calor pela exposição das células a 42 ° C durante 45 seg.
- Pressão refrigerar os tubos em gelo durante 5 min; em seguida, adicionar 400 ul de meio SOC e incubar a 37 ° C durante 45 minutos num incubador com agitação.
- Espalhe 100 ul de células DH5a de uma placa para a selecção positiva de células transformadas LB-canamicina (50 ug / ml) e incuba-O / N a 37 ° C.
- A triagem positiva E.coli colónias para gRNA Inserir.
- Escolher uma colónia resistente a canamicina e ressuspender em 3 ml de LB contendo 50 ug / ml de canamicina. Repetir o mesmo para 6-8 colónias e incubar todos os tubos a 37 ° CO / N em um incubador com agitação.
- Extrair os plasmídeos a partir da cultura crescida O / N utilizando o plasmídeo mini-kit de preparação, seguindo o manual do fabricante.
- Preparar um EcoR I mix reacção de digestão para verificar se há gRNA inserção sequênciano plasmídeo. Para cada amostra, preparar a mistura da reacção como se segue: 2 jul de tampão de enzima, 1 ul de EcoRI, 15 ul de água. Alíquota da mistura de reacção para tubos de 1,5 ml e adicionar 2 ul de plasmídeo. Incubar os tubos a 37 ° C durante 2 h.
- Executar o produto digerido num gel de agarose a 1,5%.
Nota: As amostras com inserção irá exibir um tamanho de banda de 475 pb que está 100 pb maior do que os clones sem inserções.
Observação: Como alternativa, os clones positivos podem ser rastreados por um PCR de colónias utilizando Sp6 (para a frente) e os iniciadores de T7 (reverso) (Tabela 7) que se ligam à sequência de vector para dar um fragmento de tamanho 642 pb na presença de um gRNA de inserção. A abordagem de PCR de colónias é vantajoso quando existe um sítio de restrição EcoR I dentro da sequência gRNA.
- Confirmar a sequência do gRNA Insert pelo DNA Sequencing Usando o T7 Primer.
2. Transfecção
Nota:A electroporação é um método eficiente de transfecção de plasmídeos em células ES. O método descrito aqui utiliza a tecnologia de microporos transfecção.
- Cultivar células F1 ES em um prato revestido com gelatina a 10 cm, que inclua 10 ml de meio de células ES (Tabela 1), a 37 ° C / 5% de CO 2. Quando as células chegar a 85% de confluência remova a mídia e adicionar 2 ml de tripsina. Incubar a 37 ° C na incubadora de CO 2 durante 5 min.
Nota: células F1 ES foram obtidos a partir de Barbara Panning 24 e estão disponíveis a pedido. - Neutraliza-se a tripsina por adição de 10 ml de meio de centrifugação (Tabela 2). Pipeta várias vezes para retirar as células completamente.
- Recolha todas as células em um tubo de 15 ml e centrifugação a 300 xg durante 5 min. Ressuspender em 3 ml de PBS e contar as células utilizando um hemocitómetro ou contador de células automatizado.
- Pellet 1 x 10 6 células ES em um tubo de 1,5 ml por centrifugação a 300 xg durante 5 min e ressuspender em 100 ul de R (ressuspensão) como tampão fornecido pelo fabricante do kit.
- Adicionam-se 5 ug de cada pCas9_GFP (uma oferta do Kiran Musunuru; Addgene plasmídeo # 44719) 31, 5 'e 3' plasmídeos gRNA de deleção da região alvo e misturar cuidadosamente com uma pipeta para evitar a introdução de bolhas.
- Utilizar a pipeta de ponta electrónico para aspirar 100 ul da mistura de electroporação, tendo o cuidado de evitar uma bolha na ponta.
- Programa da volts, largura e pulsos para eletroporação. Para as células F1 ES, usar 1400 V, 10 ms para 3 pulsos.
- Enquanto a eletroporação está sendo executado observar a ponta para ver qualquer faíscas na solução. Uma faísca indica a presença de uma bolha de ar e irá interferir com a transfecção.
- Ejectar as células ES transfectados para um prato revestido com gelatina a 10 cm, que inclua 10 ml ES meio celular (Tabela 1) e incuba-se a 37 ° C / 5% de CO 2.
3. separação por FACS células transfectadas
- Após 48 horas, separar as células por adição de 2 ml de tripsina e incubar a 37 ° C na incubadora de CO 2 durante 5 min.
- Neutraliza-se a placa por adição de 10 ml de tampão de recolha (Tabela 3). Recolher as células num tubo de 15 mL e centrifugação a 300 xg durante 5 min.
- Descartar o sobrenadante e ressuspender as células em 1 ml de tampão de triagem (Tabela 4). Contar as células e dilui-se com base na plataforma de triagem. Dilui-se as células de 0,5-1 x 10 6 células / ml para a separação em tubos de 15 ml e para classificar células individuais directamente em placas de 96 poços, diluir as células a 2-5 x 10 6 culas / ml.
- Ordenar células Cas9-GFP + ES utilizando um citómetro de fluxo FACS 32. Recolher as células em grandes quantidades em tubos com 2 mL de meio de recuperação (Tabela 5) e de placas como descrito em 3.5 para a colheita de colónias ou de células individuais tipo directamente em placas de 96 poços revestidas com gelatina contendo 100 ul de meio celular ES / poço ( Tabela 1).
- Semente de 1-1,5 x 10 4 GFP + células ES em um prato revestido com gelatina a 10 cm, que inclua meios de células 10 mL ES (Tabela 1). Chapeamento neste baixa densidade facilitará escolher colônias de células ES individuais.
4. Clones Cultivar para genotipagem, Análise de Expressão e Stocks cela gelada
- No dia 4-5, após separação, marcar cada poço de placas de 96 poços ordenadas directos para a presença de colónias de células ES.
- Dissociar colônias de células ES, removendo a mídia e adicionando 30 ul de tripsina. Incubar a 37 ° C durante 5 min. Neutraliza-se a tripsina por adição de 170 ul de meios de células ES (Tabela 1), e pipeta-se para baixo e para a dissociação completa da colónia em células individuais. Crescer as células a 37 ° C / 5% de CO2 até que a maioria dos poços são mais de 70% confluentes (normalmente 2-3 dias).
- Alternativamente pegar colônias de células ES individuais de placas de 10 cm usandoum microscópio invertido. Após aspiração da colónia no seguimento passo ponta da pipeta 4.1.1 colocando cada colónia em um poço de uma placa de 96 poços, pré-tratadas com gelatina e contendo 30 ul de tripsina.
Nota: Colonies pode sentar-se em tripsina a RT, enquanto uma linha inteira de colônias é escolhido. - Uma vez que todas as colónias foram repicadas e dissociados em meio de crescer as células a 37 ° C na incubadora de CO 2 até que a maioria dos poços são mais de 70% confluentes (geralmente 2 dias).
- Quando as placas de 96 poços estão prontas para a separação, remoção dos suportes, adicionar 30 ul de tripsina e incubar a 37 ° C durante 5 min. Neutraliza-se a tripsina por adição de 180 ul de meios de células ES (Tabela 1) a cada poço e pipeta-se para baixo e para a dissociação completa em células individuais.
- A partir do 210 ul resultante, sementes 70 ul em três revestidos com gelatina placas de 96 poços, cada um contendo 130 ul de meio celular ES / cavidade (Quadro 1). Use essas placas para genotyping, análise e expressão de congelação reservas de células, para cada clone, tal como descrito abaixo.
- Quando a placa de genotipagem atinge 70-85% de confluência, tratar a placa conforme descrito no capítulo 6 "genotipagem a exclusão".
- Quando a placa de análise de expressão atinge 70-85% de confluência, remova a mídia, selar a placa com fita de vedação e armazenar a -80 ° C até que os clones foram genotipados.
Nota: A placa de análise de expressão é útil para analisar as alterações na expressão de genes em passagens precoces dos clones. A análise da expressão do gene a partir da placa de 96 poços é possível, mas como os números de células são baixo um kit de micro-extração de RNA é recomendado. - Preparação de Congelar placa de 96 poços de Stock:
- Quando a placa de reservas de células congeladas (stock-1) atinge 70-85% de confluência, aspirar os meios de comunicação, adicionar 30 ul de tripsina e incubar a 37 ° C durante 5 min.
- Neutraliza-se a tripsina por adição de 100 ul de meios de células ES (Tabela 1) tO cada poço e pipeta-se para baixo e para a dissociação completa em células individuais.
- Transferir 15 uL de suspensão de células de cada poço para duas placas de 96 poços revestidas com gelatina, cada uma contendo 185 ul de meios de células ES (Tabela 1) e deixa-se crescer a 37 ° C / 5% de CO 2.
Nota: Isto é para estoque-2 e -3 placas que são clones de back-up adicionais no caso de reviver as células do estoque-1 não for bem sucedida.
- Entretanto, para os 100 ul de células remanescentes na placa de 96 poços (estoque-1), adicionar 100 uL de meio de congelação 2x (Tabela 6). Selar a placa com uma fita de vedação e rapidamente inverter a placa 4-5 vezes para a mistura adequada. Armazenar a placa à temperatura de -80 ° C até os clones são genotipados.
- Quando as placas de estoque-2 e da 3-estão prontos para congelar os meios aspirado, adicionar 30 ul de tripsina e incubar a 37 ° C durante 5 min. Neutralizar a tripsina, adicionando 70 mL de meios de células ES (Tcapaz 1) a cada poço e pipeta-se para baixo e para a dissociação completa em células individuais.
- Adicione 100 ml de mídia congelamento 2x, selar a placa com fita de vedação e rapidamente inverter a placa 4-5 vezes para a mistura adequada. Armazenar a placa à temperatura de -80 ° C até serem necessários são estas placas.
5. desenho de primers específicos de alelo
- Design de 4 conjuntos de iniciadores (Figura 1C) para o rastreio dos clones para a supressão desejada: dentro iniciadores, os iniciadores externos e iniciadores flanqueando gRNA (para ambos 5 'e 3' sítios alvo gRNA) como descrito abaixo.
- Obter a faixa SNP correspondentes aos 129 e elenco genótipos em http://labs.csb.utoronto.ca/mitchell/crispr.html. A pista dada mostra substituições de base entre 129 e atirarem coordenadas na montagem do genoma do rato MM9.
Nota: O link no site acima irá redirecionar para navegador genoma UCSC e adicionar uma faixa personalizada que contém o SNPs entre o 129 e elenco genomos. - Digite as coordenadas da região a ser excluído. Zoom em uma região de cerca de 500 pb no meio da eliminação desejado contendo> 3 SNPs.
- Vá em Exibir> DNA na barra de opção e clique em DNA começar a baixar a sequência alvo em todos os formatos de maiúsculas.
- Criar duas sequências FASTA; um para 129 e outro para Elenco pela substituição de base na posição do SNP. Marcar os SNPs por um caso inferior.
- Ir para Primer3 mais (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi/) e cole o SNP substituído 129 sequência. Use as configurações padrão para projetar primers.
- Para projetar os primers específicos de alelo dentro selecionar Primer_List no menu drop tarefas e clique em Escolher Primers. Escolha um primer para a frente ou para trás que tem um SNP quer no "fim ou dentro de 4 bases de 3 'a 3 e cai dentro da região a ser excluído.
Nota: Os iniciadores que têm um SNP na extremidade 3 'de exibição especificidade aumentada alelo em qPCR. - Para selecionar o segundo retorno de primer para a página principal, selecione Detecção no menu drop tarefa e colar a primeira sequência de iniciador na caixa apropriada na parte inferior da página. Na aba Configurações Gerais alterar a configuração de escala do tamanho do produto para 80-200 pb e clique em Selecionar Primers. Escolheu um conjunto de primers a partir dos pares de primers listados; estes serão os 129 iniciadores específicos de alelo dentro.
- Repita os passos 5.1.5 a 5.1.7 para projetar primers para o alelo Cast.
- Voltar ao navegador genoma UCSC e digite as coordenadas da região a ser excluído. Vá em Exibir> DNA na barra de opção, em Opções Sequence Retrieval Região adicionar 1.000 pb a montante ea jusante, clique em obter o DNA para baixar a sequência alvo.
- Marque a sequência alvo gRNA entre parênteses. Guardar esta sequência inteira antes de prosseguir.
- Para desenhar os iniciadores fora remover a sequência entre as duas sequências alvo gRNA. Repita o passo 5.1.4-5.1.8 para projetar o primer alelo-específico foras, mas mudar o tamanho do produto 400-800 pb.
- Dividir a sequência obtida no passo 5.1.10 em duas sequências, cada uma com 500 bp 5 'e 3' da sequência alvo gRNA. Repita os passos 5.1.5 a 5.1.7 projetar primers específicos não-alélicas para regiões flanqueando gRNA mas mudar o tamanho do produto 400-800 pb.
Nota: Para iniciadores não específicos de alelo, ou 129 ou uma sequência de molde pode ser usada e os iniciadores devem ser escolhidos que não contêm um SNP. É aconselhável definir um tamanho de produto de 400-800 bp para a concepção de fora e gRNA flanqueando primers. Isto permite a amplificação mesmo que pequenas indels estão presentes.
- Obter a faixa SNP correspondentes aos 129 e elenco genótipos em http://labs.csb.utoronto.ca/mitchell/crispr.html. A pista dada mostra substituições de base entre 129 e atirarem coordenadas na montagem do genoma do rato MM9.
- Teste os primers dentro de especificidade alelo por qPCR utilizando 129 e Elenco DNA genômico pura tensão a 2 ng / mL. Siga o passo 6,2-6,4 para configurar a reação qPCR.
Nota: Se o genótipo 129 na região alvo é o mesmo que murganhos C57BL / 6J, ADN de ratinhos C57BL / 6J pode ser utilizado no lugar de 129 de ADN. iniciadores específicos de alelo deve exibir pelo menos 5 ciclosdiferença entre o valor de Ct (limiar de ciclo) sobre a correcta em relação ao genótipo incorrecta. Os iniciadores externos podem ser testados para garantir que amplificam a eliminação utilizando Cas9 / gRNA transfectadas células ES, e os controlos F1 ADN genómico, respectivamente, como positivos e negativos. O alelo de especificidade dos iniciadores externos podem ser testados uma vez que os clones monoalélicos foram identificados.
6. A genotipagem de eliminação
- Extracto de ADN genómico a partir da genotipagem placa de 96 poços utilizando a placa do passo 4.6, que é gerado após a expansão colónia.
- Preparar a mistura de extracção de ADN genómico: 89 ul de água, 10 ul de tampão 10x e 1 ul de reagente de extracção (fornecido pelo fabricante). Adicionar 100 ul de ADN genómico mistura de extracção para cada cavidade e selar a placa com uma fita de vedação.
- Incubar a placa a 75 ° C durante 5 minutos, seguido de 95 ° C durante 5 min.
- Permitir que a placa arrefecer por incubação em gelo durante alguns minutos a umad, em seguida, centrifugar brevemente para resolver qualquer condensação para o fundo do poço. Isto serve como a placa de DNA molde para o rastreio de eliminação.
- Defina-se as reacções de qPCR em duplicado para cada clone como se segue: 5 ul de 2x SYBR qPCR mistura, para a frente e iniciador (3 uM) a cada 1 ml e 1 ml de água reversa. Usar uma pipeta de canais múltiplos para adicionar 2 ul de ADN molde, seguido de 8 ul de mistura reaccional a cada poço de uma placa de 384 poços.
- Selar a placa com fita de vedação e centrifugação a 600 xg durante 2 min para misturar o conteúdo. Colocar a matriz placa de 384 poços no reciclador tempo real.
- Programa o reciclador tempo real para um 2-passo de PCR seguido por análise de curva de fusão com detecção como se segue: 1 ciclo a 95 ° C durante 10 min, 40 ciclos de 95 ° C durante 15 s, 62 ° C durante 30 seg, com leitura da placa e 95 ° C durante 10 s, 65 ° C a 95 ° C com incrementos de 5 ° C durante 5 seg + placa de ler.
Nota: Além de cartilha design, o qPCR miparâmetros x e o ciclo também contribuem para a cartilha especificidade. Os parâmetros descritos acima e reagentes listados nos materiais mais frequentemente se obter a amplificação específica do alelo. - Analisando os resultados qPCR
- Verifique cada alelo para a amplificação com primers específicos de alelo dentro. Sem amplificação de um alelo ou diferenças de alto valor Ct (> 5 ciclos) entre alelos sugerem que estes clones realizar uma deleção heterozigótica do alelo com o alto valor / ausentes Ct. Sem amplificação de ambos os alelos sugere que eles carregam uma deleção homozigótica.
- Verificar cada alelo por amplificação com iniciadores específicos de alelo fora. Quando a eliminação do alvo é maior do que 1 kb de amplificação com iniciadores externos apenas quando ocorre uma deleção está presente. Um valor Ct de 22-28 confirma a exclusão. Para deleções alvo menor do que 1 kb, confirmar o tamanho do fragmento amplificado por electroforese.
Nota: Se os iniciadores externos mostrar apenas alelo especificidade moderado (ver FiguRE 2), podem ser obtidos produtos de amplificação com os dois conjuntos de iniciadores alélicas nos clones monoalélicos devido à amplificação do alvo fora dos iniciadores externos. Neste caso, um valor de Ct diferença entre os dois alelos de, pelo menos, cinco ciclos deverá confirmar o correcto alelo (menor valor de Ct) é eliminada com base nos resultados obtidos a partir dos iniciadores dentro. Se o no alvo contra off alelo alvo Ct diferença é inferior a cinco ciclos de projetar novos alelo iniciadores externos específicos. - Em clones de deleção monoalélicos verificar a integridade do alelo não-excluídos usando a triagem secundária, gRNA flanqueando primers.
Nota: Indels de> 25 bp tamanho em torno do local de destino a gRNA podem ser identificados por meio da observação de um deslocamento da curva de derreter na qPCR para 400-800 amplicons pb. Alternativamente, os produtos de amplificação a partir de iniciadores que flanqueiam as gRNA pode ser sequenciado para detectar pequenas indels de <25 pb.- Realizar qPCR com 2 conjuntos de iniciadores que flanqueiam gRNA isto é, 5 'e 3' gARN utilizado na geração de supressão CRISPR. Sem amplificação com estes conjuntos de iniciadores indica indels maiores do que o amplicão qPCR estão presentes no local alvo do gRNA no alelo não apagados de clones de deleção monoalélicos. Descartar clones contendo estes grandes indels de posterior análise como os resultados podem ser difíceis de interpretar sem conhecer a extensão da deleção.
- Purificar dos amplicões obtidos a partir do exterior primário reação qPCR utilizando um PCR limpar kit seguindo as instruções do fabricante.
- Confirmar a sequência do alelo suprimido por sequenciação de ADN do produto de PCR purificado a partir da etapa anterior. Use os iniciadores de amplificação qPCR para a frente e sequenciação reversa.
Nota: Neste estágio SNPs dentro do ato amplicon como uma confirmação secundária do genótipo do alelo excluído.
7. Analisar Expressão com o alelo primers específicos
- Descongelar o 96 poços pl estoque de célulascomeu armazenadas a -80 ° C (estoque-1 a partir do passo 4.9), colocando-o em um banho de talão quente. Quando mais de metade dos poços da placa são descongeladas, centrifugação a 300 xg durante 5 min.
- Cuidadosamente, remover a fita de vedação e transferir rapidamente as células a partir das cavidades positivas de deleção, em placas de 12 poços revestidas com gelatina contendo 1 ml de meio de células ES (Tabela 1) e incuba-se a 37 ° C / 5% de CO 2.
- Quando a placa atinge 70-85% de confluência, as células de passagem e dividi-los em três poços de uma placa de 6 poços revestidos com gelatina, cada uma contendo 2 ml de meio de células ES (Tabela 1). Usar dois poços para preparar 2 frascos de estoques de células congeladas para armazenagem de longo prazo em azoto líquido (descrito no passo 8) e o terceiro poço para extracção de ARN.
- Extrair ARN utilizando um kit de extracção de ARN.
- Converter 100-500 ng de ARN para ADNc através de transcrição reversa (RT) do ARN utilizando o kit de síntese de cDNA, seguindo o protocolo do fabricante. Incluir um n RTegative reacção para cada amostra de ARN para monitorizar a quantidade de ADN contaminante nas amostras de ARN.
- Dilui-se o ADNc antes de qPCR em uma proporção compreendida entre 1: 2 e 1: 4; dependendo do nível do gene alvo em células ES expressão.
- Defina o qPCR como descrito acima, incluindo DNA genómico F1 como curva padrão (5 diluições de 250 a 0,08 ng / mL) para a quantificação absoluta dos níveis de transcrição. Comparar a expressão de cada alelo do gene de interesse em cada clone confirmou suprimido para um gene de controlo adequados, por exemplo, de GAPDH (iniciadores listados na Tabela 7).
Nota: primers gene de controle não precisa ser alelo específico. concepção de iniciadores específicos de alelo é o mesmo para os iniciadores de RT-qPCR como descrito para os iniciadores de genotipagem com a excepção da região alvo para amplificação. A sequência do gene deve ser utilizado; Se utilizando iniciadores para um único exão ou uma fronteira exão-intrão (para monitorizar transcrito primário) F1 ADN genómico pode ser usado paraa curva padrão. Para mais detalhes sobre RT-qPCR consulte Forlenza et al. 2012 33.
8. Congelar da Preparação para o armazenamento de longo prazo de células ES
- Adicionar 300 ul de tripsina a cada um de 6 poços (a partir do passo 7.3) e incubar durante 5 min a 37 ° C. Adicionar 2 ml de meio de centrifugação (Tabela 2) para neutralizar a tripsina e pipeta cima e para baixo várias vezes a dissociar-se em células individuais.
- Transferir as células para um tubo de 15 mL e centrifugação a 300 xg durante 5 min.
- Aspirar o sobrenadante e adicionar 500 ul de meios de células ES (Tabela 1). Pipeta cima e para baixo para voltar a suspender as células.
- Transferir o conteúdo para um tubo de 1,5 ml criotubo e adicionar 500 mL de meio de congelação de células ES 2x (Tabela 3). Misture bem invertendo o tubo e colocar o tubo em um recipiente de congelamento celular livre de álcool. Coloque esta célula congelamento recipientes a -80 ° C durante pelo menos 12 horas antes de transferrinag para um tanque de armazenamento de azoto líquido.
Access restricted. Please log in or start a trial to view this content.
Resultados
O protocolo aqui descrito utiliza células F1 ES para estudar -regulação cis da expressão do gene em células estimuladoras suprimido monoalélicos gerados usando CRISPR / edição Cas9 genoma (Figura 1). O gRNA e desenho de primers específicos de alelo para genotipagem e expressão do gene são os fatores-chave para esta abordagem. Cada conjunto de primers específicos de alelo devem ser validados por qPCR para confirmar a especificidade alelo. Iniciadores ...
Access restricted. Please log in or start a trial to view this content.
Discussão
CRISPR / Cas9 tecnologia de edição genoma mediada fornece um método simples, rápido e barato para a modificação do genoma. O método aqui descrito para gerar e analisar eliminação monoalélicos potenciador para potenciador caracterização funcional tira vantagem de SNPs em células de ratinho F1. As vantagens deste tipo de abordagem são os seguintes: 1) as supressões potenciadoras monoalélicos não produzem efeitos de confusão que ocorrem quando um intensificador crítica é suprimida de ambos os alelos,
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores leram as políticas de Jove dos conflitos de interesses e não têm conflitos de divulgar.
Agradecimentos
We would like to thank all the members of the Mitchell lab for helpful discussions. This work was supported by the Canadian Institutes of Health Research, the Canada Foundation for Innovation and the Ontario Ministry of Research and Innovation (operating and infrastructure grants held by JAM).
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S | high fidelity DNA polymerase used in gRNA assembly |
| Gibson Assembly Master Mix | NEB | E2611L | |
| gRNA_Cloning Vector | Addgene | 41824 | A target sequence is cloned into this vector to create the gRNA plasmid |
| pCas9_GFP | Addgene | 44719 | Codon-optimized SpCas9 and EGFP co-expression plasmid |
| AflII | NEB | R0520S | |
| EcoRI | NEB | R3101S | |
| Neon Transfection System 100 µL Kit | Life Technologies | MPK10096 | Microporator transfection technology |
| prepGEM | ZyGEM | PT10500 | genomic DNA extraction reagent |
| Nucleo Spin Gel & PCR Clean-up | Macherey-Nagel | 740609.5 | |
| High-Speed Plasmid Mini Kit | Geneaid | PD300 | |
| Maxi Plasmid Kit Endotoxin Free | Geneaid | PME25 | |
| SYBR select mix for CFX | Life Technologies | 4472942 | qPCR reagent |
| iScript cDNA synthesis kit | Bio-rad | 170-8891 | Reverse transcription reagent |
| 0.25% Trypsin with EDTA | Life Technologies | 25200072 | |
| PBS without Ca/Mg2+ | Sigma | D8537 | |
| 0.5 M EDTA | Bioshop | EDT111.500 | |
| HBSS | Life Technologies | 14175095 | |
| 1 M HEPES | Life Technologies | 13630080 | |
| BSA fraction V (7.5%) | Life Technologies | 15260037 | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258012 | |
| FBS | ES cell qualified | FBS is subjected to a prior testing in mouse ES cells for pluripotency | |
| DMSO | Sigma | D2650 | |
| Glutamax | Invitrogen | 35050 | |
| DMEM | Life Technologies | 11960069 | |
| Pencillin/Streptomycin | Invitrogen | 15140 | |
| Sodium pyruvate | Invitrogen | 11360 | |
| Non-essential aminoacid | Invitrogen | 11140 | |
| β-mercaptoethanol | Sigma | M7522 | |
| 96-well plate | Sarstedt | 83.3924 | |
| Sealing tape | Sarstedt | 95.1994 | |
| CoolCell LX | Biocision | BCS-405 | alcohol-free cell freezing container |
| CHIR99021 | Biovision | 1748-5 | Inhibitor for F1 ES cell culture |
| PD0325901 | Invivogen | inh-pd32 | Inhibitor for F1 ES cell culture |
| LIF | Chemicon | ESG1107 | Inhibitor for F1 ES cell culture |
Referências
- Sagai, T., Hosoya, M., Mizushina, Y., Tamura, M., Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 132 (4), 797-803 (2005).
- Kleinjan, D. A., Lettice, L. A. Long-range gene control and genetic disease. Adv Genet. 61, 339-388 (2008).
- Visel, A., Rubin, E. M., Pennacchio, L. A. Genomic views of distant-acting enhancers. Nature. 461 (7261), 199-205 (2009).
- Maurano, M. T., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 337 (6099), 1190-1195 (2012).
- Heintzman, N. D., et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 459 (7243), 108-112 (2009).
- Shen, Y., et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 488 (7409), 116-120 (2012).
- Johnson, D. S., Mortazavi, A., Myers, R. M., Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 316 (5830), 1497-1502 (2007).
- Rhee, H. S., Pugh, B. F. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 147 (6), 1408-1419 (2011).
- Whyte, W. A., et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 153 (2), 307-319 (2013).
- Chen, C. Y., Morris, Q., Mitchell, J. A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 13 (1), 152(2012).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 30 (3), 265-270 (2012).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 30 (3), 271-277 (2012).
- Jinek, M., et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337 (6096), 816-821 (2012).
- Cong, L., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 339 (6121), 819-823 (2013).
- Mali, P., et al. RNA-guided human genome engineering via Cas9. Science. 339 (6121), 823-826 (2013).
- Hsu, P. D., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 31 (9), 827-832 (2013).
- Cho, S. W., et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 24 (1), 132-141 (2014).
- Zhou, H. Y., et al. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev. 28 (24), 2699-2711 (2014).
- Fujii, W., Kawasaki, K., Sugiura, K., Naito, K. Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res. 41 (20), e187(2013).
- Tuan, D. Y., Solomon, W. B., London, I. M., Lee, D. P. An erythroid-specific, developmental-stage-independent enhancer far upstream of the human 'beta-like globin' genes. Proc Natl Acad Sci U S A. 86 (8), 2554-2558 (1989).
- Amano, T., et al. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell. 16 (1), 47-57 (2009).
- Li, Y., et al. CRISPR reveals a distal super-enhancer required for Sox2 expression in mouse embryonic stem cells. PLoS One. 9 (12), e114485(2014).
- Canver, M. C., et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem. 289 (31), 21312-21324 (2014).
- Mlynarczyk-Evans, S., et al. X chromosomes alternate between two states prior to random X-inactivation. PLoS Biol. 4 (6), e159(2006).
- Lefever, S., Pattyn, F., Hellemans, J., Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin Chem. 59 (10), 1470-1480 (2013).
- Huang, M. M., Arnheim, N., Goodman, M. F. Extension of base mispairs by Taq DNA polymerase: implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 20 (17), 4567-4573 (1992).
- Keane, T. M., et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 477 (7364), 289-294 (2011).
- Yalcin, B., et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 477 (7364), 326-329 (2011).
- Gibson, D. G., et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 6 (5), 343-345 (2009).
- Gibson, D. G., Smith, H. O., Hutchison, C. A., Venter, J. C., Merryman, C. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 7 (11), 901-903 (2010).
- Ding, Q., et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell. 12 (4), 393-394 (2013).
- Basu, S., Campbell, H. M., Dittel, B. N., Ray, A. Purification of specific cell population by fluorescence activated cell sorting (FACS). J Vis Exp. (41), (2010).
- Forlenza, M., Kaiser, T., Savelkoul, H. F., Wiegertjes, G. F. The use of real-time quantitative PCR for the analysis of cytokine mRNA levels. Methods Mol Biol. 820, 7-23 (2012).
- Wu, J. H., Hong, P. Y., Liu, W. T. Quantitative effects of position and type of single mismatch on single base primer extension. J Microbiol Methods. 77 (3), 267-275 (2009).
- Sanyal, A., Lajoie, B. R., Jain, G., Dekker, J. The long-range interaction landscape of gene promoters. Nature. 489 (7414), 109-113 (2012).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados