
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
جيل المحددة والمسوخ Markerless في أنواع السيانوبكتيريا نموذج
In This Article
Summary
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Abstract
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Introduction
البكتيريا الزرقاء هي أحد الأسرة في اللغات تطويريا القديمة ومتنوعة من البكتيريا الموجودة في البيئة الطبيعية تقريبا على الأرض. في النظم الإيكولوجية البحرية أنها وفيرة للغاية ولعب دورا رئيسيا في العديد من الدورات المواد الغذائية، وهو ما يمثل ما يقرب من نصف تثبيت الكربون 1، والغالبية العظمى من تثبيت النيتروجين 2 ومئات الملايين من الأطنان من إنتاج النفط والغاز 3 في المحيطات سنويا. البلاستيدات الخضراء، العضية المسؤولة عن عملية التمثيل الضوئي في الطحالب والنباتات حقيقية النواة، من المرجح أن تطورت من cyanobacterium الذي كان محاطا الكائن المضيف 4. وقد ثبت أن البكتيريا الزرقاء الكائنات نموذج مفيدة لدراسة التمثيل الضوئي، نقل الإلكترون 5 والمسارات البيوكيميائية، وكثير منها يتم حفظها في النباتات. وبالإضافة إلى ذلك البكتيريا الزرقاء تستخدم بشكل متزايد لإنتاج الغذاء والوقود الحيوي 6، 7 الكهرباء والصناعية مركبات 8، بسبب مرحبا بهمتحويل كفاءة ghly من الماء وثاني أكسيد الكربون 2 إلى الكتلة الحيوية باستخدام الطاقة الشمسية 9. العديد من الأنواع يمكن زراعتها في الأراضي غير الصالحة للزراعة مع الحد الأدنى من المواد الغذائية ومياه البحر، مما يوحي بأن البكتيريا الزرقاء يحتمل زراعتها في نطاق واسع دون أن يؤثر ذلك على الإنتاج الزراعي. بعض الأنواع هي أيضا مصادر للمنتجات الطبيعية، بما في ذلك فطريات، مضاد للجراثيم ومضاد للسرطان المركبات 10،11.
القدرة على توليد المسوخ هي المفتاح لفهم السيانوبكتيريا الضوئي، والكيمياء الحيوية وعلم وظائف الأعضاء، وضرورية لتطوير سلالات للأغراض الصناعية. معظم الدراسات التي نشرت توليد سلالات معدلة وراثيا عن طريق إدخال الكاسيت المقاومة للمضادات الحيوية في الموقع من الفائدة. وهذا يحد من عدد من الطفرات التي يمكن إدخالها إلى سلالة، كما ليست سوى عدد قليل من الأشرطة المقاومة للمضادات الحيوية المتاحة للاستخدام في البكتيريا الزرقاء. سلالات تحتوي على جينات تمنح إعادة المضادات الحيويةمقاومة لأ لا يمكن استخدامها للإنتاج الصناعي في البرك المفتوحة، والتي من المرجح أن تكون وسيلة فعالة من حيث التكلفة الوحيدة لإنتاج الوقود الحيوي وغيرها من المنتجات ذات القيمة المنخفضة 12. جيل من المسوخ تحمل علامات يتغلب على هذه القيود. المسوخ تحمل علامات لا تحتوي على الحمض النووي الأجنبية، إلا إذا تضمن عمدا، ويمكن التلاعب بها عدة مرات. ولذلك فمن الممكن توليد العديد من التعديلات في سلالة كما تريد. وبالإضافة إلى ذلك، والآثار القطبية على الجينات المصب للموقع تعديل يمكن أن يكون الحد الأدنى، مما يسمح التعديل أكثر دقة للكائن الحي (13).
لتوليد سلالات متحولة، البلازميدات الانتحارية التي تحتوي على اثنين شظايا الحمض النووي متطابقة إلى مناطق في الكروموسوم السيانوبكتيريا المرافقة الجين المراد حذفه (وهو ما يسمى 5 'و 3' المناطق المحيطة) هي التي شيدت أولا. ثم يتم إدراج اثنين من الجينات بين هذه المناطق المحيطة. واحدة من هذه يشفر بروتين المقاومة للمضادات الحيوية. ثاني بترميز هيئة تنسيق المعونة، التي همزuces ليفانسوكراز، وهو مركب يمنح الحساسية للالسكروز. في المرحلة الأولى من العملية، المسوخ ملحوظ، سلالات أي تحتوي على بعض الحمض النووي الأجنبية، يتم إنشاء. يتم خلط بناء البلازميد مع الخلايا السيانوبكتيريا وأخذ الحمض النووي بشكل طبيعي من قبل الكائن الحي. ويتم اختيار Transformants النمو على لوحات أجار التي تحتوي على المضادات الحيوية المناسبة وراثى المسخ التحقق من PCR. البلازميدات الانتحارية لا يمكن نسخ ضمن سلالة من الفائدة. لذا فإن أي مستعمرات المقاومة للمضادات الحيوية سوف تنجم عن حدث إعادة التركيب حيث الجينات في المصالح في إدراجها في كروموسوم. لتوليد المسوخ لا تحمل علامات مميزة، متحولة ملحوظ ثم يتم خلط مع البلازميد انتحاري ثان يحتوي فقط على المناطق المحيطة 5 "و 3". ومع ذلك، إذا كان مطلوبا إدخال دنا غريب، البلازميد التي تتكون من 5 'و 3' المرافقة المناطق مع الكاسيت التي تحتوي على الجينات التي تهم تدرج بين هذه الشظايا الحمض النووي، ويمكن استخدامها. سيليction هو عن طريق النمو على لوحات أجار التي تحتوي على السكروز. كما السكروز قاتلة للخلايا عندما يتم التعبير عن الجين المنتج هيئة تنسيق المعونة، والخلايا الوحيدة التي البقاء على قيد الحياة هي تلك التي وقوع حدث إعادة التركيب الثاني، حيث الجين حساسية السكروز، بالإضافة إلى الجينات المقاومة للمضادات الحيوية، وقد معاد للخروج من كروموسوم وعلى البلازميد. ونتيجة لتبادل recombinational، يتم إدراج المناطق المحيطة وأي الحمض النووي بينهما في كروموسوم.
لقد استخدمت بنجاح هذه الأساليب لتوليد الطفرات الكروموسومية متعددة في نفس السلالة من Synechocystis ليرة سورية. PCC6803 (يشار إليها فيما Synechocystis) 13،14، ليعرض الطفرات نقطة واحدة في الجين 13 و للتعبير عن أشرطة الجينات. بينما الجيل بالضربة القاضية تحمل علامات وقد تجلى مسبق لعملنا في Synechocystis 15،16، وهي طريقة مفصلة، وساعد علىعرض مرئي من الخطوات الحاسمة، ليست متاحة للجمهور. لقد طبقنا أيضا نفس الأسلوب لتوليد بالضربة القاضية ملحوظة في cyanobacterium آخر نموذج، متعاقبات حبيبية ليرة سورية. PCC7002 (يشار إليها فيما متعاقبات حبيبية). يوفر هذا البروتوكول طريقة بسيطة واضحة لتوليد المسوخ وبروتوكول السريع للتحقق من صحة وتخزين هذه السلالات.
Protocol
1. إعداد وسائل الإعلام الثقافة
- إعداد BG11 المتوسطة وفقا لCastenholz، 1988 17.
- إعداد الحلول الأسهم من BG11 100X، العناصر النزرة والأسهم الحديد (الجدول 1).
- إعداد الحلول منفصلة من الأسهم الفوسفات، نا 2 CO 3 الأسهم، N - [تريس (hydroxymethyl) الميثيل] حامض -2-aminoethanesulfonic (TES) العازلة وNaHCO 3 (الجدول 1).
- الأوتوكلاف الفوسفات والصوديوم 2 الأسهم CO 3. تصفية تعقيم TES العازلة وNaHCO 3 مع 0.2 ميكرون مرشحات.
- إعداد BG11 من خلال الجمع بين 976 مل من الماء، 10 مل من 100X BG11، 1 مل من العناصر النزرة و1 مل من الأسهم الحديد وتعقيم الحل. بعد هذا الحل قد برد إلى درجة حرارة الغرفة، إضافة 1 مل من الأسهم الفوسفات، 1 مل من نا 2 CO الأسهم 3 و 10 مل من NaHCO 3.
- لBG11 المتوسطة الصلبة، إضافة 15 غراما من أجار و 700 مل من الماء لرابطة العمل المنصف واحدةكورونا. إلى القارورة الثانية، إضافة 3 غرام من نا 2 S 2 O 3، 226 مل من الماء، 10 مل من 100X BG11، 1 مل من العناصر النزرة و1 مل من الأسهم الحديد. الأوتوكلاف كلا الحلول. بعد هذه الحلول التي يتم تبريده الى درجة حرارة الغرفة، والجمع بينها وإضافة 1 مل من الأسهم الفوسفات، 1 مل من نا 2 CO الأسهم 3، 10 مل من العازلة TES، و 10 مل من NaHCO 3.
يتم إعداد الحلول على حدة لتجنب ترسب بعض الأملاح: مذكرة.
- لاختيار على السكروز، وإعداد 50٪ (ث / ت) محلول السكروز. فلتر تعقيم الحل مع 0.2 ميكرون الفلاتر وإضافة إلى BG11 (100 مل من 50٪ سكروز إلى 900 مل من BG11) لإنتاج BG11 / 5٪ لوحات السكروز.
ملاحظة: لا تقم بإضافة NaHCO 3 إلى BG11 / 5٪ لوحات السكروز أجار. إضافة نا 2 CO 3 كالمعتاد. - للزراعة من متعاقبات حبيبية إضافة 10 مل من 1 M 4- (2-هيدروكسي) بيبيرازين-1-ethanesulfonic حامض، N - (2-هيدروكسي) piperazine- NR42؛ - (حمض 2-ethanesulfonic) (HEPES) و 1 مل من فيتامين ب 12 (الجدول 1) إلى 1 لتر من المتوسط BG11.
ملاحظة: التحول من سلالات مثقف في وسائل الإعلام BG11 متوفرة تجاريا بشكل ملحوظ أقل كفاءة مما كانت عليه في وصفات وسائل الإعلام BG11 الموصوفة هنا، وبالتالي لا ينصح.
2. نمو السيانوبكتيريا سلالات
- ثقافة سلالات في 100 مل قوارير المخروطية مع الحد الأقصى لحجم 50 مل ويهز في 120 دورة في الدقيقة. ختم لوحات BG11 مع بارافيلم وثقب ثلاثة ثقوب صغيرة في جانب من لوحة لتتيح تبادل الغاز. احتضان جميع سلالات عند 30 درجة مئوية تحت المصابيح الفلورية في مفاعل حيوي ضوئي في شدة الضوء بين 20-40 مكرومول الفوتونات م -2 ثانية -1.
- استخدام أفضل التقنيات معقمة. التعامل مع جميع سلالات السيانوبكتيريا في غطاء تدفق الصفحي.
ملاحظة: هذا مهم بشكل خاص عند سلالات يتم تربيتها مع وسائل الإعلام التي تحتوي على السكروز، والتي يمكن بسهولة كونتامانATED.
3. الجيل البلازميد التركيبات
- مجموعات تصميم الاشعال، بما في ذلك مواقع الانزيم تقييد المطلوبة، وذلك باستخدام التمهيدي تصميم البرمجيات مثل Primer3 (http://frodo.wi.mit.edu/primer3/)، لتضخيم اثنين ~ 1 مناطق كيلوبايت 5 "و 3" لل الجينات في المصالح. الرجوع إلى تسلسل جينوم الأنواع السيانوبكتيريا عبر Cyanobase (http://genome.kazusa.or.jp/cyanobase). انظر الجدول 2 لجميع البادئات المستخدمة هنا. عند تصميم الاشعال النظر في العوامل التالية:
- تأكد من أن مناطق تضخيم تشمل 5 مناطق "و 3" من الجينات التي سيتم تحور، مثل الشكل 1.
- لا يتحور المناطق بين الجينات لتجنب طفرة غير مقصودة من العقاقير والرنا غير الترميز. لجيل من المسوخ في Synechocystis، الرجوع إلى قائمة المواقع بداية النسخي موثقة في Mitschke وآخرون، 2011 18، وذلك لتجنب طفرة من العقاقيرأو غير الترميز الرنا.
- عند اختيار المناطق المحيطة لا تشمل إطار القراءة مفتوحة بالكامل من الجينات المجاورة كتعبير عن هذه الجينات في كولاي قد تتداخل مع الاستنساخ.
- تضخيم المنتجات PCR باستخدام عالية الدقة الحمض النووي بوليميريز وفقا لتعليمات الشركة الصانعة.
ملاحظة: في تجربتنا هذا الانزيم تنتج بعض الأخطاء.- إعداد 50 ميكرولتر تفاعلات PCR يحتوي عازلة HF وإما 0، 1.5 أو 3 ميكرولتر من DMSO. استخدام 100 نانوغرام من الحمض النووي الجيني في رد الفعل. استخدام برنامج يتألف من خطوة تمسخ الأولية من 98 درجة مئوية لمدة 30 ثانية، 35 طلقة من 98 درجة مئوية لمدة 10 ثانية، 67 درجة مئوية لمدة 30 ثانية، 72 درجة مئوية لمدة 30 ثانية، تليها خطوة التمديد النهائي من 72 درجة مئوية لمدة 5 دقائق. هذا عادة ما يعطي المنتجات متسقة.
- تحقق من المنتجات PCR وعينات يتفاعل مع انزيمات نوكلياز داخلية لحجم الصحيح عن طريق الكهربائي للهلام. تشغيل 1٪ (ث / ت) والمواد الهلامية الاغاروز تحتوي على 0.02٪(ت / ت) بروميد إيثيديوم لمدة 45 دقيقة في 100 V.
تنبيه: إيثيديوم بروميد هو المغير المحتملين، وينبغي التعامل مع الحماية الملائمة. - منتجات تنقية PCR باستخدام طقم تنقية الحمض النووي وفقا لتعليمات الشركة الصانعة. أيضا استخدام هذه المجموعة لتنقية شظايا البلازميد، بما في ذلك قطع قطع من المواد الهلامية الاغاروز. أزل الحمض النووي المنقى في 14 ميكرولتر من الماء.
- للحصول على خطوات الاستنساخ، واحتضان خليط من رد فعل تقييد نوكلياز داخلية عند 37 درجة مئوية لمدة> 1 ساعة في الحجم الكلي لل30 ميكرولتر وفقا لتعليمات الشركة الصانعة.
- للحصول على خطوات الربط، شظايا الحمض النووي ligate في درجة حرارة الغرفة ل> 1 ساعة في الحجم الكلي لل20 ميكرولتر، تحتوي على 5 ميكرولتر من البلازميد هضمها تنقيته، 12 ميكرولتر من إدراج هضمها المنقى، 2 ميكرولتر من العازلة و 1 ميكرولتر من يغاز.
- إعداد الخلايا المحولة كولاي DH5α وفقا لطريقة التالي.
- تنمو E. بين عشية وضحاها القولونية الثقافة في 10 مل لوريا Bertani (LB) وسائل الاعلام.
- تطعيم 400 مل LB في قارورة مخروطي 1 لتر تحتوي على 6 مل 1 M MgCl 2 (الجدول 1) مع 1 مل من الثقافة بين عشية وضحاها.
- تنمو ثقافة في 37 درجة مئوية في 220 دورة في الدقيقة لحوالي 4 ساعات أو حتى OD ل 600Nm يصل 0،4-0،6.
- وضع الخلايا على الجليد لمدة 1 ساعة.
- أجهزة الطرد المركزي في 2800 x ج لمدة 10 دقيقة لتكوير الخلايا في 4 درجات مئوية.
- إزالة وطاف resuspend في 160 مل من محلول ألف (الجدول 1)، واحتضان على الجليد لمدة 20 دقيقة.
- أجهزة الطرد المركزي في 2800 x ج لمدة 10 دقيقة لتكوير الخلايا في 4 درجات مئوية.
- إزالة وطاف resuspend في 4 مل من محلول A + الجلسرين (الجدول 1).
- تجهيز 50 مكل، وتجميد في السائل N 2، مخزن في -80 درجة مئوية.
- مزيج 5 ميكرولتر من خليط الربط مع 50 ميكرولتر من الخلايا المختصة واحتضان لمدة 1 ساعة على الجليد.
- حرارة صدمة الخلايا في درجة حرارة 42 درجة مئوية لمدة 90 ثانية، واتباعهتزوجا قبل الحضانة على الجليد لمدة 2 دقيقة.
- إضافة 950 ميكرولتر من وسائل الاعلام LB (الجدول 1)، واحتضان عند 37 درجة مئوية لمدة 1 ساعة.
- قسامة 50 و 200 ميكرولتر على لوحات مع المضادات الحيوية المناسبة، إما الأمبيسلين (100 ميكروغرام / مل) و / أو كانامايسين (30 ميكروغرام / مل).
تنبيه: كل كانامايسين والأمبيسلين سامة، وينبغي التعامل مع الحماية الملائمة. - اختيار واحتضان المستعمرات واحد في 2 مل الإعلام LB تلقيح مع المضادات الحيوية المناسبة.
- تنقية جميع البلازميدات استخدام عدة تنقية miniprep البلازميد وفقا لتعليمات الشركة الصانعة.
- توليد البلازميدات، في هذا المثال معينة لضرب الجينات cpcC1C2، وفقا للخطوات التالية.
- تضخيم المنطقة المحيطة 1012 بي بي 5 "(جزء الأيسر) باستخدام بادئات cpcC1C2leftfor وcpcC1C2leftrev (انظر الخطوة 3.2، الجدول 2). إزالة كمية صغيرة من رد فعل PCR وتأكيد ما إذا كانتم تضخيمه الصحيح حجم المنتج عن طريق الكهربائي للهلام (الخطوة 3.3). هضم هذه القطعة وpUC19 مع Xba الأول وبام مرحبا (الخطوة 3.5).
- تنقية كل من الاستعدادات (الخطوة 3.4)، ligate (الخطوة 3.6)، وتحويل (الخطوة 3.7) وتشكيل أربع ل2 مل LB الثقافات السائل مع الأمبيسلين (100 ميكروغرام / مل) من مستعمرات منفصلة لتنقية البلازميد عبر minipreps (الخطوة 3.8).
- تحقق من إدخال جزء في pUC19 عبر Xba I / بام مرحبا الهضم والكهربائي للهلام (الخطوة 3.3). عصابات من 2660 سنة مضت و1،012 سنة مضت وتشير مقدمة الصحيح للإدراج في البلازميد.
- تضخيم المنطقة المحيطة 1016 بي بي 3 "(جزء الأيمن) باستخدام بادئات cpcC1C2rightfor وcpcC1C2rightrev (انظر الخطوة 3.2، الجدول 2). إزالة كمية صغيرة من رد فعل PCR وتأكيد ما إذا كان حجم المنتج الصحيح تم تضخيمه عبر الكهربائي للهلام (الخطوة 3.3). هضم هذه القطعة وpUC19 مع الحويصلة الأول وايكو RI (شارعالجيش الشعبي 3.5).
- تنقية كل من الاستعدادات (الخطوة 3.4)، ligate (الخطوة 3.6)، وتحويل (الخطوة 3.7) وتشكيل أربع ل2 مل LB الثقافات السائل مع الأمبيسلين (100 ميكروغرام / مل) من مستعمرات منفصلة لتنقية البلازميد عبر minipreps (الخطوة 3.8).
- تحقق من إدخال جزء في pUC19 عبر الحويصلة I / ايكو RI الهضم (الخطوة 3.5) والكهربائي للهلام (الخطوة 3.3). عصابات من 2660 سنة مضت و1،016 سنة مضت وتشير مقدمة الصحيح للإدراج في البلازميد.
ملاحظة: المواقع Xba I / بام مرحبا لاستنساخ 5 'المنطقة والحويصلة I / ايكو RI لاستنساخ 3' المنطقة إلى pUC19 تستخدم أينما. إذا كان ذلك ممكنا، وتشمل دائما موقع بام مرحبا على التمهيدي العكسي ل 'المنطقة أو التمهيدي إلى الأمام ل3' 5 المنطقة للتأكد من أن خطوات الاستنساخ في وقت لاحق من الأسهل القيام بها. - تسلسل كلا إدراج لتحديد ما إذا كان التسلسل الصحيح باستخدام بادئات تمتد الموقع الإدراج، على سبيل المثال M13 إلى الأمام وM13 العكس (الجدول 2). يجب أن يكون التسلسل الصحيح لضمان تقديم أية أخطاء في المناطق المحيطة.
- استئصال جزء الأيسر من pUC19 عبر Xba I / بام مرحبا الهضم. هضم pUC19 + جزء الصحيح مع Xba I / بام مرحبا (الخطوة 3.5).
- تنقية 1،012 سنة مضت ترك جزء و3،676 سنة مضت pUC19 + جزء الأيمن من agarose هلام (الخطوة 3.3) عن طريق الاستئصال من الحمض النووي باستخدام شفرة المشرط.
- تنقية كل من الاستعدادات (الخطوة 3.4)، ligate (الخطوة 3.6)، وتحويل (الخطوة 3.7) وتشكيل أربع ل2 مل LB الثقافات السائل مع الأمبيسلين (100 ميكروغرام / مل) من مستعمرات منفصلة لتنقية البلازميد عبر minipreps (الخطوة 3.8).
- تحقق من إدخال جزء في pUC19 + جزء الصحيح عبر Xba I / بام مرحبا الهضم (الخطوة 3.5) والكهربائي للهلام (الخطوة 3.3). عصابات من 3676 سنة مضت و1،012 سنة مضت تشير الإدراج الصحيح للإدراج في البلازميد (أشير إلى هذا النحو البلازميد B).
- المكوس وNPT1 / هيئة تنسيق المعونة كاسيت من pUM24cm 19 عبر بام مرحبا الهضم. هضم البلازميد B مع بام مرحبا (الخطوة 3.5).
ملاحظة: ليس لدى NPT1 / هيئة تنسيق المعونة الكاسيت إلى أن تنقيته من المواد الهلامية الاغاروز منذ pUM24cm يشفر بروتين يمنح مقاومة الكلورامفينيكول. لذلك إذا نمت المستعمرات على LB / الأمبيسلين / لوحات أجار كانامايسين مزيج الوحيد الممكن من شأنها أن تؤدي إلى المستعمرات المقاومة هي إدماج NPT1 / كاسيت هيئة تنسيق المعونة في البلازميد B. - تنقية كل من الاستعدادات (الخطوة 3.4)، ligate (الخطوة 3.6)، وتحويل (الخطوة 3.7) وأقامت أربعة 2 مل LB الثقافات السائل مع الأمبيسلين (100 ميكروغرام / مل)، وكانامايسين (30 ميكروغرام / مل) من مستعمرات منفصلة لتنقية البلازميد عبر minipreps (الخطوة 3.8).
- تحقق لادراج NPT1 / كاسيت هيئة تنسيق المعونة في البلازميد B عبر بام مرحبا الهضم (الخطوة 3.5) والكهربائي للهلام (الخطوة 3.3). عصابات من 4688 سنة مضت و3،894 سنة مضت تشير الإدراج الصحيح من الالبريد تضاف إلى البلازميد (أشير إلى هذا النحو البلازميد A).
- بدلا من ذلك، نهاية حادة وNPT1 / كاسيت هيئة تنسيق المعونة واستنساخ في موقع مختلف تقييد نوكلياز داخلية بين شظايا اليسار واليمين في البلازميد B. يجب المستنسخة وNPT1 / كاسيت هيئة تنسيق المعونة بين اليسار وشظايا الصحيحة.
ملاحظة: إذا كانت هناك حاجة تعبير عن كاسيت الخارجية ثم وهذا ينبغي أن تدرج بين شظايا اليسار واليمين البلازميد B. ثم يتم استخدام هذا البلازميد في الخطوات خروج المغلوب لا تحمل علامات.
4. توليد ملحوظ Synechocystis ومتعاقبات حبيبية المسوخ
- إنشاء ثقافة جديدة، فحقن حلقة كاملة من الخلايا في 30-50 مل من المتوسط BG11. تنمو ثقافة لمدة 2-3 أيام إلى 750nm OD = 0،2-0،6.
ملاحظة: عادة المستعمرات الفردية هي صغيرة جدا لاستخدامها في التلقيح وتعرض الخلايا الفردية حتى لمستويات منخفضة من الضوء سوف يؤدي إلى photoinhibition واختيارلطفرات مقاومة الخفيفة. - الطرد المركزي 1-2 مل من ثقافة في 2300 x ج لمدة 5 دقائق والتخلص من طاف. لا أجهزة الطرد المركزي أي الثقافات السيانوبكتيريا في> 2300 x ج لأن ذلك قد يؤدي إلى تلف الخلايا. يغسل بيليه مرة واحدة مع متوسط BG11.
ملاحظة: لا و resuspend الخلايا قبل vortexing لأن هذا قد يؤدي إلى فقدان الشعرة التي تعتبر ضرورية لامتصاص الحمض النووي. و resuspend الخلايا طيف من قبل pipetting. - إضافة المتوسطة BG11 إلى الحجم النهائي من 100 ميكرولتر. نقل الخلايا إلى 14 مل أنبوب جولة القاع.
- إضافة 1 ميكروغرام البلازميد ألف من الخلايا ومزيج من خلال استغلال طيف. إضافة <10 ميكرولتر من البلازميد.
ملاحظة: يفضل أن يكون البلازميد بتركيز> 100 نانوغرام / ميكرولتر ولكن بتركيزات أقل من ذلك هي كافية للتحول الناجح. - وضع أنابيب أسفل أفقيا في الحاضنة. احتضان الثقافات لمدة 4-6 ساعة.
ملاحظة: خلايا يمكن أن تكون مختلطة لفترة وجيزة من خلال استغلال كل 1-2 ساعة ولكن هذا ليس ضروريا. عينات يمكن وضعها فيهز حاضنة وإن كان هذا لا يحسن إلى حد كبير كفاءة. - نشر aliquots من الخليط الحمض النووي خلية ثقافة / البلازميد على لوحات BG11 أجار دون المضادات الحيوية. وعادة ما ينتشر 20 ميكرولتر و 80 مكل على لوحات منفصلة.
- ~ بعد 24 ساعة، إضافة 2.5-3 مل من 0.6٪ حل أجار في المياه التي تحتوي على كانامايسين (لكل 20 مل: 0.12 غرام من أجار، و 100 ميكرولتر من 100 ملغ / مل الكانامايسين) على لوحة أجار. تبريد هذا الحل إلى ~ 42 درجة مئوية، وإضافة إلى حافة صفيحة أجار. إمالة لوحة بحيث يشكل حلا وحتى طبقة "كبار أجار" على السطح.
- احتضان لوحات أجار لفترة أخرى من الزمن. وينبغي أن تكون مستعمرات مرئية بعد حوالي 7 أيام.
ملاحظة: لوحات أجار يمكن أن تكون مكدسة 3 عالية في حاضنة. وعادة ما يتم الحصول على مئات من المستعمرات في التحول. - خط المستعمرات الفردية على BG11 + كانامايسين (30 ميكروغرام / مل) أجار لوحات. تقسيم لوحة أجار في 6 قطاعات واستخدام المسواك نهاية حادة إلى خط خارجالمستعمرات على كل قطاع على حدة. الحصول على المستعمرات واحد ليس مهما، والنمو العادل للtransformants.
- تأكيد خروج المغلوب ملحوظا من قبل PCR باستخدام البلمرة طق الحمض النووي وفقا لتعليمات الشركة الصانعة. إضافة 2 ميكرولتر من MgCl 2 (25 ملم) في رد الفعل.
- إزالة نسبة صغيرة من الخلايا ونقل في أنبوب يحتوي على 50 ميكرولتر المياه و~ 20 425-600 ميكرون الخرز الزجاجي. هزة في هزاز لمدة 5 دقائق في ~ 2000 دورة في الدقيقة. أجهزة الطرد المركزي في 15700 x ج لمدة 5 دقائق واستخدام 5 ميكرولتر من طاف في 50 ميكرولتر تفاعل PCR.
ملاحظة: لا resuspend والحل. حطام خلية يحتاج الى البقاء في الجزء السفلي من الأنبوب.
- إزالة نسبة صغيرة من الخلايا ونقل في أنبوب يحتوي على 50 ميكرولتر المياه و~ 20 425-600 ميكرون الخرز الزجاجي. هزة في هزاز لمدة 5 دقائق في ~ 2000 دورة في الدقيقة. أجهزة الطرد المركزي في 15700 x ج لمدة 5 دقائق واستخدام 5 ميكرولتر من طاف في 50 ميكرولتر تفاعل PCR.
- تحقق من المسوخ
- الاشعال التصميم الذي تمتد هذه المنطقة بالضربة القاضية باستخدام برامج التصميم التمهيدي (مثل Primer3). الاشعال تصميم ابتداء من الساعة ~ 200 شركة بريتيش بتروليوم جانبي المنطقة بالضربة القاضية.
وترد الاشعال للتحقق من المسخ cpcC1C2 في الجدول 2: مذكرةووصف cpcC1C2for وcpcC1C2rev. - تضخيم المنتجات باستخدام برنامج يتألف من خطوة تمسخ الأولية من 95 درجة مئوية لمدة 2 دقيقة، 35 طلقة من 95 درجة مئوية لمدة 1 دقيقة، 60 درجة مئوية لمدة 1 دقيقة، 72 درجة مئوية لمدة 1 دقيقة لكل كيلو من تسلسل، تليها خطوة التمديد النهائي من 72 درجة مئوية لمدة 5 دقائق. تشمل عنصر تحكم من النوع البري. هذا عادة ما يعطي المنتجات متسقة.
- تحقق من النمط الجيني عن طريق الكهربائي للهلام. سوف transformants خروج المغلوب ملحوظ تظهر عصابة من ~ 4 كيلوبايت (0.2 كيلوبايت من كل من اليسار واليمين شظايا بالإضافة إلى NPT1 / كاسيت هيئة تنسيق المعونة) وعدم وجود الفرقة البرية من نوع (الشكل 2).
ملاحظة: في بعض الحالات ولم يلاحظ الفرقة كيلوبايت ~ 4 في متحولة ملحوظ نظرا لحجم كبير من هذا المنتج PCR. ومع ذلك، إذا فرقة الموافق الحجم المتوقع من النوع البري ليست لاحظ ثم عادة هذه السلالة هو خروج المغلوب ملحوظا.
- الاشعال التصميم الذي تمتد هذه المنطقة بالضربة القاضية باستخدام برامج التصميم التمهيدي (مثل Primer3). الاشعال تصميم ابتداء من الساعة ~ 200 شركة بريتيش بتروليوم جانبي المنطقة بالضربة القاضية.
- إذا فرقة من النوع البري لا تزال موجودة ثم إعادة خط الضغط علىBG11 طازج + كانامايسين (30 ميكروغرام / مل) لوحة أجار وتكرار PCR. تكرار عملية تسليط الضوء على إعادة حتى متحولة هو الفصل بين الجنسين بحيث يتم احترام أي فرقة من النوع البري في رد فعل PCR.
ملاحظة: زيادة كمية كانامايسين إلى تركيز 50 ميكروغرام / مل، ثم 100 ميكروغرام / مل ضروري في بعض الأحيان من أجل التفريق بين المسخ ملحوظ تماما. - إذا أظهر سلالة متحولة لمحة ملحوظ عن طريق PCR، ثم إعادة متتالية على BG11 طازج + كانامايسين (30 ميكروغرام / مل) لوحة أجار. استخدام هذه السلالة لتوليد خروج المغلوب لا تحمل علامات.
ملاحظة: بروتوكول يمكن استخدامها لتوليد المسوخ التي تحمل مجرد كاسيت المقاومة للمضادات الحيوية أي عن طريق استبدال NPT1 / كاسيت هيئة تنسيق المعونة فقط مع كاسيت NPT1 من pUC18K 20 بين اليسار واليمين شظايا.
5. الجيل التي لا تحمل علامات Synechocystis المسوخ
- إنشاء ثقافة جديدة من دور الستة عشر تميزت بتلقيح حلقة كاملة من جملتعلمي اللغة اإلنكليزية إلى 30-50 مل من المتوسط BG11. تنمو ثقافة لمدة 2-3 أيام إلى 750nm OD = 0،2-0،6.
- الطرد المركزي 10 مل من ثقافة في 2300 x ج لمدة 5 دقائق والتخلص من طاف. يغسل مرة واحدة مع متوسط BG11.
ملاحظة: لا و resuspend الخلايا قبل vortexing لأن هذا قد يؤدي إلى فقدان الشعرة التي تعتبر ضرورية لامتصاص الحمض النووي. و resuspend الخلايا طيف من قبل pipetting. - إضافة BG11 إلى الحجم النهائي من 200 ميكرولتر. نقل الخلايا إلى 14 مل أنبوب جولة القاع.
- إضافة 1 ميكروغرام من البلازميد B إلى الخلايا ومزيج من خلال استغلال طيف.
- احتضان هذه العينات لمدة 4-6 ساعة. وضع أنابيب أسفل أفقيا.
ملاحظة: خلايا يمكن أن تكون مختلطة لفترة وجيزة من خلال استغلال كل 1-2 ساعة ولكن هذا ليس ضروريا. عينات يمكن وضعها في حاضنة تهتز على الرغم من أن هذا لا يحسن كفاءة. - إضافة 1.8 مل من المتوسط BG11 واحتضان عينات ليصبح المجموع 4 أيام مع اهتزاز. وهذا هو ما يكفي من الوقت للسماح إعادة التركيب للتحدث في نسخ الكروموسومات متعددة.
- قسامات لوحة من خليط التحول على BG11 / 5٪ لوحات السكروز أجار. لوحة 50 ميكرولتر، 10 ميكرولتر و 1 ميكرولتر في لوحة أجار. في حالة ظهور العشب مستعمرة على كل هذه وحات أجار تمييع الحل أبعد من ذلك وقسامة على لوحات جديدة. وينبغي أن تكون مستعمرات مرئية بعد حوالي 7 أيام.
- التصحيح 30-50 المستعمرات الفردية على BG11 + كانامايسين (30 ميكروغرام / مل) أجار لوحات أولا وBG11 / 5٪ لوحات السكروز أجار الثانية، وذلك باستخدام مسواك نهاية حادة. أي البكتيريا التي تنمو على BG11 / 5٪ لوحات السكروز ولكن لوحات لا BG11 + كانامايسين وبالضربة القاضية تحمل علامات محتملة. ومن المرجح أن تكون مقاومة السكروز البكتيريا تنمو على حد سواء لوحات بسبب طفرة في جين هيئة تنسيق المعونة.
- تحقق بالضربة القاضية تحمل علامات باستخدام نفس الاشعال والأسلوب كما تم استخدامها للتحقق من بالضربة القاضية ملحوظة. على سبيل المثال cpcC1C2for وcpcC1C2rev (الجدول 2) للتحقق من خروج المغلوب لا تحمل علامات cpcC1C2. ومن شأن خروج المغلوب لا تحمل علامات تدل على الفرقة على الاغاروز ر جل المقابلةس البرية من نوع حجم ناقص المنطقة حذف (الشكل 2).
- إذا أظهر سلالة ملف تعريف تحمل علامات متحولة عن طريق PCR (خطوة 4.11.2) والكهربائي للهلام (الشكل 2)، ثم إعادة متتالية على لوحة BG11 أجار جديدة بدون المضادات الحيوية.
6. التخزين على المدى الطويل من سلالات
- إنشاء ثقافة جديدة من سلالة، فحقن حلقة كاملة من الخلايا في 30-50 مل من المتوسط BG11. تنمو ثقافة لمدة 3-4 أيام إلى 750nm OD = 0،4-0،7.
- غسل خلايا مرة واحدة مع BG11 وresuspend في ~ 2 مل من BG11.
- إضافة 0.8 مل من الخلايا تتركز في أنبوب واحد. ثم يضاف 0.2 مل من 80٪ تصفية تعقيمها الجلسرين.
- اختياري: إضافة 0.93 مل من الخلايا المركزة إلى أنبوب آخر. إضافة 0.07 مل من DMSO لهذا الأنبوب.
تنبيه: DMSO غير سامة، وينبغي التعامل معها الحماية المناسبة. - تخزين كل أنابيب في -80 درجة مئوية. لإحياء سلالات إزالة الأنبوب وكشط بعض الخلايا مع الأسنان حادةاختيار على لوحة آغار دون المضادات الحيوية. خط خارج كالمعتاد باستخدام حلقة عقيمة.

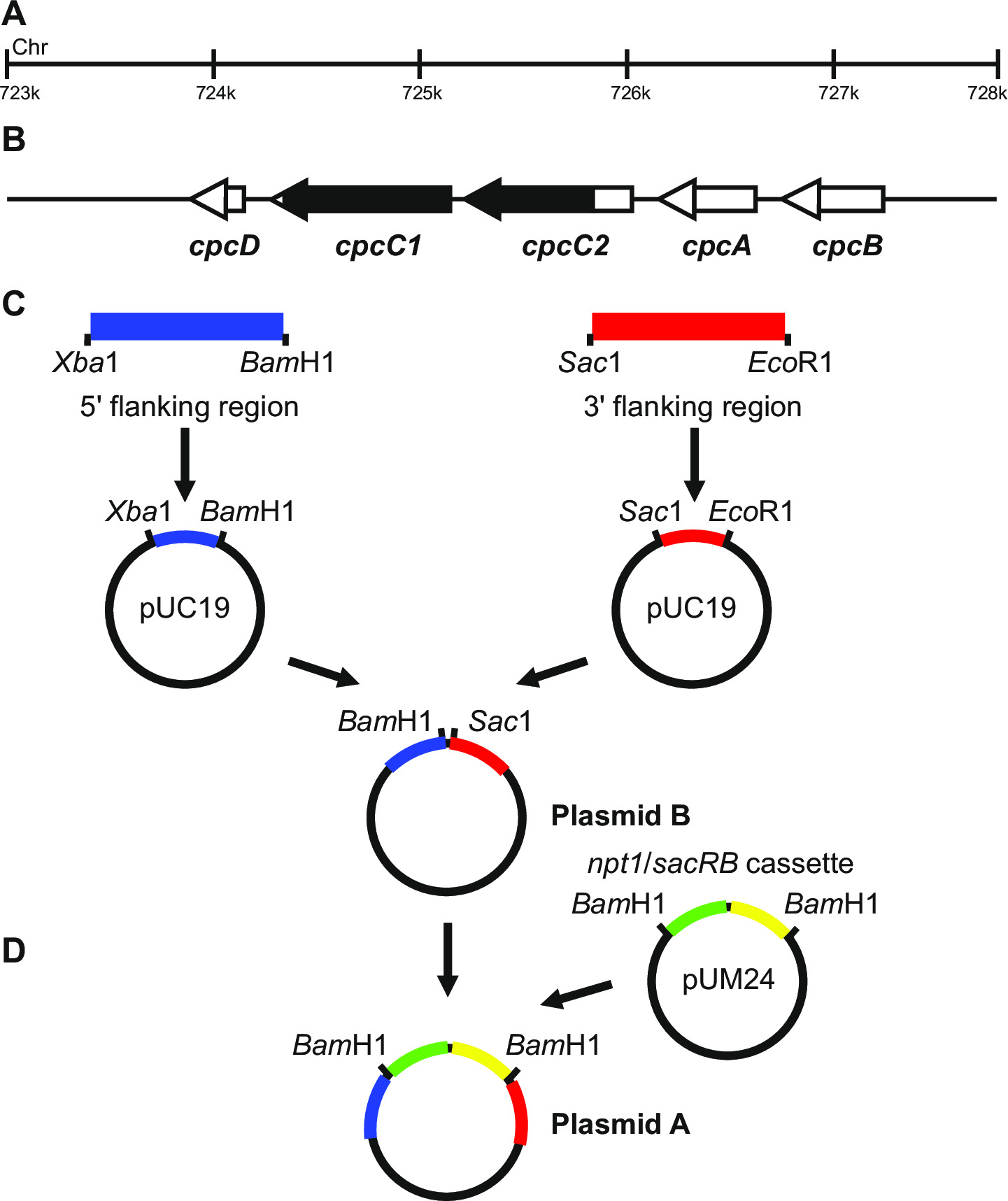
الشكل 1: بناء البلازميد لتوليد بالضربة القاضية ملحوظ وغير مميزة، على سبيل المثال cpcC1 وcpcC2 في Synechocystis (A) المنطقة من الجينوم Synechocystis حيث توجد (ب) cpcC1 وcpcC2 والجينات المجاورة. سلط الضوء باللون الأسود هي المنطقة من الجينوم المراد حذفه في متحولة. (C) مواقع من الجينوم التي يتم تضخيمها من قبل PCR. وتتضخم في "المنطقة المحيطة (المشار إليها باللون الأزرق) و 3 '5 المنطقة المحيطة (المشار إليها باللون الأحمر) مع تقييد مواقع نوكلياز داخلية للاستنساخ في pUC19. 5 '(أو 3') المرافقة المنطقة يتم استئصاله من pUC19 وإدراجها في pUC19 + 3 "(أو 59؛) المرافقة المنطقة البلازميد توليد بلازميد B. (D) يتم استئصاله NPT1 / كاسيت هيئة تنسيق المعونة من pUM24 عبر بام مرحبا الهضم وتدرج بين 5 'و 3' المرافقة المناطق لتوليد البلازميد A. الرجاء انقر هنا لعرض أكبر نسخة من هذا الرقم.
{kind=link}
النتائج
تصميم البلازميد هو أمر حاسم لجيل ناجح من كل من المسوخ ملحوظ وغير مميزة الشكل 1 يعطي مثالا البلازميد ألف وباء استخدامها لتوليد متحولة الحذف في الجينات Synechocystis cpcC1 وcpcC2 13. في كل حالة المناطق المحيطة 5 "و 3" ما يقرب من 900-1?...
Discussion
الخطوات الأكثر أهمية في جيل من المسوخ تحمل علامات مميزة هي: 1) تصميم البلازميد دقيق لضمان فقط يتم تبديل المنطقة المستهدفة. 2) التأكد من أن العينات لا تزال ممحوضة، وخصوصا عندما تربيتها على السكروز. 3) الطلاء تحول الخلايا لتوليد متحولة ملحوظ في البداية على لوحات أجار BG11 ت?...
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
ونحن ممتنون للخدمات البيئية رابطة التعليم الثقة، وعلم الأحياء الاصطناعية في كامبريدج صندوق SynBio وزارة العدل الاجتماعي والتمكين، حكومة الهند، للحصول على الدعم المالي.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis?. Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved