
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Erzeugung von Marked und Markerless Mutants in Modell Cyanobakterienarten
In diesem Artikel
Zusammenfassung
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Zusammenfassung
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Einleitung
Cyanobakterien sind eine evolutionär alten und unterschiedlichen Stamm der Bakterien in fast allen natürlichen Umwelt auf der Erde gefunden. In marinen Ökosystemen sind sie besonders reichlich vorhanden und spielen eine Schlüsselrolle in vielen Nährstoffkreisläufe spielen, für etwa die Hälfte der Kohlenstoff - Fixierung entfallen 1, die Mehrheit der Stickstofffixierung 2 und Hunderte von Millionen von Tonnen an Kohlenwasserstoffproduktion 3 in den Ozeanen jährlich. Chloroplasten, die Organell verantwortlich für die Photosynthese in eukaryotischen Algen und Pflanzen, sind wahrscheinlich von einem Cyanobakterium entwickelt zu haben , die vier von einem Wirtsorganismus verschlungen wurde. Cyanobakterien haben sich als nützlich erwiesen Modellorganismen für die Untersuchung der Photosynthese, Elektronentransport 5 und biochemischen Wege, von denen viele in Pflanzen konserviert sind. Darüber hinaus Cyanobakterien werden zunehmend für die Herstellung von Lebensmitteln verwendet werden, Biokraftstoffe 6, Strom 7 und Industrieverbindungen 8, die aufgrund ihrer halloghly effiziente Umwandlung von Wasser und CO 2 in Biomasse Solarenergie 9 verwenden. Viele Arten können auf Nicht-Ackerland mit minimalen Nährstoffe und Meerwasser gezüchtet werden, dass Cyanobakterien was darauf hindeutet, möglicherweise in großem Umfang angebaut werden könnten, ohne die landwirtschaftliche Produktion zu beeinträchtigen. Bestimmte Arten sind auch Quellen von natürlichen Produkten, einschließlich Antimykotika, antibakterielle und Anti-Krebs - Verbindungen 10,11.
Die Fähigkeit, Mutanten zu erzeugen, ist der Schlüssel zum Verständnis cyanobakteriellen Photosynthese, der Biochemie und Physiologie und essentiell für die Entwicklung von Stämmen für industrielle Zwecke. durch Einfügen eines Antibiotikaresistenzkassette in den Ort von Interesse, die Mehrheit der veröffentlichten Studien erzeugen genetisch Stämme modifiziert. Dies begrenzt die Anzahl von Mutationen, die in einen Stamm eingeführt werden kann, da nur wenige Antibiotikaresistenz Kassetten zur Verwendung in Cyanobakterien verfügbar sind. Die Stämme Gene, Antibiotika re enthält,Beständigkeit kann nicht für die industrielle Produktion in offenen Teichen verwendet werden, die die einzigen kosteneffektiven Mittel , um wahrscheinlich 12 Biokraftstoffe und andere geringwertige Produkte zu erzeugen. Die Erzeugung von nicht markierten Mutanten überwindet diese Einschränkungen. Nicht markierte Mutanten enthalten keine Fremd-DNA, sofern nicht absichtlich enthalten und kann mehrfach manipuliert werden. Daher ist es möglich, so viele Veränderungen in einem Stamm zu erzeugen, wie gewünscht. Zusätzlich nachgeschalteten polaren Effekte auf Gene der Modifikationsstelle minimiert werden, präziser Modifikation des Organismus 13 ermöglicht.
In den Mutantenstämmen, suicide Plasmiden, die zwei DNA-Fragmente identisch zu Regionen im cyanobakteriellen Chromosom flankieren das Gen deletiert werden (bezeichnet als die 5'- und 3'-flankierende Bereiche) zuerst konstruiert erzeugen. Zwei Gene werden dann zwischen diesen flankierenden Regionen eingeführt. Eines davon codiert eine antibiotische Resistenz-Protein; der zweite kodiert SacB, die produces Levansucrase, verleihen eine Verbindung Empfindlichkeit gegenüber Saccharose. In der ersten Stufe des Verfahrens, markierte Mutanten, enthält also Stämme einige fremde DNA, erzeugt. Das Plasmid-Konstrukt wird mit den Cyanobakterien-Zellen gemischt, und die DNA wird auf natürliche Weise durch den Organismus aufgenommen. Transformanten werden durch Wachstum auf Agar-Platten, die das entsprechende Antibiotikum und das mutierte Genotyp durch PCR verifiziert ausgewählt. Suicide Plasmide können nicht innerhalb der Stamm von Interesse replizieren. Daher irgendwelche Antibiotika-resistente Kolonien werden aus einem Rekombinationsereignis führen, wobei das interessierende Gen in das Chromosom eingefügt in. Zur Erzeugung nicht markierten Mutanten wird die markierte Mutante dann mit einem zweiten Selbstmord Plasmid gemischt, die nur die 5 'und 3' flankierenden Regionen. wenn Insertion von Fremd-DNA jedoch erforderlich ist, ein Plasmid, bestehend aus den 5'- und 3'-flankierenden Regionen mit einer Kassette, die die Gene von Interesse zwischen diesen DNA-Fragmente eingefügt enthalten, können verwendet werden. Selection ist über Wachstum auf Agar-Platten, die Saccharose. Wie Saccharose auf Zellen letal ist , wenn das sacB Gen - Produkt exprimiert wird, daß die nur Zellen überleben , sind jene , in denen ein zweites Rekombinationsereignis stattgefunden hat, wobei die Saccharose Empfindlichkeitsgens, zusätzlich zu dem Antibiotika - Resistenzgen, aus dem rekombinierten wurde Chromosom und auf dem Plasmid. Als Folge der rekombinatorischen Austausch, die flankierenden Regionen und jede DNA zwischen ihnen werden in das Chromosom inseriert.
Wir haben erfolgreich diese Methoden zu generieren mehrere chromosomale Mutationen in dem gleichen Stamm von Synechocystis sp. PCC6803 (nachfolgend als Synechocystis) 13,14, einzelne Punktmutationen in einem Gen von Interesse 13 und für die Expression von Genkassetten einzuführen. Während Generation von unmarkierten Vorprägungen hat vor unserer Arbeit in Synechocystis 15,16, eine detaillierte Methode nachgewiesen wurde, unterstützt durcheine visuelle Darstellung der kritischen Schritte, ist nicht öffentlich verfügbar. Wir haben galt auch die gleiche Methode zur Erzeugung von markierten Vorprägungen in einem anderen Modell Cyanobakterium, sp Synechococcus. PCC7002 (nachstehend als Synechococcus). Dieses Protokoll stellt eine klare, einfache Methode für Mutanten und ein schnelles Protokoll zur Validierung und Speicherung dieser Stämme zu erzeugen.
Protokoll
1. Herstellung von Kulturmedien
- Bereiten Sie BG11 Medium nach Castenholz, 1988 17.
- Stocklösungen von 100x BG11, Spurenelemente und Eisen Lager (Tabelle 1).
- Bereiten Sie separate Lösungen von Phosphat Lager, Na 2 CO 3 Lager, N - [Tris (hydroxymethyl) methyl] -2-aminoethansulfonsäure (TES) Puffer und NaHCO 3 (Tabelle 1).
- Autoklave mit Phosphat und Na 2 CO 3 Aktien. Filter-sterilisieren TES - Puffer und NaHCO 3 mit 0,2 & mgr; m - Filter.
- Bereiten Sie BG11 durch die Kombination von 976 ml Wasser, 10 ml 100x BG11, 1 ml Spurenelemente und 1 ml Eisen Lager und im Autoklaven die Lösung. Nachdem diese Lösung auf Raumtemperatur abgekühlt ist, fügen Sie 1 ml Phosphat - Lager, 1 ml Na 2 CO 3 Lager und 10 ml NaHCO 3.
- Für BG11 festes Medium, fügen Sie 15 g Agar und 700 ml Wasser zu einer flask. Zum zweiten Kolben gemischt, 3 g Na 2 S 2 O 3, 226 ml Wasser, 10 ml 100x BG11, 1 ml Spurenelemente und 1 ml Eisen Lager. Autoclave beide Lösungen. Nachdem diese Lösungen auf Raumtemperatur abgekühlt sind, sie zu kombinieren und 1 ml Phosphat - Lager hinzufügen, 1 ml Na 2 CO 3 Lager, 10 ml TES - Puffer und 10 ml NaHCO 3.
Hinweis: Die Lösungen werden separat zu vermeiden Ausfällen bestimmter Salze hergestellt.
- Für die Selektion auf Saccharose, bereiten eine 50% (w / v) Saccharose-Lösung. Filter sterilisieren, die Lösung mit 0,2 & mgr; m-Filter und in den BG11 (100 ml von 50% Saccharose zu 900 ml BG11) BG11 / 5% Sucrose-Platten herzustellen.
Hinweis: Nicht NaHCO 3 bis BG11 / 5% Saccharose - Agar - Platten hinzufügen. Hinzufügen , Na 2 CO 3 als normal. - Für die Kultivierung von Synechococcus 10 ml 1 M 4- (2-Hydroxyethyl) piperazin-1-ethansulfonsäure, N - (2-hydroxyethyl) NR Piperazin-42; - (2-ethansulfonsäure) (HEPES) und 1 ml Vitamin B 12 ( siehe Tabelle 1) auf 1 L des BG11 - Medium.
Hinweis: Transformation von kultivierten Stämmen in handelsüblichen BG11 Medien wesentlich weniger effizient ist als in den BG11 Medienrezepte hier beschriebenen und wird deshalb nicht empfohlen.
2. Wachstum von Cyanobakterien Dehnungen
- Kultur-Stämme in 100 ml Erlenmeyerkolben mit einem maximalen Volumen von 50 ml und schütteln bei 120 Umdrehungen pro Minute. Siegel BG11-Platten mit Parafilm und Durchlöcherung drei kleine Löcher in der Seite der Plattengasaustausch zu ermöglichen. Inkubieren alle Stämme bei 30 ° C unter Leuchtstoffröhren in einem Photobioreaktor bei einer Lichtintensität zwischen 20 bis 40 & mgr; mol Photonen m -2 s -1.
- Verwenden Sie am besten sterile Techniken. Behandeln Sie alle Cyanobakterien-Stämme in einer laminaren Strömung Haube.
Anmerkung: Dies ist besonders wichtig, wenn die Stämme mit Saccharose enthaltenden Medien kultiviert werden, die leicht ist Contaminated.
3. Erzeugung von Plasmid-Konstrukten
- Design Sätze von Primern, einschließlich der erforderlichen Restriktionsenzymstellen, unter Verwendung von Primer-Design-Software wie Primer3 (http://frodo.wi.mit.edu/primer3/), zu amplifizieren zwei ~ 1 kb Regionen 5 'und 3' der Gen von Interesse. Lesen Sie die Genomsequenz der Cyanobakterien über Cyanobase (http://genome.kazusa.or.jp/cyanobase). Siehe Tabelle 2 für alle Primer zum Einsatz. Bei der Gestaltung von Primer die folgenden Faktoren berücksichtigen:
- Stellen Sie sicher , dass verstärkt die Regionen, 5 'und 3' - Regionen des Gens , die mutiert werden, zB 1.
- mutieren intergenischen Regionen nicht unbeabsichtigte Mutation von Antisense und nicht-kodierenden RNAs zu vermeiden. Für die Erzeugung von Mutanten in Synechocystis finden Sie in der Liste der Transkriptionsstartstellen dokumentiert in Mitschke et al., 2011 18, um die Mutation von Antisense zu vermeidenoder nicht-kodierenden RNAs.
- Bei der Auswahl der flankierenden Regionen enthalten nicht den gesamten offenen Leserahmen von benachbarten Genen die Expression dieser Gene in Escherichia coli mit Klonierung stören können.
- Amplify Produkte von PCR High Fidelity DNA-Polymerase gemäß den Anweisungen des Herstellers.
Hinweis: Nach unserer Erfahrung dieses Enzym einige Fehler produziert.- Stellen Sie bis zu 50 & mgr; l PCR-Reaktionen HF-Puffer, und entweder 0, 1,5 oder 3 ul DMSO. Verwenden von 100 ng genomischer DNA pro Reaktion. Verwenden eines Programms aus einem anfänglichen Denaturierungsschritt von 98 ° C für 30 sec, 35 Runden von 98 ° C für 10 sec, 67 ° C für 30 sec, 72 ° C für 30 sec, gefolgt von einem letzten Verlängerungsschritt von 72 & deg; C für 5 min. Dies gibt normalerweise konsistente Produkte.
- Stellen Sie sicher, PCR-Produkte und Proben mit Endonuklease Enzyme für die richtige Größe über Gelelektrophorese verdaut. Versuch 1% (w / v) Agarose-Gelen, enthaltend 0,02%(V / v) Ethidiumbromid für 45 min bei 100 V.
VORSICHT: Ethidiumbromid ist ein potentieller mutagen und sollte mit der entsprechenden Schutz behandelt werden. - Reinige PCR-Produkte eine DNA Purification Kit nach den Anweisungen des Herstellers verwendet wird. Verwenden Sie auch dieses Kit für die Aufreinigung von Plasmid-Fragmente, darunter Stücke aus Agarosegelen geschnitten. Eluieren gereinigte DNA in 14 ul Wasser.
- Für Klonierungsschritte, Restriktionsendonuclease Reaktionsgemische bei 37 ° C für> 1 Stunde in einem Gesamtvolumen von 30 & mgr; l gemß den Anweisungen des Herstellers inkubiert.
- Zur Ligation Schritte Ligat DNA-Fragmente bei Raumtemperatur> 1 Stunde in einem Gesamtvolumen von 20 & mgr; l, enthaltend 5 & mgr; l des gereinigten verdauten Plasmid, 12 & mgr; l des gereinigten verdauten insert, 2 & mgr; l Puffer und 1 ul Ligase.
- Bereiten Escherichia coli DH5 & alpha; transformanten Zellen gemäß dem folgenden Verfahren.
- Wachsen Sie eine Nacht E. coli Kultur in 10 ml Luria Bertani (LB) Medien.
- Beimpfen von 400 ml LB in einem 1 L - Erlenmeyerkolben , enthaltend 6 ml 1 M MgCl 2 (Tabelle 1) mit 1 ml der Übernacht - Kultur.
- Die Kultur bei 37 ° C bei 220 Upm für etwa 4 Stunden oder bis die OD 600nm erreicht 0,4-0,6.
- Legen Sie Zellen auf Eis für 1 Stunde.
- Zentrifuge bei 2.800 xg für 10 min Zellen bei 4 ° C pelletiert.
- Überstand entfernen und resuspendieren in 160 ml Lösung A (Tabelle 1) und Inkubation auf Eis für 20 min.
- Zentrifuge bei 2.800 xg für 10 min Zellen bei 4 ° C pelletiert.
- Überstand entfernen und resuspendieren in 4 ml Lösung A + Glycerin (Tabelle 1).
- Bereiten Sie 50 ul Aliquots, gefrieren in flüssigem N 2, bei -80 ° C.
- Mischen Sie 5 ul Ligierungsgemisch mit 50 ul kompetente Zellen und Inkubation für 1 Stunde auf Eis.
- Hitzeschock der Zellen bei 42 ° C für 90 sec, followed durch Inkubation auf Eis für 2 min.
- Hinzufügen , 950 & mgr; l LB - Medium (Tabelle 1) und Inkubation bei 37 ° C für 1 Stunde.
- Aliquotieren 50 und 200 & mgr; l auf Platten mit dem entsprechenden Antibiotikum, entweder Ampicillin (100 ug / ml) und / oder Kanamycin (30 ug / ml).
ACHTUNG: Sowohl Kanamycin und Ampicillin sind giftig und sollten mit angemessenen Schutz behandelt werden. - Pick-and-Inkubation einzelne Kolonien in 2 ml LB-Medium mit dem entsprechenden Antibiotikum geimpft.
- Reinige Alle Plasmide eine Miniprep-Plasmid-Reinigungskits gemß der Anweisungen des Herstellers.
- Erzeugen Plasmide, in diesem speziellen Beispiel für das Klopfen die cpcC1C2 Gene aus, entsprechend den folgenden Schritten.
- Amplify das 1.012 bp 5' - flankierenden Region (linke Fragment) unter Verwendung von Primern cpcC1C2leftfor und cpcC1C2leftrev (siehe Schritt 3.2, Tabelle 2). Entfernen Sie eine kleine Menge der PCR-Reaktion und bestätigen, ob dierichtige Größe Produkt wurde über Gelelektrophorese (Schritt 3.3) verstärkt. Digest dieses Fragment und pUC19 mit Xba I und Bam HALLO (Schritt 3.5).
- Reinige beide Präparate (Schritt 3.4), Ligat (Schritt 3.6), Transformation (Schritt 3.7) und vier 2 ml LB Flüssigkulturen mit Ampicillin (100 ug / ml) aus einzelnen Kolonien für Plasmidaufreinigung über Minipreps (Schritt 3.8) eingerichtet.
- Prüfen zum Einsetzen des Fragments in pUC19 über Xba I / Bam HALLO Verdauung und Gelelektrophorese (Schritt 3.3). Bands von 2660 bp und 1.012 bp zeigen korrekte Einführung des Einsatzes in das Plasmid.
- Amplify das 1.016 bp 3 'flankierenden Region (rechts Fragment) unter Verwendung von Primern cpcC1C2rightfor und cpcC1C2rightrev (siehe Schritt 3.2, Tabelle 2). Entfernen Sie eine kleine Menge der PCR-Reaktion und bestätigen, ob die richtige Größe Produkt über Gelelektrophorese verstärkt wurde (Schritt 3.3). Digest dieses Fragment und pUC19 mit Sac I und Eco RI (step 3.5).
- Reinige beide Präparate (Schritt 3.4), Ligat (Schritt 3.6), Transformation (Schritt 3.7) und vier 2 ml LB Flüssigkulturen mit Ampicillin (100 ug / ml) aus einzelnen Kolonien für Plasmidaufreinigung über Minipreps (Schritt 3.8) eingerichtet.
- Prüfen zum Einsetzen des Fragments in pUC19 über Sac I / Eco RI - Verdau (Schritt 3.5) und Gelelektrophorese (Schritt 3.3). Bands von 2660 bp und 1.016 bp zeigen korrekte Einführung des Einsatzes in das Plasmid.
Anmerkung: Xba I / Bam HALLO Stellen zur Klonierung des 5' - Region und Sac I / Eco RI zur Klonierung des 3' - Region in pUC19 werden , wo immer möglich , eingesetzt. Falls möglich, immer auch eine Bam HALLO auf dem Reverse - Primer für die 5' - Region oder der Vorwärts - Primer für die 3' - Region zu gewährleisten , dass spätere Klonierungsschritte sind einfacher durchzuführen. - Sequence beide Einsätze zu bestimmen , ob die Sequenz korrekt ist unter Verwendung von Primern , die die Insertionsstelle Spanning zB M13 nach vorne und M13 reverse (Tabelle 2). Die Reihenfolge muss stimmen keine Fehler zu gewährleisten, werden in flankierenden Regionen eingeführt.
- Excise die linke Fragment aus pUC19 über Xba I / Bam HALLO Verdauung. Digest das pUC19 + rechts Fragment mit Xba I / Bam HALLO (Schritt 3.5).
- Man reinige das 1.012 bp linke Fragment und 3676 bp pUC19 + rechts Fragment aus einem Agarosegel (Schritt 3.3) über Exzision der DNA, die ein Skalpell verwendet wird.
- Reinige beide Präparate (Schritt 3.4), Ligat (Schritt 3.6), Transformation (Schritt 3.7) und vier 2 ml LB Flüssigkulturen mit Ampicillin (100 ug / ml) aus einzelnen Kolonien für Plasmidaufreinigung über Minipreps (Schritt 3.8) eingerichtet.
- Überprüfen Sie zum Einfügen des Fragments in pUC19 + rechts Fragment über Xba I / Bam HALLO Verdauung (Schritt 3.5) und Gelelektrophorese (Schritt 3.3). Bands von 3676 bp und 1.012 bp zeigen korrekte Einsetzen des Einsatzes in das Plasmid (dies als Plasmid B beziehen).
- Verbrauch die NPT1 / sacB Kassette aus pUM24cm 19 über Bam HALLO Verdauung. Digest Plasmid B mit Bam HALLO (Schritt 3.5).
Hinweis: Die NPT1 / sacB Kassette nicht aus Agarosegelen gereinigt werden muss , da pUM24cm eines Proteins Chloramphenicol - Resistenz kodiert. Deshalb , wenn Kolonien auf LB / Ampicillin / Kanamycin - Agar - Platten , die einzig mögliche Kombination angebaut werden , die zu resistenten Kolonien führen wird , ist der Einbau von NPT1 / sacB Kassette in das Plasmid B. - Reinige beide Präparate (Schritt 3.4), Ligat (Schritt 3.6), Transformation (Schritt 3.7) und vier 2 ml LB Flüssigkulturen mit Ampicillin eingerichtet (100 ug / ml) und Kanamycin (30 ug / ml) aus einzelnen Kolonien für Plasmidaufreinigung über Minipreps (Schritt 3.8).
- Prüfen zum Einführen des NPT1 / sacB Kassette in das Plasmid B über Bam HALLO - Verdau (Schritt 3.5) und Gelelektrophorese (Schritt 3.3). Bands von 4688 bp und 3894 bp zeigen ein richtiges Einsetzen von the einfügen in das Plasmid (bezeichnen dies als Plasmid A).
- Alternativ stumpfen Ende der NPT1 / sacB Kassette und Klon in eine andere Restriktionsendonuklease - Stelle zwischen den linken und rechten Fragmente in Plasmid B. Die NPT1 / sacB Kassette muss zwischen den linken und rechten Fragmente kloniert werden.
Hinweis: Wenn die Expression eines fremden Kassette erforderlich ist, dann sollte dies Dieses Plasmid wird dann in den nicht markierten Knockout Schritten zwischen den linken und rechten Fragmente von Plasmid B. eingesetzt werden.
4. Erzeugung von Marked Synechocystis und Synechococcus Mutants
- Stellen Sie eine neue Kultur, indem Sie eine Schleife voll von Zellen in 30-50 ml BG11 Medium Inokulation. Die Kultur für 2-3 Tage auf OD 750nm = 0,2 bis 0,6.
Hinweis: In der Regel einzelne Kolonien zu klein sind für die Impfung und die Belichtung einzelner Zellen zu verwenden, um auch geringe Mengen an Licht führen in Photoinhibition und Auswahlfür Licht resistenten Mutanten. - Zentrifuge 1-2 ml der Kultur bei 2.300 xg für 5 Minuten und der Überstand verworfen. Zentrifuge Sie keine Cyanobakterien Kulturen bei> 2300 xg, da dies die Zellen schädigen können. Waschen Sie das Pellet einmal mit BG11-Medium.
Hinweis: Die Zellen nicht durch Verwirbelung, da dies zu einem Verlust der Pili führen können, die für die DNA-Aufnahme wesentlich sind. Die Zellen durch vorsichtiges Pipettieren. - Hinzufügen BG11-Medium bis zu einem Endvolumen von 100 ul. Transfer-Zellen zu einem 14 ml-Rundbodenrohr.
- 1 ug Plasmid-A zu den Zellen und durch leichtes Klopfen mischen. In <10 & mgr; l Plasmid.
Anmerkung: Vorzugsweise wird das Plasmid in einer Konzentration von> 100 ng / & mgr; l sein sollte, aber Konzentrationen unter diese sind ausreichend für eine erfolgreiche Transformation. - Legen Sie Rohre horizontal in den Inkubator nach unten. Inkubieren Kulturen für 4-6 Std.
Hinweis: Zellen durch Klopfen alle 1-2 h kurz gemischt werden kann, aber dies ist nicht wesentlich. Proben können in einem platziert werdenSchüttelinkubator obwohl dies die Effizienz zu verbessern nicht wesentlich. - Verbreiten Aliquots der Zellkultur / Plasmid-DNA-Mischung auf BG11-Agarplatten ohne Antibiotika. Typischerweise 20 ul und 80 ul Aliquots werden auf getrennten Platten verteilt.
- später ~ 24 h, hinzufügen 2,5-3 ml 0,6% Agar-Lösung in Wasser, enthaltend Kanamycin (je 20 ml: 0,12 g Agar, 100 & mgr; l von 100 mg / ml Kanamycin) auf der Agarplatte. Kühlen dieser Lösung auf ~ 42 ° C, und an den Rand der Agar-Platte hinzuzufügen. Kippen Sie die Platte, so dass die Lösung bildet eine noch "Top-Agar" Schicht auf der Oberfläche.
- Inkubieren Agarplatten für einen weiteren Zeitraum. Kolonien sollte nach ca. 7 Tage sichtbar sein.
Hinweis: Agar-Platten 3 hoch in einem Inkubator gestapelt werden können. Typischerweise Hunderte von Kolonien pro Transformation erhalten. - Streak einzelne Kolonien auf BG11 + Kanamycin (30 ug / ml) Agar-Platten. Teilen Sie die Agar-Platte in 6 Sektoren und ein stumpfes Ende Zahnstocher Streifen verwenden ausdie Kolonien über jeden einzelnen Sektor. Einzelkolonien zu erhalten ist nicht wichtig, nur das Wachstum der Trans.
- Bestätigen markierten knockout durch PCR unter Verwendung von Taq DNA-Polymerase gemäß den Anweisungen des Herstellers. In 2 ul MgCl 2 (25 mM) pro Reaktion.
- Entfernen Sie einen kleinen Teil der Zellen und Transfer in ein Rohr 50 & mgr; l Wasser enthält und ~ 20 425-600 & mgr; m Glasperlen. Schütteln in einem Vibrator für 5 min bei ~ 2.000 Umdrehungen pro Minute. Zentrifuge bei 15.700 xg für 5 min und mit 5 & mgr; l Überstand pro 50 & mgr; l PCR-Reaktion.
Hinweis: Verwenden Sie die Lösung nicht suspendieren. Die Zelltrümmer muss am Boden des Röhrchens zu bleiben.
- Entfernen Sie einen kleinen Teil der Zellen und Transfer in ein Rohr 50 & mgr; l Wasser enthält und ~ 20 425-600 & mgr; m Glasperlen. Schütteln in einem Vibrator für 5 min bei ~ 2.000 Umdrehungen pro Minute. Zentrifuge bei 15.700 xg für 5 min und mit 5 & mgr; l Überstand pro 50 & mgr; l PCR-Reaktion.
- Validate-Mutanten
- Design-Primern, die den knockout Region unter Verwendung von Primer-Design-Software (wie Primer3) überspannen. Design-Primer beginnend bei ~ 200 bp auf jeder Seite der K.-o.-Region.
Hinweis: Die Primer für die cpcC1C2 Mutante Überprüfung sind in Tabelle 2 skizziertenund cpcC1C2for und cpcC1C2rev bezeichnet. - Amplifizieren Produkte unter Verwendung eines Programms, bestehend aus einem anfänglichen Denaturierungsschritt von 95 ° C für 2 min, 35 Runden von 95 ° C für 1 min, 60 ° C für 1 min, 72 ° C 1 min per kb der Sequenz, gefolgt von einem endgültige Verlängerungsschritt von 72 ° C für 5 min. Fügen Sie ein Wildtyp-Kontrolle. Dies gibt normalerweise konsistente Produkte.
- Überprüfen Sie den Genotyp über Gelelektrophorese. Markierte Knockout Trans zeigt eine Bande von ~ 4 kb (0,2 kb von der linken und rechten Fragmente sowie die NPT1 / sacB - Kassette) und das Fehlen des Wildtyp - Band (Abbildung 2).
Anmerkung: In bestimmten Fällen a ~ 4-kb-Bande in dem markierten Mutante aufgrund der großen Größe von PCR-Produkt nicht beobachtet. Wenn jedoch ein Band der erwarteten Größe des Wildtyp beobachtet entspricht, wird nicht dann in der Regel dieser Stamm ist eine deutliche Knockout.
- Design-Primern, die den knockout Region unter Verwendung von Primer-Design-Software (wie Primer3) überspannen. Design-Primer beginnend bei ~ 200 bp auf jeder Seite der K.-o.-Region.
- Wenn ein Wildtyp-Band immer noch vorhanden ist, dann wieder Streifen die Belastung auf einfrisch BG11 + Kanamycin (30 ug / ml) Agar-Platte und die PCR wiederholen. Wiederholen Sie den Wieder Ausstreichen Prozess, bis die Mutante getrennt ist, so dass kein Wildtyp-Bande in der PCR-Reaktion beobachtet wird.
Anmerkung: Die Erhöhung der Menge von Kanamycin in einer Konzentration von 50 ug / ml, dann 100 & mgr; g / ml ist manchmal notwendig, um eine markierte Mutante vollständig zu trennen. - Wenn der Stamm eine markierte Mutante Profil mittels PCR zeigt, dann wieder Streifen auf einem frischen BG11 + Kanamycin (30 ug / ml) Agar-Platte. Verwenden Sie diese Belastung auf den nicht markierten Knockout erzeugen.
Hinweis: Das Protokoll kann verwendet werden , um markierte Mutanten erzeugen mit nur einem Antibiotikum - Resistenzkassette dh durch die NPT1 / sacB Kassette ersetzt nur mit dem NPT1 Kassette aus pUC18K 20 zwischen der linken und rechten Fragmente..
5. Generation von Unmarked Synechocystis Mutants
- Legen Sie eine neue Kultur des markierten Knockout, indem Sie eine Schleife voll von c Impfenells in 30-50 ml BG11-Medium. Die Kultur für 2-3 Tage auf OD 750nm = 0,2 bis 0,6.
- Zentrifuge 10 ml der Kultur bei 2300 × g für 5 min und den Überstand verwerfen. Waschen Sie einmal mit BG11-Medium.
Hinweis: Die Zellen nicht durch Verwirbelung, da dies zu einem Verlust der Pili führen können, die für die DNA-Aufnahme wesentlich sind. Die Zellen durch vorsichtiges Pipettieren. - Hinzufügen BG11 bis zu einem Endvolumen von 200 & mgr; l. Transfer-Zellen zu einem 14 ml-Rundbodenrohr.
- 1 ug Plasmid-B-DNA in die Zellen und mischen durch leichtes Klopfen.
- Inkubieren Sie die Proben für 4-6 Std. Legen Sie Rohre horizontal nach unten.
Hinweis: Zellen durch Klopfen alle 1-2 h kurz gemischt werden kann, aber dies ist nicht wesentlich. Die Proben können in einem Schüttelinkubator gestellt werden, obwohl diese Effizienz nicht verbessert. - In 1,8 ml BG11-Medium und Inkubation Proben für insgesamt 4 Tage mit Schütteln. Dies ist eine ausreichende Zeit Rekombination zu erlauben, in den mehrere chromosomale Kopien auftreten.
- Platte Aliquots des Transformationsgemisches auf BG11 / 5% Sucrose-Agar-Platten. Tafel 50 & mgr; l, 10 & mgr; l und 1 & mgr; l pro Agar-Platte. Wenn eine Kolonie Rasen wird auf alle diese Agarplatten die Lösung weiter und aliquoten auf frische Platten verdünnen. Kolonien sollte nach ca. 7 Tage sichtbar sein.
- Patch 30-50 einzelne Kolonien auf BG11 + Kanamycin (30 ug / ml) Agarplatten ersten und BG11 / 5% Sucrose-Agar-Platten zweite, ein stumpfes Ende Zahnstocher. Alle Bakterien, die auf BG11 / 5% Saccharose-Platten wachsen, aber nicht BG11 + Kanamycin-Platten sind potentielle unmarkierten Vorprägungen. Bakterien auf beiden Platten wachsen wahrscheinlich aufgrund einer Mutation in dem Gen sacB Sucrose resistent.
- Stellen Sie sicher , nicht markierten Vorprägungen der gleichen Primer und Verfahren wie verwendet wurde , um die markierten Vorprägungen zu überprüfen. ZB cpcC1C2for und cpcC1C2rev (Tabelle 2) für die cpcC1C2 unmarkierten Knockout zu überprüfen. Ein unmarkierten Knockout wird ein Band auf einem Agarose-Gel entspricht t zeigeno die Wildtyp - Größe minus der gelöschte Bereich (Abbildung 2).
- Wenn der Stamm eine unmarkierte Mutant Profil mittels PCR zeigt (Schritt 4.11.2) und Gelelektrophorese (Abbildung 2), dann wieder Streifen auf einem frischen BG11 - Agar - Platte ohne Antibiotika.
6. Langzeitlagerung von Dehnungen
- Legen Sie eine neue Kultur des Stammes nach oben durch eine Schleife voll von Zellen in 30-50 ml BG11 Medium Inokulation. Die Kultur für 3-4 Tage auf OD 750nm = 0,4 bis 0,7.
- Waschen Sie die Zellen einmal mit BG11 und resuspendieren in ~ 2 ml BG11.
- Fügen 0,8 ml konzentrierte Zellen zu einem Röhrchen. Dann fügen Sie 0,2 ml 80% Filter sterilisiert Glycerin.
- Optional: Fügen Sie 0,93 ml konzentrierte Zellen in ein anderes Röhrchen. Hinzufügen 0,07 ml DMSO zu dieser Röhre.
ACHTUNG: DMSO ist giftig und sollte mit der entsprechenden Schutz behandelt werden. - Bewahren Sie beide Röhrchen bei -80 ° C. Zur Wiederbelebung Stämme, die das Rohr zu entfernen und einige Zellen mit einem stumpfen Zahn abkratzenPick auf eine Agar-Platte ohne Antibiotika. Streak aus als normal, dass eine sterile Schleife.

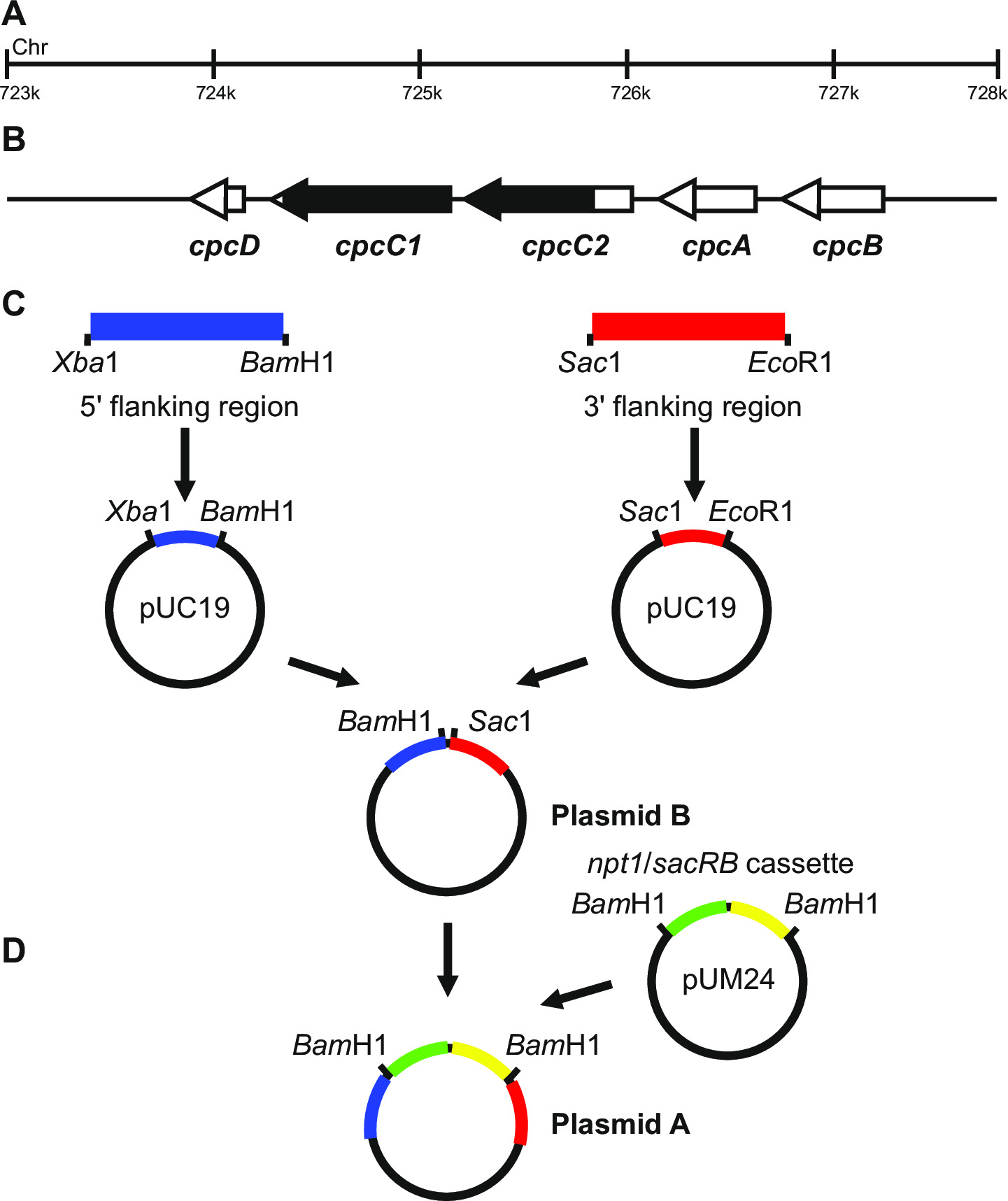
Abbildung 1: Plasmidkonstruktion für die Erzeugung von markierten und nicht markierten Vorprägungen, zB cpcC1 und cpcC2 in Synechocystis (A) Region des Synechocystis - Genoms , wobei (B) cpcC1 und cpcC2 und benachbarte Gene befinden.. Hervorgehoben in schwarz ist die Region des Genoms, in dem mutanten gelöscht werden. (C) Seiten des Genoms , die durch PCR amplifiziert werden. Die 5'-flankierende Region (blau gekennzeichnet) und 3 'flankierenden Region (in rot dargestellt) mit Restriktionsendonukleasestellen verstärkt für in pUC19 klonen. Die 5 '(oder 3') flankierende Region ist aus pUC19 ausgeschnitten und in die pUC19 + 3 '(oder 59;) flankierende Region Plasmid Plasmid B. (D) Die NPT1 / sacB Kassette aus pUM24 wird ausgeschnitten über Bam HALLO Verdauung und zwischen den 5 'und 3' flankierenden Regionen zu erzeugen Plasmid A zu erzeugen Bitte klicken hier , um eine größere Version dieser Figur.
{kind=link}
Ergebnisse
Plasmid - Design ist entscheidend für die erfolgreiche Erzeugung der beiden markierten und nicht markierten Mutanten. Figur 1 gibt ein Beispiel von Plasmid A und B verwendet , um eine Deletionsmutante in den Synechocystis Gene 13 cpcC1 und cpcC2 zu erzeugen. In jedem Fall sind die 5 'und 3' flankierenden Regionen sind etwa 900-1.000 bp. Reduzierte flankierenden Regionen können verwendet werden, obwohl das kleinste wir erfol...
Diskussion
Die wichtigsten Schritte bei der Erzeugung von nicht markierten Mutanten sind: 1) vorsichtig Plasmid-Design, um sicherzustellen, nur die Zielregion verändert wird; 2) sicherzustellen, dass die Proben bleiben axenic, vor allem, wenn die Kultur auf Saccharose; 3) plating Zellen für markierte Mutante Generation zunächst auf Agarplatten BG11 transformiert Antibiotika, gefolgt von der Zugabe von Agar und Antibiotika 24 Stunden später fehlen; 4) markierte Kultivierung Mutanten für 4 volle Tage vor auf BG11 und Saccharose...
Offenlegungen
The authors declare that they have no competing financial interests.
Danksagungen
Wir sind dankbar, dass der Umwelt Services Association Education Trust, der Synthetischen Biologie in Cambridge SynBio Fonds und des Ministeriums für soziale Gerechtigkeit und Übertragung von Verantwortung, Regierung von Indien, für die finanzielle Unterstützung.
Materialien
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
Referenzen
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis?. Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten