
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Generación de mutantes sin marcadores marcados y en el modelo de especies de cianobacterias
En este artículo
Resumen
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Resumen
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Introducción
Las cianobacterias son un phylum antigua y diversa evolutivamente de bacterias que se encuentran en casi todos los medio natural en la Tierra. En los ecosistemas marinos son particularmente abundantes y juegan un papel clave en muchos ciclos de nutrientes, lo que representa aproximadamente la mitad de la fijación de carbono 1, la mayoría de la fijación de nitrógeno 2 y cientos de millones de toneladas de producción de hidrocarburos 3 en los océanos anualmente. Cloroplastos, el orgánulo responsable de la fotosíntesis de las algas y las plantas eucariota, es probable que hayan evolucionado a partir de una cianobacteria que fue engullido por un organismo huésped 4. Las cianobacterias han demostrado organismos modelo útil para el estudio de la fotosíntesis, transporte de electrones y 5 vías bioquímicas, muchos de los cuales se conservan en las plantas. Además cianobacterias cada vez se utilizan para la producción de alimentos, los biocombustibles 6, 7 y electricidad compuestos industriales 8, debido a su altaghly conversión eficiente del agua y CO 2 a la biomasa utilizando la energía solar 9. Muchas especies pueden cultivarse en tierras no cultivables con nutrientes mínimos y el agua de mar, lo que sugiere que las cianobacterias potencialmente podrían ser crecido a gran escala sin afectar la producción agrícola. Ciertas especies son también fuentes de productos naturales, incluidos los compuestos antifúngicos, antibacterianos y anti-cáncer 10,11.
La capacidad de generar mutantes es clave para entender la fotosíntesis de cianobacterias, bioquímica y fisiología, y esencial para el desarrollo de cepas con fines industriales. La mayoría de los estudios publicados generate modificado genéticamente cepas mediante la inserción de un casete de resistencia a los antibióticos en el sitio de interés. Esto limita el número de mutaciones que pueden introducirse en una cepa, como sólo unos casetes de resistencia a antibióticos están disponibles para su uso en cianobacterias. Las cepas que contienen los genes que confieren re antibióticoresistencia no puede ser utilizado para la producción industrial en estanques abiertos, lo que es probable que sea el único medio rentables para la producción de biocombustibles y otros productos de bajo valor 12. La generación de mutantes no marcados supera estas limitaciones. mutantes no marcados no contienen ADN extraño, excepto si se incluye intencionalmente, y se pueden manipular varias veces. Por lo tanto es posible generar tantas alteraciones en una cepa como se desee. Además, los efectos polares de genes aguas abajo del sitio de modificación pueden minimizarse, lo que permite la modificación más preciso del organismo 13.
Para generar cepas mutantes, plásmidos suicidas que contienen dos fragmentos de ADN idéntica a las regiones en el cromosoma de cianobacterias que flanquean el gen a eliminar (denominado el 5 'y 3' flanqueantes) se construyó primero. Dos genes se insertan entonces entre estas regiones flanqueantes. Uno de ellos codifica una proteína de resistencia a los antibióticos; la segunda codifica SacB, que produces levansucrasa, un compuesto que confiere sensibilidad a la sacarosa. En la primera etapa del proceso, los mutantes marcados, es decir, las cepas que contienen algo de ADN externa, se generan. La construcción de plásmido se mezcla con las células de cianobacterias y el ADN se recogió de forma natural por el organismo. Los transformantes se seleccionaron por crecimiento en placas de agar que contienen el antibiótico apropiado y el genotipo mutante verificada por PCR. plásmidos suicidas no pueden replicarse dentro de la cepa de interés. Por lo tanto cualquier colonias resistentes a los antibióticos serán el resultado de un evento de recombinación mediante el cual el gen de interés en insertado en el cromosoma. Para generar mutantes no marcados, el mutante marcada se mezcla a continuación con un segundo plásmido suicida que contiene sólo las regiones flanqueantes 5 'y 3'. Sin embargo, si se requiere la inserción de ADN extraño, un plásmido que consiste en el 5 'y 3' que flanquean las regiones con un casete que contiene los genes de interés insertado entre estos fragmentos de ADN, se puede utilizar. selección es a través del crecimiento en placas de agar que contienen sacarosa. Como la sacarosa es letal para las células cuando se expresa el producto del gen sacB, las únicas células que sobreviven son aquellos en los que se ha producido un segundo suceso de recombinación, con lo cual el gen de sensibilidad sacarosa, además de el gen de resistencia a los antibióticos, se ha recombinado de la cromosoma y en el plásmido. Como consecuencia del intercambio de recombinación, las regiones flanqueantes y cualquier ADN entre ellos se insertan en el cromosoma.
Hemos utilizado con éxito estos métodos para generar múltiples mutaciones cromosómicas en la misma cepa de Synechocystis sp. PCC6803 (en adelante como Synechocystis) 13,14, para introducir mutaciones puntuales en un gen de interés y 13 para la expresión de cassettes de genes. Mientras que la generación de knockouts sin marcar se ha demostrado con anterioridad a nuestro trabajo en Synechocystis 15,16, un método detallado, con la ayuda deuna presentación visual de los pasos críticos, no está disponible públicamente. También hemos aplicado el mismo método para la generación de knockouts marcadas en otro modelo de cianobacteria, Synechococcus sp. PCC7002 (en lo sucesivo, Synechococcus). Este protocolo proporciona un método claro y sencillo para la generación de mutantes y un protocolo rápido para la validación y almacenamiento de estas cepas.
Protocolo
1. Preparación de Medios de Cultivo
- Preparar medio BG11 según Castenholz, 1988 17.
- Preparar soluciones madre de BG11 100x, elementos y hierro stock (Tabla 1) rastrear.
- Preparar soluciones separadas de madre de fosfato, Na 2 CO 3 stock, N - [Tris (hidroximetil) metil] -2-aminoetanosulfónico (TES) de amortiguación y NaHCO3 (Tabla 1).
- Autoclave el fosfato y Na 2 CO 3 poblaciones. Filter-esterilizar tampón TES y NaHCO 3 con 0,2 micras filtros.
- Preparar BG11 combinando 976 ml de agua, 10 ml de 100x BG11, 1 ml de oligoelementos y 1 ml de hierro stock y autoclave la solución. Después de esta solución se ha enfriado a temperatura ambiente, añadir 1 ml de Stock fosfato, 1 ml de Na 2 CO 3 stock y 10 ml de NaHCO3.
- Para BG11 medio sólido, añadir 15 g de agar y 700 ml de agua a una flask. Para el segundo frasco, añadir 3 g de Na 2 S 2 O 3, 226 ml de agua, 10 ml de 100x BG11, 1 ml de oligoelementos y 1 ml de hierro stock. Autoclave ambas soluciones. Después de estas soluciones se hayan enfriado a temperatura ambiente, combinarlos y se añade 1 ml de madre de fosfato, 1 ml de Na 2 CO 3 stock, 10 ml de tampón TES, y 10 ml de NaHCO3.
Nota: Las soluciones se preparan por separado para evitar la precipitación de ciertas sales.
- Para la selección de la sacarosa, preparar un 50% (w / v) de solución de sacarosa. Filtro de esterilizar la solución con 0,2 micras filtros y añadir a BG11 (100 ml de 50% de sacarosa a 900 ml de BG11) para producir BG11 / 5% de sacarosa placas.
Nota: No agregar NaHCO3 a placas de agar sacarosa BG11 / 5%. Añadir Na 2 CO 3 como normal. - Para el cultivo de Synechococcus añadir 10 ml de H 4- (2-hidroxietil) piperazina-1-etanosulfónico ácido 1, N - (2-hidroxietil) piperazina NR42; - (ácido 2-etanosulfónico) (HEPES) y 1 ml de vitamina B 12 (Tabla 1) a 1 l de medio BG11.
Nota: Transformación de las cepas cultivadas en medios BG11 comercialmente disponible es significativamente menos eficiente que en las recetas de los medios BG11 descritos aquí y por lo tanto no se recomienda.
2. Crecimiento de cepas de cianobacterias
- Cultura cepas en matraces de 100 ml cónicos con un volumen máximo de 50 ml y agitar a 120 rpm. Sellar las placas con Parafilm y BG11 punción tres pequeños agujeros en el lado de la placa para permitir el intercambio de gases. Incubar todas las cepas a 30 ° C bajo bombillas fluorescentes en un fotobiorreactor a una intensidad de luz entre 20-40 mmol fotones m-2 s-1.
- Utilizar las mejores técnicas estériles. Manejar todas las cepas de cianobacterias en una campana de flujo laminar.
Nota: Esto es especialmente importante cuando las cepas se cultivan con medio que contiene sacarosa, que puede ser fácilmente Contaminado.
3. Generación de Plásmido construcciones
- conjuntos de diseño de cebadores, incluyendo los sitios de enzimas de restricción requerido, utilizando el software de diseño de cebadores, tales como Primer3 (http://frodo.wi.mit.edu/primer3/), para amplificar dos ~ 1 kb regiones 5 'y 3' de la gen de interés. Consulte la secuencia del genoma de las especies de cianobacterias través Cyanobase (http://genome.kazusa.or.jp/cyanobase). Véase la Tabla 2 para todos los cebadores utilizados aquí. En el diseño de cebadores consideran los siguientes factores:
- Asegúrese de que las regiones amplificadas se compone de 5 regiones 'y 3' del gen mutado que será, por ejemplo, la Figura 1.
- No mutar regiones intergénicas para evitar mutación involuntaria de antisentido y ARN no codificantes. Para la generación de mutantes de Synechocystis, consulte la lista de los sitios de inicio de la transcripción documentados en Mitschke et al., 2011 18, con el fin de evitar la mutación del antisentidoo ARN no codificante.
- Al elegir las regiones flanqueantes no incluyen el marco de lectura abierto de genes adyacentes como la expresión de estos genes en Escherichia coli pueden interferir con la clonación.
- Amplificar productos por PCR de alta fidelidad utilizando ADN polimerasa de acuerdo con las instrucciones del fabricante.
Nota: En nuestra experiencia de esta enzima produce pocos errores.- Configurar 50 l reacciones de PCR contenían tampón HF y 0, 1,5 o 3 l de DMSO. Utilice 100 ng de ADN genómico por reacción. Utilice un programa que consta de una etapa de desnaturalización inicial de 98 ° C durante 30 seg, 35 rondas de 98 ° C durante 10 s, 67 ° C durante 30 seg, 72 ° C durante 30 seg, seguido de una etapa de extensión final de 72 ° C durante 5 min. Esto normalmente da a los productos consistentes.
- Verificar productos de PCR y muestras digeridas con enzimas endonucleasas para el tamaño correcto mediante electroforesis en gel. Ejecutar 1% (w / v) de geles de agarosa que contiene 0,02%(V / v) de bromuro de etidio durante 45 min a 100 V.
PRECAUCIÓN: El bromuro de etidio es un mutágeno potencial y debe ser manejado con una protección adecuada. - Purificar los productos de PCR utilizando un kit de purificación de ADN de acuerdo con las instrucciones del fabricante. También utilice este kit para la purificación de fragmentos de plásmidos, incluyendo piezas cortadas a partir de geles de agarosa. Eluir el ADN purificado en 14 l de agua.
- Para etapas de clonación, se incuban las mezclas de reacción de endonucleasa de restricción a 37 ° C durante> 1 h en un volumen total de 30 l de acuerdo con las instrucciones del fabricante.
- Para obtener los pasos de ligación, los fragmentos de ADN Se ligan a temperatura ambiente durante> 1 h en un volumen total de 20 l, que contiene 5 l de plásmido digerido purificado, 12 l de inserto digerido purificado, 2 l de tampón y 1 l de ligasa.
- Preparar células transformantes de Escherichia coli DH5a de acuerdo con el método siguiente.
- Crecer durante toda la noche una E. coli Cultura en 10 ml de Luria Bertani (LB) los medios de comunicación.
- Inocular 400 ml de LB en un matraz cónico de 1 l que contenía 6 ml 1 M MgCl 2 (Tabla 1) con 1 ml de cultivo de una noche.
- Mantener el cultivo a 37 ° C a 220 rpm durante aproximadamente 4 horas o hasta 600 nm alcanza 0,4-0,6.
- Colocar las células en hielo durante 1 hr.
- Se centrifuga a 2.800 g durante 10 min para sedimentar las células a 4 ° C.
- Eliminar el sobrenadante y resuspender en 160 ml de solución A (Tabla 1) y se incuba en hielo durante 20 min.
- Se centrifuga a 2.800 g durante 10 min para sedimentar las células a 4 ° C.
- Eliminar el sobrenadante y resuspender en 4 ml Solución A + glicerol (Tabla 1).
- Preparar 50 ml de alícuotas, congelarse en N2 líquido, se almacena a -80 ° C.
- Mezclar 5 l de mezcla de ligación con 50 l de células competentes y se incuba durante 1 hora en hielo.
- Choque térmico de las células a 42 ° C durante 90 segundos, Folloconducido por incubación en hielo durante 2 min.
- Añadir 950 l de medio LB (Tabla 1) y se incuba a 37 ° C durante 1 hr.
- Alícuota de 50 y 200 l en placas con el antibiótico apropiado, ya sea a la ampicilina (100 mg / ml) y / o kanamicina (30 mg / ml).
PRECAUCIÓN: Tanto la kanamicina y ampicilina son tóxicos y deben manejarse con la protección adecuada. - Recoger e incubar colonias individuales en 2 ml de medio LB inoculados con el antibiótico apropiado.
- Purificar todos los plásmidos utilizando un kit de purificación de plásmido miniprep de acuerdo con las instrucciones del fabricante.
- Generar plásmidos, en este ejemplo específico para la anulación de los genes cpcC1C2, de acuerdo con los siguientes pasos.
- Amplificar la región que flanquea 1.012 pb 5 '(fragmento izquierda) usando cebadores cpcC1C2leftfor y cpcC1C2leftrev (Ver el paso 3.2, Tabla 2). Extraer una pequeña cantidad de la reacción de PCR y confirmar si elproducto del tamaño correcto ha sido amplificada por medio de electroforesis en gel (paso 3.3). Digerir este fragmento y pUC19 con Xba I y Bam HI (paso 3.5).
- Purificar ambas preparaciones (paso 3.4), ligado (paso 3.6), transformación (paso 3.7) y establecer cuatro 2 ml de cultivos líquidos LB con ampicilina (100 mg / ml) a partir de colonias separadas para la purificación del plásmido mediante minipreparaciones (paso 3.8).
- Consultar la inserción del fragmento en pUC19 mediante digestión Xba I / Bam HI y electroforesis en gel (paso 3.3). Bandas de 2.660 pb y 1.012 pb indican introducción correcta del inserto en el plásmido.
- Amplificar la región que flanquea 1.016 pb 3 '(fragmento derecha) usando cebadores cpcC1C2rightfor y cpcC1C2rightrev (Ver el paso 3.2, Tabla 2). Retirar una pequeña cantidad de la reacción de PCR y confirmar si el producto de tamaño correcto se ha amplificado mediante electroforesis en gel (paso 3.3). Digerir este fragmento y pUC19 con Sac I y Eco RI (step 3.5).
- Purificar ambas preparaciones (paso 3.4), ligado (paso 3.6), transformación (paso 3.7) y establecer cuatro 2 ml de cultivos líquidos LB con ampicilina (100 mg / ml) a partir de colonias separadas para la purificación del plásmido mediante minipreparaciones (paso 3.8).
- Consultar la inserción del fragmento en pUC19 mediante digestión Sac I / Eco RI (etapa 3.5) y la electroforesis en gel (paso 3.3). Bandas de 2.660 pb y 1.016 pb indican introducción correcta del inserto en el plásmido.
Nota: los sitios Xba I / Bam HI para la clonación de la región 5 'y Sac I Eco RI / para la clonación de la región 3' en pUC19 se usan siempre que sea posible. Si es factible, siempre incluir un sitio Bam HI en el cebador inverso para la 'región o el cebador directo para el 3' 5 región para asegurar que los pasos después de clonación son más fáciles de realizar. - Secuencia de las dos tapas para determinar si la secuencia es correcta utilizando cebadores que abarcan el sitio de inserción, por ejemplo M13 hacia delante y M13 inverso (Tabla 2). La secuencia debe ser correcto para asegurar que no se introducen errores en regiones que flanquean.
- Cortar el fragmento de pUC19 izquierda a través de la digestión XbaI / BamHI. Digerir el fragmento de pUC19 + derecha con XbaI / BamHI (paso 3.5).
- Se purifica el fragmento de la izquierda 1012 pb y 3676 pb de pUC19 + fragmento de derecho de un gel de agarosa (paso 3.3) a través de la escisión del ADN usando una hoja de bisturí.
- Purificar ambas preparaciones (paso 3.4), ligado (paso 3.6), transformación (paso 3.7) y establecer cuatro 2 ml de cultivos líquidos LB con ampicilina (100 mg / ml) a partir de colonias separadas para la purificación del plásmido mediante minipreparaciones (paso 3.8).
- Compruebe para la inserción del fragmento en el fragmento derecho pUC19 + a través de la digestión Xba I / Bam HI (paso 3.5) y la electroforesis en gel (paso 3.3). Bandas de 3.676 pb y 1.012 pb indican la inserción correcta del inserto en el plásmido (referirse a esto como plásmido B).
- Impuestos Especiales de la NPT1 / sacB casete de pUM24cm 19 a través de la digestión Bam HI. Se digiere el plásmido B con Bam HI (paso 3.5).
Nota: El casete NPT1 / sacB no tiene que ser purificado a partir de geles de agarosa desde pUM24cm codifica una proteína que confiere resistencia a cloranfenicol. Por lo tanto, si las colonias se cultivan en LB / ampicilina / kanamicina placas de agar la única combinación posible que dará lugar a colonias resistentes es la incorporación de la NPT1 casete / sacB en el plásmido B. - Purificar ambas preparaciones (paso 3.4), ligado (paso 3.6), la transformada (paso 3.7) y estableció cuatro 2 ml de cultivos líquidos LB con ampicilina (100 mg / ml) y kanamicina (30 mg / ml) a partir de colonias separadas para la purificación del plásmido a través de minipreparaciones (paso 3.8).
- Compruebe para la inserción del casete NPT1 / sacB en el plásmido B a través de la digestión Bam HI (paso 3.5) y la electroforesis en gel (paso 3.3). Bandas de 4.688 pb y 3.894 pb indican la correcta inserción de THe insertar en el plásmido (referirse a esto como plásmido A).
- Alternativamente, el extremo romo NPT1 / cassette sacB y el clon en un sitio de endonucleasa de restricción diferentes entre los fragmentos izquierdo y derecho en el plásmido B. El / cassette sacB NPT1 debe ser clonado entre los fragmentos izquierdo y derecho.
Nota: Si se requiere la expresión de un casete extranjera entonces esto debe ser insertado entre los fragmentos izquierdo y derecho de plásmido B. Este plásmido se usa después en los pasos knockout sin marcar.
4. Generación de mutantes Synechocystis señalizados y Synechococcus
- Establecer un cultivo fresco mediante la inoculación de un bucle completo de las células en 30-50 ml de medio BG11. Mantener el cultivo durante 2-3 días a 750 nm OD = 0,2 a 0,6.
Nota: Por lo general las colonias individuales son demasiado pequeñas para ser usadas para la inoculación y la exposición de las células individuales que incluso niveles bajos de luz resultarán en fotoinhibicion y selecciónpara los mutantes resistentes a la luz. - Centrifugar 1-2 ml del cultivo a 2300 xg durante 5 min y descartar el sobrenadante. No centrifugar los cultivos de cianobacterias en> 2.300 xg ya que esto puede dañar las células. Lavar el sedimento una vez con medio BG11.
Nota: No volver a suspender las células mediante agitación ya que esto puede resultar en la pérdida de pili, que son esenciales para la absorción de ADN. Resuspender las células mediante pipeteo suave. - Añadir medio BG11 a un volumen final de 100 l. La transferencia de células a un tubo de fondo redondo de 14 ml.
- Añadir 1 g de plásmido A a las células y se mezcla con unos golpecitos suaves. Añadir <10 l de plásmido.
Nota: Preferiblemente, el plásmido debe estar a una concentración de> 100 ng / l, pero concentraciones inferiores a esta son adecuados para la transformación exitosa. - Colocar los tubos en posición horizontal en la incubadora. Se incuban los cultivos durante 4-6 horas.
Nota: Las células se pueden mezclar brevemente tocando cada 1-2 horas, pero esto no es esencial. Las muestras pueden ser colocados en unasacudiendo incubadora aunque esto no mejora significativamente la eficiencia. - Spread alícuotas de la mezcla de ADN de cultivo de células / plásmido en placas de agar BG11 sin antibióticos. Típicamente 20 l y 80 ml de alícuotas se extienden en placas separadas.
- ~ 24 horas más tarde, añadir 2,5 a 3 ml de 0.6% de solución de agar en agua que contiene kanamicina (por cada 20 ml: 0,12 g de agar, 100 l de 100 mg / ml de kanamicina) a la placa de agar. Enfriar esta solución a ~ 42 ° C, y añadir a la borde de la placa de agar. Inclinar la placa de modo que la solución forma una capa uniforme 'agar top' en la superficie.
- Incubar las placas de agar por un nuevo período de tiempo. Las colonias deben ser visibles después de aproximadamente 7 días.
Nota: Las placas de agar se pueden apilar 3 de alta en una incubadora. Típicamente cientos de colonias se obtienen por transformación. - Streak colonias individuales en BG11 + kanamicina (30 mg / ml) en placas de agar. Divida la placa de agar en 6 sectores y usar un palillo de dientes extremo romo a la raya a cabolas colonias más de cada sector individual. La obtención de colonias individuales no es sólo un crecimiento importante, de los transformantes.
- Confirmar marcada knockout por PCR usando Taq ADN polimerasa de acuerdo con las instrucciones del fabricante. Añadir 2 l de MgCl 2 (25 mM) por reacción.
- Retirar una pequeña proporción de las células y transferir a un tubo que contiene 50 l de agua y 20 ~ 425-600 micras perlas de vidrio. Agitar en un vibrador durante 5 min a ~ 2.000 rpm. Centrifugar a 15.700 xg durante 5 min y el uso de 5 l de sobrenadante por 50 l de reacción de PCR.
Nota: No volver a suspender la solución. Los restos celulares necesita para mantenerse en la parte inferior del tubo.
- Retirar una pequeña proporción de las células y transferir a un tubo que contiene 50 l de agua y 20 ~ 425-600 micras perlas de vidrio. Agitar en un vibrador durante 5 min a ~ 2.000 rpm. Centrifugar a 15.700 xg durante 5 min y el uso de 5 l de sobrenadante por 50 l de reacción de PCR.
- validar los mutantes
- Diseño cebadores que abarcan la región de eliminación utilizando el software de diseño de cebadores (como Primer3). cebadores de diseño a partir de ~ 200 pb cada lado de la región de final.
Nota: Los cebadores para la verificación de la mutante cpcC1C2 se resumen en la Tabla 2y se denominan cpcC1C2for y cpcC1C2rev. - Amplificar productos utilizando un programa que consta de una etapa de desnaturalización inicial de 95 ° C durante 2 min, 35 ciclos de 95 ° C durante 1 min, 60 ° C durante 1 min, 72 ° C durante 1 min por kb de la secuencia, seguida de una paso de extensión final de 72 ° C durante 5 min. Incluir un control de tipo salvaje. Esto normalmente da a los productos consistentes.
- Verificar el genotipo mediante electroforesis en gel. Transformantes knockout marcados mostrarán una banda de ~ 4 kb (0,2 kb tanto de los fragmentos izquierdo y derecho más el NPT1 / cassette sacB) y la ausencia de la banda de tipo salvaje (Figura 2).
Nota: En algunos casos una banda kb ~ 4 no se observa en el mutante marcada debido a la gran tamaño de este producto de PCR. Sin embargo, si una banda correspondiente al tamaño esperado de la de tipo salvaje no se observa a continuación, típicamente esta cepa es un knockout marcada.
- Diseño cebadores que abarcan la región de eliminación utilizando el software de diseño de cebadores (como Primer3). cebadores de diseño a partir de ~ 200 pb cada lado de la región de final.
- Si una banda de tipo salvaje todavía está presente a continuación, volver a raya la tensión en unaBG11 fresco + kanamicina (30 mg / ml) en placa de agar y repetir la PCR. Repetir el proceso de re-rayas hasta que el mutante es segregada de modo que no se observa ninguna banda de tipo salvaje en la reacción de PCR.
Nota: El aumento de la cantidad de kanamicina a una concentración de 50 mg / ml, a continuación, 100 mg / ml a veces es esencial con el fin de segregar un mutante marcada completamente. - Si la cepa mutante muestra un perfil marcado a través de PCR, a continuación, volver a rayas o manchas de BG11 fresca + kanamicina (30 mg / ml) en placa de agar. Utilice esta cepa para generar el golpe de gracia sin marcar.
Nota: El protocolo puede ser utilizado para generar mutantes marcados con sólo un casete de resistencia a antibiótico es decir, mediante la sustitución de la NPT1 / cassette sacB con sólo el casete de NPT1 pUC18K 20 entre los fragmentos izquierdo y derecho..
5. Generación de mutantes Synechocystis sin marcar
- Establecer un cultivo fresco de la eliminatoria marcada por la inoculación de una vuelta completa del cells en 30-50 ml de medio BG11. Mantener el cultivo durante 2-3 días a 750 nm OD = 0,2 a 0,6.
- Centrifugadora de 10 ml del cultivo a 2.300 xg durante 5 minutos y descartar el sobrenadante. Lavar una vez con medio BG11.
Nota: No volver a suspender las células mediante agitación ya que esto puede resultar en la pérdida de pili, que son esenciales para la absorción de ADN. Resuspender las células mediante pipeteo suave. - Añadir BG11 a un volumen final de 200 l. La transferencia de células a un tubo de fondo redondo de 14 ml.
- Añadir 1 g de ADN plásmido a las células B y mezclar con unos golpecitos suaves.
- Se incuban las muestras durante 4-6 horas. Colocar los tubos en posición horizontal.
Nota: Las células se pueden mezclar brevemente tocando cada 1-2 horas, pero esto no es esencial. Las muestras pueden ser colocados en una incubadora de agitación, aunque esto no mejora la eficiencia. - Añadir 1,8 ml de medio BG11 e incubar las muestras para un total de 4 días con agitación. Este tiempo es suficiente para permitir la recombinación que se produzca en las múltiples copias cromosómicas.
- alícuotas de placa de la mezcla de transformación en placas de agar sacarosa BG11 / 5%. Plate 50 l, 10 l y 1 l por placa de agar. Si un césped colonia aparece en todas estas placas de agar diluyen la solución adicional y alícuota en placas frescas. Las colonias deben ser visibles después de aproximadamente 7 días.
- Parche 30-50 colonias individuales en BG11 + kanamicina (30 mg / ml) de agar placas primeras y BG11 / 5% placas de agar sacarosa segundo lugar, utilizando un palillo de dientes extremo romo. Cualquier bacteria que crecen en BG11 / 5% de sacarosa placas placas pero no BG11 + kanamicina son posibles golpes de gracia sin marcar. Las bacterias que crecen en ambas placas es probable que sean resistentes a la sacarosa debido a una mutación en el gen sacB.
- Verificar nocauts sin marcar utilizando los mismos cebadores y el método que se utilizó para comprobar los agujeros ciegos marcadas. Ej cpcC1C2for y cpcC1C2rev (Tabla 2) para la verificación de los octavos de final sin marcar cpcC1C2. Un golpe de gracia sin marcar mostrará una banda en un gel correspondiente t agarosao el tamaño de tipo salvaje, menos la región eliminada (Figura 2).
- Si la cepa muestra un perfil sin marcar mutante a través de PCR (paso 4.11.2) y electroforesis en gel (Figura 2), a continuación, volver a la racha en una placa de agar BG11 fresco sin antibióticos.
6. El almacenamiento a largo plazo de las cepas
- Establecer un cultivo fresco de la cepa mediante la inoculación de un bucle completo de las células en 30-50 ml de medio BG11. Mantener el cultivo durante 3-4 días a 750 nm OD = 0,4 a 0,7.
- Lavar las células una vez con BG11 y resuspender en ~ 2 ml de BG11.
- Añadir 0,8 ml de células concentradas a un tubo. A continuación, añadir 0,2 ml de 80% de glicerol esterilizado por filtración.
- Opcional: Añadir 0,93 ml de células concentradas a otro tubo. Añadir 0,07 ml de DMSO a este tubo.
PRECAUCIÓN: El DMSO es tóxico y debe ser manejado con una protección adecuada. - Almacenar ambos tubos a -80 ° C. Para reactivar las cepas quitar el tubo y raspar algunas células con un diente romorecoger en una placa de agar sin antibióticos. Consecutivas a cabo de manera normal utilizando un asa estéril.

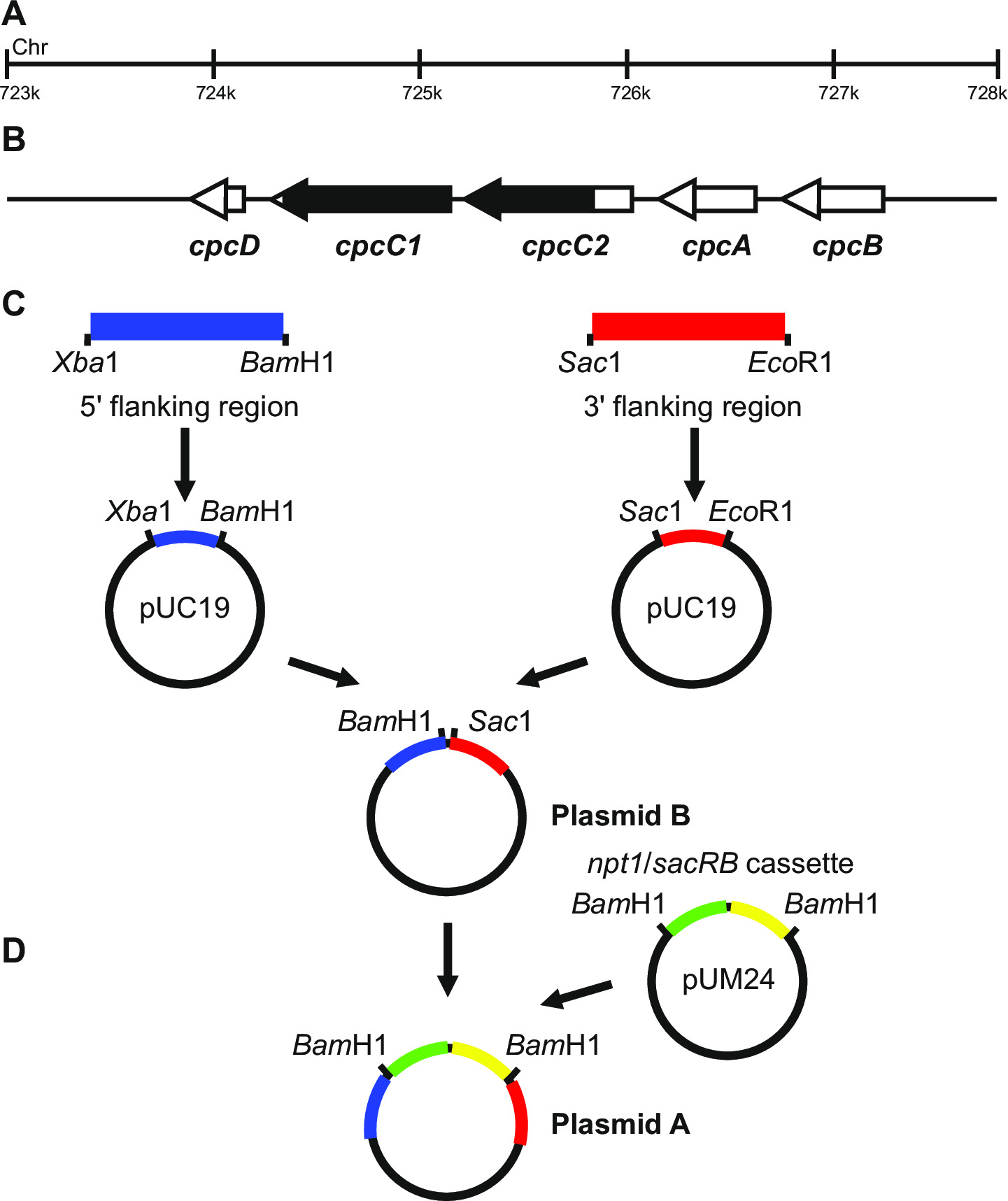
Figura 1: Construcción de plásmidos para la generación de knockouts marcados y sin marcar, por ejemplo cpcC1 y cpcC2 en Synechocystis (A) Región del genoma de Synechocystis donde se encuentran (B) y cpcC1 cpcC2 y los genes adyacentes.. Destacado en negro es la región del genoma a eliminar en el mutante. (C) Los sitios de genoma que son amplificadas por PCR. La región flanqueante (indicada en azul) y 3 '5 región flanqueante (indicado en rojo) se amplifican con los sitios de endonucleasas de restricción para la clonación en pUC19. El 5 '(o 3') región flanqueante se escinde de pUC19 e insertado en el pUC19 + 3 '(o 59;) que flanquean la región para generar plásmido El NPT1 / casete sacB de pUM24 se extirpa a través de la digestión Bam HI y se insertó entre el 5 'y 3' de acompañamiento regiones para generar el plásmido A. B. plásmido (D) Haga clic aquí para ver una más grande versión de esta figura.
{kind=link}
Resultados
Diseño plásmido es crítica para la generación exitosa de ambos mutantes marcados y sin marcar. Figura 1 da un ejemplo del plásmido A y B utiliza para generar un mutante de deleción en los genes Synechocystis cpcC1 y cpcC2 13. En cada caso, las regiones flanqueantes 5 'y 3' son de aproximadamente 900-1.000 pb. regiones que flanquean reducidos se pueden utilizar aunque el más pequeño que hemos ensayado con éxito ha sid...
Discusión
Los pasos más importantes en la generación de mutantes no marcados son: 1) Diseño de plásmido cuidado para asegurar que sólo se altera la región objetivo; 2) garantizar que las muestras permanezcan axénicos, especialmente cuando se cultivan en sacarosa; 3) placas células de marcado generación mutante transformada inicialmente en placas de agar BG11 que carecen de antibióticos, seguido de la adición de agar más antibióticos 24 horas más tarde; 4) cultivar marcado mutantes para 4 días completos antes de la ...
Divulgaciones
The authors declare that they have no competing financial interests.
Agradecimientos
Estamos muy agradecidos a la Asociación de Servicios Medioambientales Fiduciario para la Educación, la biología sintética en el fondo de la Biología Sintética Cambridge y el Ministerio de Justicia Social y Empoderamiento, Gobierno de la India, por el apoyo financiero.
Materiales
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
Referencias
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis?. Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados