
Bu içeriği görüntülemek için JoVE aboneliği gereklidir. Oturum açın veya ücretsiz deneme sürümünü başlatın.
Method Article
Model siyanobakteri Türlerinde İşaretli ve belirtisiz Mutants Üretimi
Bu Makalede
Özet
Introducing multiple genomic alterations into cyanobacteria is an essential tool in the development of strains for industrial and basic research purposes. We describe a system for generating unmarked mutants in the model cyanobacterial species Synechocystis sp. PCC6803 and marked mutants in Synechococcus sp. PCC7002.
Özet
Cyanobacteria are ecologically important organisms and potential platforms for production of biofuels and useful industrial products. Genetic manipulation of cyanobacteria, especially model organisms such as Synechocystis sp. PCC6803 and Synechococcus sp. PCC7002, is a key tool for both basic and applied research. Generation of unmarked mutants, whereby chromosomal alterations are introduced into a strain via insertion of an antibiotic resistance cassette (a manipulatable fragment of DNA containing one or more genes), followed by subsequent removal of this cassette using a negative selectable marker, is a particularly powerful technique. Unmarked mutants can be repeatedly genetically manipulated, allowing as many alterations to be introduced into a strain as desired. In addition, the absence of genes encoding antibiotic resistance proteins in the mutated strain is desirable, as it avoids the possibility of 'escape' of antibiotic resistant organisms into the environment. However, detailed methods for repeated rounds of genetic manipulation of cyanobacteria are not well described in the scientific literature. Here we provide a comprehensive description of this technique, which we have successfully used to generate mutants with multiple deletions, single point mutations within a gene of interest and insertion of novel gene cassettes.
Giriş
Siyanobakteriler yeryüzünde hemen her doğal ortamda bulunan bakteri evrimsel eski ve çeşitli filum vardır. Deniz ekosistemlerinin onlar özellikle bol ve karbon fiksasyonu 1 yaklaşık yarısında, okyanuslardaki azot fiksasyonu 2 ve hidrokarbon üretimi 3 ton yüzmilyonlarca çoğunluğu yıllık muhasebe, birçok besin döngüleri önemli bir rol oynamaktadır. Kloroplastlar, ökaryot alglerin ve bitkilerde fotosentez için sorumlu organel bir konak organizmada 4 tarafından yuttu bir siyanobakteriye evrimleşmiş olması muhtemeldir. Siyanobakteri tesislerinde muhafaza birçoğu fotosentez, elektron taşıma 5 ve biyokimyasal yollar çalışma için faydalı model organizmalar kanıtlamıştır. Ilave siyanobakteriler giderek dolayı hi, elektrik 7 ve endüstriyel bileşikleri, 6 biyoyakıt, gıda üretimi için 8 kullanılmaktadırsu ve CO 2 ghly verimli dönüşüm güneş enerjisi 9 kullanarak biyokütle. Birçok tür potansiyel tarımsal üretim etkilemeden büyük ölçekli yetiştirilen olabileceğini siyanobacteria düşündüren, en az besin ve deniz suyu olmayan ekilebilir arazi üzerinde yetiştirilebilir. Bazı türler de mantar önleyici, anti-bakteriyel ve anti-kanser bileşikleri 10,11 gibi doğal ürünler, kaynaklarıdır.
mutantlar oluşturmak için yeteneği endüstriyel amaçlı suşları geliştirilmesi için siyanobakteri fotosentez, biyokimya ve fizyoloji, anlamanın anahtarı ve esastır. yayınlanan çalışmaların çoğunluğu genetik ilgi siteye bir antibiyotik direnç kasetinin sokulmasıyla suşları modifiye oluşturun. Bu sadece birkaç antibiyotik direnç kaseti siyanobakteriler kullanım için uygun olarak bir hücreye dahil edilebilir mutasyon sayısını sınırlar. Antibiyotik yeniden kazandıran genler ihtiva eden suşlardirencin biyoyakıt ve diğer düşük değerli ürünler 12 üretmek için sadece maliyet-etkili bir araç olması muhtemeldir açık havuzlarda sanayi üretimi için kullanılamaz. işaretsiz mutantların nesil bu kısıtlamaların üstesinden. Işaretsiz mutantlar kasten dahil olmadıkça, hiçbir yabancı DNA'yı içerir ve birden çok kez manipüle edilebilir. Nedenle arzu edilen bir suş olarak birçok değişiklik elde etmek mümkündür. Buna ek olarak, modifikasyon bölgesinden akım-aşağı genin kutup etkisi organizma 13 daha hassas modifikasyonu sağlayan en aza indirilebilir.
Silinecek gene komşu siyanobakteri kromozomda bölgelere İki DNA parçası aynı içeren mutant suşları, intihar plasmidleri oluşturmak için ilk inşa edilir (5 've 3' yan bölge olarak da adlandırılır). Iki gen daha sonra bu yan bölgeler arasına yerleştirilir. Bunlardan biri, bir antibiyotik direnç proteini kodlar İkinci SacB, hangi eşya kodlarlevansükraz uces, bir bileşik sukroz duyarlılık kazandıran. Işlemin birinci aşamasında, işaretlenmiş mutantlar, bazı yabancı DNA'yı ihtiva eden, yani suşlar oluşturulur. plazmid yapılarında siyanobakteriyel hücreleri ile karıştırılır, ve DNA organizmanın doğal olarak alınır. Transformantlar uygun antibiyotiği ve PCR ile teyit mutant genotip ihtiva eden agar plakaları üzerinde büyüme ile seçilir. İntihar plazmidler ilgi suşu içinde çoğaltma yapamaz. Bu nedenle, herhangi bir antibiyotik dirençli koloniler ilgi konusu genin kromozom içine yerleştirilir, böylece bir rekombinasyon olayı ile sonuçlanacaktır. İşaretlenmemiş mutantlan oluşturmak için, belirgin bir mutantı sadece 5 've 3' yan bölgelerini içeren ikinci bir intihar plasmidi ile karıştırılır. Yabancı DNA'nın sokulması gereklidir, ancak, söz konusu DNA fragmanları arasındaki yerleştirilen ilgili genleri içeren bir kaset ile yandan kuşatma bölgeleri, 5 've 3' içeren bir plazmid, kullanılabilir. Selection sukroz içeren agar plakaları üzerinde büyüme yoluyladır. SacB gen ürünü ifade edildiğinde sükroz hücreleri için öldürücü olduğu için, hayatta sadece hücreleri sükroz duyarlılığı geni, antibiyotik direnç genine ek olarak, takım rekombine edilmiş ve böylece ikinci bir rekombinasyon olayı hangi olanlar vardır kromozom ve plazmid üzerine. rekombinasyonel değişiminin bir sonucu olarak, kuşatıcı bölge ve bunların arasında herhangi bir DNA kromozom içine yerleştirilir.
Biz başarıyla Synechocystis sp aynı suşu birden kromozomal mutasyon üretmek için bu yöntemleri kullandık. (Bundan sonra Synechocystis olarak anılacaktır) PCC6803 13,14 çıkar 13 bir gen içine ve gen kasetleri ekspresyonu için tek nokta mutasyonların katılması için. İşaretlenmemiş tırnakları üretilmesi Synechocystis 15,16, detaylı yöntemde çalışmalarımızda önce gösterilmiştir birlikte, destekliKritik adımlar görsel sunum, kamuya açık değildir. Biz de başka bir model siyanobakterisine Synechococcus sp belirgin Knockouts üretimi için aynı yöntemi uyguladık. PCC7002 (bundan sonra Synechococcus olarak anılacaktır). Bu protokol mutantlar ve doğrulama ve bu suşları saklamak için hızlı bir protokol oluşturmak için bir açık, basit bir yöntem sağlar.
Protokol
Kültür Medya 1. Hazırlık
- Castenholz, 1988 17'ye göre BG11 orta hazırlayın.
- Elemanları ve demir stoku (Tablo 1) iz, 100x BG11 stok çözümleri hazırlayın.
- Fosfat stok ayrı çözeltilerinin hazırlanması, Na 2 CO 3 hazır, N - [Tris (hidroksimetil) metil] -2-aminoetansülfonik asit (TES), tampon ve NaHCO 3 (Tablo 1).
- Fosfat ve Na 2 CO 3 stokları otoklavlayın. -Filtre sterilize 0.2 um filtreleri ile NaHCO 3 TES tampon ve.
- su 976 ml, 100 x BG11 10 ml demir stokunun 1 eser elementler ve 1 ml birleştirerek BG11 hazırlamak ve çözelti otoklav. Bu çözelti, oda sıcaklığına kadar soğuduktan sonra, fosfat, 1 mL stok, Na 2 CO 3, 1 mL stok NaHCO 3 10 ml.
- BG11 katı ortam, agar 15 g ve bir FLA 700 ml su ilavesk. İkinci bir şişeye, Na 2 S, 3 g ilave 2 O 3, su 226 mi, 10 100 x BG11 ml 1 mi iz elementleri ve demir, 1 mL stok. Her iki çözüm otoklavlayın. Bu çözelti oda sıcaklığına soğuduktan sonra, bunları birleştirmek ve 1 fosfat stok ml, Na 2 CO 3, 1 mL stok, TES tamponu 10 ml ve NaHCO 3 10 ml.
Not: Çözümler, belirli tuzlarının çökelmesini önlemek için ayrı hazırlanır.
- sakaroz seçimi için, (ağırlık / hacim) sukroz çözümü% 50 hazırlar. Filtre 0.2 um filtre ile çözelti sterilize ve BG11 /% 5 sükroz plakaları üretmek için (BG11 900 ml% 50 sukroz 100 mi) BG11 ekle.
Not: BG11 /% 5 sakaroz agar plakalarına NaHCO 3 katmayın. Normal olarak Na 2 CO 3 ekleyin. - Synechococcus kültürlenmesi için 10 mi, 1 M 4- (2-hidroksietil) piperazin-1-etansülfonik asit, N, ekleyin - (2-hidroksietil) piperazin-NR42 - (2-etansülfonik asit) (HEPES) ve BG11 ortamı 1 L vitamin B 12 (Tablo 1) 1 mi.
Not: ticari olarak temin edilebilir BG11 ortamında kültürlenmiştir suşlarının transformasyonu, burada açıklanan BG11 ortam tarifleri göre önemli ölçüde daha az etkili olduğu ve bu nedenle de tavsiye edilmez.
Siyanobakteri Suşlarının 2. Büyüme
- Kültür 50 ml bir maksimum ses 100 ml'lik koni şişelere, şekil değişikliklerinin 120 rpm'de çalkalanır. Gaz değişimi sağlamak için levhanın yan Parafilm ve delinme üç küçük delikli BG11 plakaları Seal. 20-40 ľmol fotonlar m arasında bir ışık yoğunluğu bir foto-biyo floresan ampuller altında 30 ° C'de tüm suşları inkübe -2 sn -1.
- En iyi steril teknikleri kullanın. Laminer akış kaputu tüm Siyanobakteri suşları taşıyınız.
Not: Bu suşlar kolay olan Kirlenmiş edilebilir ortam içeren sukroz ile kültürlenir özellikle önemlidirdeğerlendirecektir.
Plazmid Yapılar 3. Nesil
- Bu Primer3 (http://frodo.wi.mit.edu/primer3/), astar tasarım yazılımı kullanarak gerekli kısıtlama enzim sitelerinin dahil olmak üzere primer, tasarımı setleri iki ~ 1 kb bölge 5 've 3' yükseltmek için ilgili gen. Cyanobase ile siyanobakteri türlerinin genom sekansı bakınız (http://genome.kazusa.or.jp/cyanobase). Burada kullanılan bütün primerler için Tablo 2'ye bakınız. tasarlarken primerler aşağıdaki faktörleri göz önünde bulundurun:
- Amplifiye bölgeleri mutasyona edilecek geni, örneğin Şekil 1, 5 've 3' bölgelerini içerir emin olun.
- antisens ve kodlayıcı olmayan RNA'lar, istenmeyen mutasyon bilmek intergenik bölgeleri mutasyona verme. Synechocystis'te mutantların nesil için, Mitschke ve ark belgelenen transkripsiyon başlangıç siteleri listesine bakınız. 2011 18, antisens mutasyon önlemek içinveya RNA'lar kodlayıcı olmayan.
- Tercih komşu bölgeleri klonlama müdahale edebilir Escherichia coli'de bu genlerin sentezlenmesi bitişik genlerin bütün açık okuma çerçevesini kapsamamaktadır zaman.
- üreticinin talimatlarına uygun olarak, yüksek doğrulukta bir DNA polimerazı kullanılarak PCR ürünlerini yükseltin.
Not: Bizim tecrübelerimize göre bu enzim az hata üretir.- HF tampon ve DMSO 0, 1.5 veya 3 ul birini içeren 50 ul PCR reaksiyonları ayarlayın. reaksiyon başına genomik DNA, 100 ng kullanımı. 72 ° 'lik bir son uzatma adımı, ardından 30 saniye, 10 saniye, 30 saniye için 30 saniye için 67 ° C, 72 ° C, 98 ° C 35 tur için 98 ° C bir başlangıç denatürasyon basamağından oluşan bir programı kullanarak 5 dakika bekletilmiştir. Bu genellikle tutarlı ürünler verir.
- jel elektroforez ile, doğru boyut endonükleaz enzimleri ile sindirilmiş PCR ürünleri ve örnekleri edin. (Ağırlık / hacim) agaroz jeli% 0,02 ihtiva eden% 1 oranında çalıştır100 V, 45 dakika boyunca (h / h) etidyum bromid
DİKKAT: Etidyum bromür potansiyel bir mutajen ve uygun koruma ile ele alınmalıdır. - üreticinin talimatlarına göre bir DNA saflaştırma kiti kullanılarak PCR ürünleri saflaştırılır. Ayrıca agaroz jellerinden kesilir parçaları dahil olmak üzere, plazmid parçalarının saflaştırılması için bu kiti kullanmak. su 14 ul saflaştırılmış DNA Zehir.
- klonlama adımları için, üreticinin talimatlarına uygun olarak 30 ul toplam hacim içinde> 1 saat süreyle 37 ° C'de sınırlama endonükleaz Reaksiyon karışımları inkübe edin.
- Ligasyon adımları için, arıtılmış, sindirilmiş plasmid 5 ul saflaştırılmış sindirilmiş geçmenin 12 ul tampon 2 ul ve ligazm 1 ul ihtiva eden 20 ul toplam hacim içinde> 1 saat süre ile, oda sıcaklığında, önceki ligate DNA fragmanları.
- Şu yönteme göre Escherichia coli DH5α transformant hücrelerin hazırlanması.
- Bir gecede E. büyütün E. coli 10 ml Luria Bertani (LB) medya kültürü.
- Bir gecelik kültürün 1 ml 6 ml 1 M MgCI2 (Tablo 1) ihtiva eden bir 1 litrelik koni şeklindeki bir şişede, 400 ml LB inoküle.
- Yaklaşık 4 saat veya OD 600nm 0.4-0.6 ulaşana kadar 220 rpm'de 37 ° C'de kültür büyütün.
- 1 saat boyunca buz üzerinde hücreleri yerleştirin.
- 10 dakika boyunca 2800 x g'de santrifüje 4 ° C 'de pelet hücreleri.
- 160 ml çözelti A (Tablo 1) supernatant ve tekrar süspansiyon çıkarın ve 20 dakika boyunca buz üzerinde inkübe edilir.
- 10 dakika boyunca 2800 x g'de santrifüje 4 ° C 'de pelet hücreleri.
- 4 mL çözelti A + gliserol (Tablo 1) supernatant ve tekrar süspansiyon çıkarın.
- -80 ° C'de, sıvı N2, deposunda dondurma, 50 ul alikotları hazırlayın.
- yetkin hücre 50 ul ligasyon karışımı 5 ul karıştırın ve buz üzerinde 1 saat süre ile inkübe edilir.
- Isı 90 sn, follo 42 ° C'de hücreler şok2 dakika için buz üzerinde kuluçkalama ile evli.
- LB ortamı (Tablo 1) 950 ul ilave edin ve 1 saat süreyle 37 ° C'de inkübe edin.
- Kısım 50, uygun antibiyotik ile plakalar üzerinde 200 ul, ampisilin (100 ug / ml) ve / veya kanamisin (ug / ml 30 ug) ya da.
DİKKAT: kanamisin ve ampisilin Hem toksik ve uygun koruma ile ele alınmalıdır. - Seçim ve uygun antibiyotik ile aşılanmış 2 ml LB ortamında tek koloniler kuluçkaya yatmaktadır.
- üreticinin talimatlarına göre bir miniprep plazmid saflaştırma kiti kullanılarak tüm plazmidler arındırın.
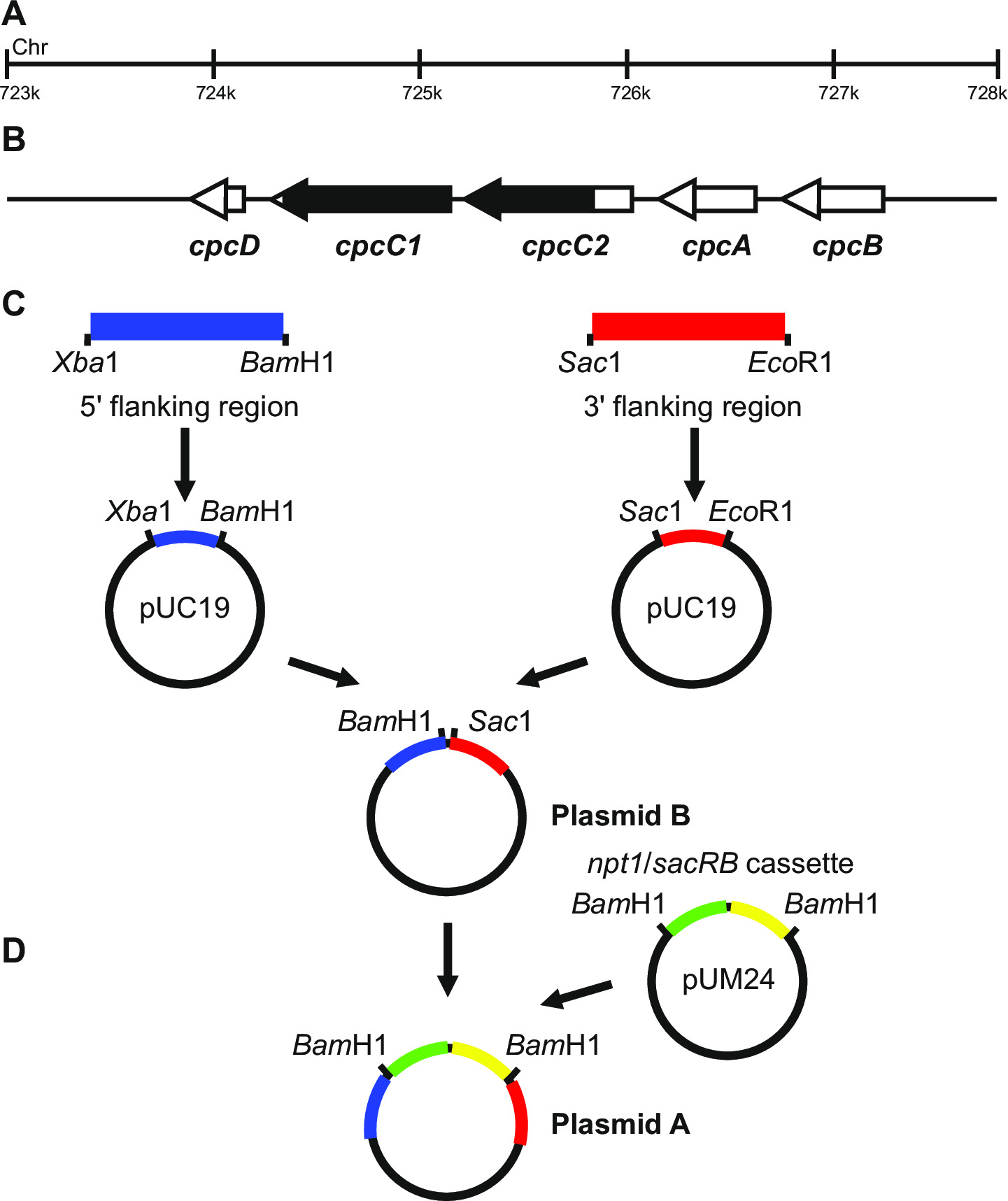
- Aşağıdaki adımlara uygun olarak, cpcC1C2 genleri nakavt için özel bir örnek olarak, plazmidler oluşturur.
- Primerler cpcC1C2leftfor ve cpcC1C2leftrev (Tablo 2, adım 3.2) kullanılarak 1.012 bp'lik 5 'yan bölge (sol kısmı) yükseltin. PCR reaksiyonunun bir miktar çıkarın ve olmadığını doğrulamakDoğru boyutlu ürünleri jel elektroforezi (aşama 3.3) ile amplifiye edilmiştir. Xba I ve Bam HI (adım 3.5) ile bu fragmanını ve pUC19 sindiremez.
- her iki preparatın (aşama 3.4), bağlanır (adım 3.6) saflaştınlır (adım 3.7) ve miniprepleri (aşama 3.8) ile plazmid arıtma için ayrı koloniler ampisilin (100 ug / mi) ile dört adet 2 mi LB sıvı kültürler ayarlamak dönüşümü.
- Xba I / BamHI sindirimi ve jel elektroforezi (aşama 3.3) ile pUC19 içine fragmanının sokulması için kontrol edin. 2660 bp ve 1012 bp bantları plazmid içine insert doğru giriş göstermektedir.
- Primerler cpcC1C2rightfor ve cpcC1C2rightrev (Tablo 2, adım 3.2) kullanılarak 1.016 bp 3 'yan bölge (sağ kısmı) yükseltin. PCR reaksiyonunun bir miktar çıkarın ve doğru boyut ürünleri jel elektroforezi (aşama 3.3) ile amplifiye edilmiş olup olmadığını teyit etmektedir. (St Sac I ve EcoRI ile fragmanını ve pUC19 DigestEP 3.5).
- her iki preparatın (aşama 3.4), bağlanır (adım 3.6) saflaştınlır (adım 3.7) ve miniprepleri (aşama 3.8) ile plazmid arıtma için ayrı koloniler ampisilin (100 ug / mi) ile dört adet 2 mi LB sıvı kültürler ayarlamak dönüşümü.
- Sac I / EcoRI sindirimi (aşama 3,5) ve jel elektroforezi (aşama 3.3) ile pUC19 içine fragmanının sokulması için kontrol edin. 2,660 bp ve 1.016 bp bantlar plazmide ucun doğru bir giriş göstermektedir.
Not: pUC19 içine bölgenin 3 'klonlanması için bölge ve Sac I / Eco RI 5' klonlanması için Xba I / BamHI siteleri mümkün kullanılır. Mümkünse, her zaman, daha sonra klonlama adımları gerçekleştirmek için daha kolay olmasını sağlamak için 5 'bölge ya da 3 ileri primeri' bölgesi için ters primer bir BamHI sitesi bulunmaktadır. - Dizisi, örneğin ekleme sitesi, M1 kapsayan primerler kullanılarak doğru olup olmadığını hem ekler dizisi belirlemek için3 İleri ve M13 Ters (Tablo 2). dizisi hata kuşatan bölgelere içine sağlamak için doğru olmalıdır.
- XbaI / Bam HI sindirimi yoluyla pUC19 sol fragmanı tüketim. XbaI / Bam HI (adım 3.5) ile pUC19 + sağ fragmanı sindiremez.
- Bir neşter bıçak kullanarak DNA'nın eksizyonu üzerinden bir agaroz jeli (adım 3.3) den 1012 bp sol fragmanı ve 3676 bp pUC19 + sağ fragmanı arındırın.
- her iki preparatın (aşama 3.4), bağlanır (adım 3.6) saflaştınlır (adım 3.7) ve miniprepleri (aşama 3.8) ile plazmid arıtma için ayrı koloniler ampisilin (100 ug / mi) ile dört adet 2 mi LB sıvı kültürler ayarlamak dönüşümü.
- Xba I / BamHI sindirimi (aşama 3,5) ve jel elektroforezi (aşama 3.3) ile pUC19 + sağ fragmanına fragmanının sokulması için kontrol edin. 3,676 bp ve 1,012 bp bantlar plazmide insertin doğru takılmasını (plazmid B olarak bakınız) göstermektedir.
- Tüketim BamHI sindirimi yoluyla pUM24cm 19 arasında NPT1 / sacB kaseti. Bam HI ile sindirmek plazmid B (adım 3.5).
Not: pUM24cm kloramfenikol direnci kazandıran bir proteini şifreleyen yana NPT1 / sacB kaseti agaroz jellerinden saflaştırılmıştır olmak zorunda değildir. Koloniler LB yetiştirilen nedenle eğer / ampisilin / kanamisin agar plakaları dirençli koloniler yol açacaktır tek olası kombinasyonu plazmid B. içine NPT1 / sacB kaset birleşme olduğunu - her iki preparatın saflaştırmak (aşama 3.4), bağlanır (adım 3.6), (aşama 3.7) ve plazmid arıtma için ayrı koloniler ampisilin (100 ug / ml) ve kanamisin (30 ug / mi) ile dört adet 2 mi LB sıvı kültürler ayarlamak dönüşümü miniprep yoluyla (adım 3.8).
- BamHI sindirimi (aşama 3,5) ve jel elektroforezi (aşama 3.3) ile plazmid B içine NPT1 / sacB kasetinin yerleştirilmesi için kontrol edin. 4688 bp ve 3894 bp bantları th doğru takılmasını gösterire plazmid (plazmid A olarak bu bakınız) takın.
- Seçenek olarak ise, kör uç plazmid B. sol ve sağ parçalar arasında farklı bir kısıtlama endonükleaz sitesine NPT1 / sacB kaseti ve klon NPT1 / sacB kaseti, sol ve sağ parçalar arasında klonlanmıştır gerekir.
Not: Yabancı kasetinin ekspresyonu ve ardından gerekli olması durumunda bu, Bu plazmid, işaretlenmemiş nakavt adımda kullanılan plazmid B. sol ve sağ fragmanlar arasında yerleştirilmelidir.
Belirgin Synechocystis ve Synechococcus Mutants 4. Nesil
- BG11 ortamının 30-50 ml hücre tam bir döngü aşılayarak taze kültür ayarlayın. OD 750nm için 2-3 gün boyunca kültür büyütün = 0.2 0.6.
Not: Genellikle tek tek koloniler ışık düşük seviyelerde bile, fotoindirgeyici ve seçim sağlanabilir, aşılama ve tek tek hücrelerin maruz kullanmak için çok küçükHafif dirençli mutantların için. - Santrifüj 01-02 Mayıs dakika boyunca 2.300 xg'de kültür ml süpernatant atın. Bu hücrelere zarar verebilir> 2.300 xg'de herhangi Siyanobakteri kültürleri santrifüj etmeyin. BG11 maddesi ile pelet yıkayın.
Not: Bu DNA alımı için gerekli olan pili kaybına neden olabilir vorteks hücreleri tekrar süspansiyon etmeyin. Nazik pipetleme hücreleri tekrar süspansiyon. - 100 ul'lik nihai bir hacme kadar BG11 orta ekleyin. 14 ml yuvarlak tabanlı bir tüpe hücreleri aktarın.
- hücrelere plazmid 1 ug ekleyin ve hafifçe vurmak suretiyle karıştırılır. Not: Tercihen plazma> 100 ng / ul'lik bir konsantrasyonda olmalıdır, ancak bundan daha düşük konsantrasyonlarda başarılı bir transformasyonu için yeterli.
- inkübatör yatay tüpler uzandı. 4-6 saat boyunca kültürlere inkübe edin.
Not: Hücreler kısaca her 1-2 saat dokunarak karıştırılabilir ancak bu şart değildir. Örnekler yerleştirilebilirbu anlamlı verimliliğini artırmak olmasa da inkübatör sallayarak. - antibiyotikler olmadan BG11 agar plakaları üzerinde hücre kültürü / plazmid DNA karışımı hacimde yayıldı. Tipik olarak 20 ul ve 80 ul tam bölünen miktarları, ayrı levhalar üzerinde yayılır.
- agar plaka: ~ 24 saat sonra, (ağar 0.12 g / ml kanamisin, 100 mg, 100 ul, 20 ml başına) kanamisin ihtiva eden su içinde% 0.6 2.5-3 ml agar çözeltisi ilave edilir. ~ 42 ° C, bu çözelti soğutulur ve agar plaka kenarına ekleyin. Plaka böylece çözüm yüzeyinde daha 'üst agar' tabaka oluşturur eğin.
- bir zaman süreler agar inkübe edin. Koloniler yaklaşık 7 gün sonra görünür olmalıdır.
Not: Ağar plakalar bir inkübatör yüksek 3 yığılmış olabilir. Tipik koloni yüzlerce dönüşüm başına elde edilir. - agar plakaları BG11 + kanamisin (30 ug / ml) üzerine leke yapmayan tek tek koloniler. 6 sektöre agar plaka bölün ve çizgi bir kör uç kürdan dışarı kullanmakher sektör üzerinde koloniler. tek koloniler elde edilmesi transformantların önemli, sadece büyüme değil.
- Üreticinin talimatlarına uygun olarak, Taq DNA polimeraz kullanılarak PCR ile belirgin nakavt teyit edin. Reaksiyon başına MgCI2 (25mM) 2 ul ekle.
- hücrelerin küçük bir oranını kaldırmak ve 50 ul su ve ~ 20 425-600 um cam boncuklar ihtiva eden bir tüp içine aktarılır. ~ 2,000 rpm'de 5 dakika için bir vibratör de çalkalanır. 5 dakika boyunca 15.700 x g'de santrifüje ve 50 ul PCR reaksiyon başına süpernatan 5 ul kullanın.
Not: çözüm tekrar süspansiyon etmeyin. Hücre enkaz tüpün dibinde kalması gerekiyor.
- hücrelerin küçük bir oranını kaldırmak ve 50 ul su ve ~ 20 425-600 um cam boncuklar ihtiva eden bir tüp içine aktarılır. ~ 2,000 rpm'de 5 dakika için bir vibratör de çalkalanır. 5 dakika boyunca 15.700 x g'de santrifüje ve 50 ul PCR reaksiyon başına süpernatan 5 ul kullanın.
- mutantlar doğrulamak
- (Örneğin Primer3 gibi) Primer tasarımı yazılımını kullanarak nakavt bölgesini kapsayan tasarım primerler. boşaltma bölgesi yaklaşık 200 bp'lik iki tarafında başlayan astar tasarımı.
Not: cpcC1C2 mutantını doğrulamak için primerler Tablo 2'de özetlenmiştirve cpcC1C2for ve cpcC1C2rev olarak adlandırılır. - Bir ardından 2 dakikada, 1 dakikada, 1 dakika boyunca 60 ° C, dizinin kb'si başına 1 dakika için 72 ° C, 95 ° C 35 tur için 95 ° C bir başlangıç denatürasyon basamağından oluşan bir programı kullanarak ürünleri yükseltin 5 dakika boyunca 72 ° C'de son uzatma adımı. vahşi tip kontrolü içerir. Bu genellikle tutarlı ürünler verir.
- jel elektroforez ile genotip doğrulayın. İşaretli nakavt transformantlar ~ 4 kb (sol ve sağ parçaları artı NPT1 / sacB kaset hem 0.2 kb) ve yabani tip bant yokluğunda (Şekil 2) bir grup gösterecektir.
Not: Bazı durumlarda, ~ 4 kb bandı nedeniyle, bu PCR ürününün büyük bir boyuta belirgin mutant gözlenmez. Yabani tip beklenen boyutuna karşı gelen bir bant daha sonra gözlenmez, ancak, tipik olarak, bu suş belirgin bir nakavt.
- (Örneğin Primer3 gibi) Primer tasarımı yazılımını kullanarak nakavt bölgesini kapsayan tasarım primerler. boşaltma bölgesi yaklaşık 200 bp'lik iki tarafında başlayan astar tasarımı.
- Bir vahşi tip bandı hala mevcut ise, o zaman bir gerginlik-Streak yenidenTaze BG11 + kanamisin (30 mg / ml) agar plaka ve PCR tekrarlayın. Resim vahşi tip bandı PCR reaksiyonunda gözlenmiştir, böylece mutant ayrılmış kadar yeniden çizgiler tekrarlayın.
Not: 50 ug / ml'lik bir konsantrasyona kadar kanamisin miktarının artırılması, daha sonra 100 ug / ml tam olarak belirgin bir mutant ayırmak için bazen gereklidir. - suşu PCR ile belirgin bir mutant profilini gösterir, sonra yeniden çizgi taze BG11 + kanamisin (30 ug / ml) agar plaka üzerinde. işaretsiz nakavt oluşturmak için bu gerginlik kullanın.
Not: protokolü, sadece bir antibiyotik direnç kaseti ile işaretlenmiş mutantlarını üretmek için kullanılabilir, örneğin, sol ve sağ fragmanları arasındaki pUC18K 20 sadece NPT1 kasetiyle NPT1 / sacB kaseti yerine..
Plakasız Synechocystis Mutants 5. Nesil
- c tam bir döngü inoküle ile işaretlenmiş nakavt taze kültür ayarlamaBG11 ortamının 30-50 ml arşın. OD 750nm için 2-3 gün boyunca kültür büyütün = 0.2 0.6.
- Santrifüj 2300 xg'de kültür 10 mi, 5 dakika ve süpernatan atılır. BG11 maddesi ile yıkanır.
Not: Bu DNA alımı için gerekli olan pili kaybına neden olabilir vorteks hücreleri tekrar süspansiyon etmeyin. Nazik pipetleme hücreleri tekrar süspansiyon. - 200 ul bir son hacme BG11 ekleyin. 14 ml yuvarlak tabanlı bir tüpe hücreleri aktarın.
- hücrelere plazmid B DNA 1 ug ekleyin ve hafif dokunarak karıştırın.
- 4-6 saat boyunca örnekleri inkübe. yatay tüpler aşağı yatırın.
Not: Hücreler kısaca her 1-2 saat dokunarak karıştırılabilir ancak bu şart değildir. Bu verimliliği artırmak olmamakla birlikte örnekler bir çalkalama inkübatöründe yerleştirilebilir. - çalkalanarak 4 günlük bir toplam numune BG11 ortamı 1,8 ml ilave edilir ve inkübe edilir. Bu rekombinasyon birden kromozomal kopya halinde yapılmasına izin vermek için yeterli bir süredir.
- BG11 /% 5 sakaroz agar plakaları üzerinde dönüşüm karışımının plaka alikotları. Plaka 50 ul, 10 ul ve agar plaka başına 1 ul. Bir koloni çim tüm bu agar plakaları taze plakaları üzerinde daha fazla çözüm ve kısım sulandırmak görünürse. Koloniler yaklaşık 7 gün sonra görünür olmalıdır.
- kör bir uç kürdan ile birinci agar plakaları BG11 + kanamisin (30 ug / ml) ile 30-50 ayrı ayrı koloniler, ikinci BG11 /% 5 sükroz agar yama. BG11 /% 5 sukroz plakalar üzerinde büyüyecek ama kanamisin değil BG11 + plakaları potansiyel işaretsiz tırnakları olan herhangi bir bakteri. Her iki levha üzerinde büyüyen bakteriler bağlı sacB genindeki bir mutasyon dirençli sükroz olması muhtemeldir.
- İşaretli Knockouts kontrol etmek için kullanılan aynı primerler ve yöntemi kullanarak işaretsiz Knockouts doğrulayın. Örneğin cpcC1C2for ve cpcC1C2rev (Tablo 2) cpcC1C2 işaretsiz nakavt doğrulamak için. Işaretlenmemiş bir nakavt bir agaroz jel gelen t bir grup gösterecektiro vahşi tip boyutu eksi silinmiş bölge (Şekil 2).
- Suşu işaretlenmemiş bir mutant PCR ile profil (adım 4.11.2) ve jel elektroforezi (Şekil 2) gösteriyorsa, o zaman yeniden çizgi taze BG11 agar plakası üzerinde antibiyotik olmadan.
Suşlarının 6. Uzun süreli depolama
- BG11 ortamının 30-50 ml hücre tam bir döngü aşılayarak soyunun taze kültürüne ayarlayın. OD 750nm 3-4 gün süreyle kültür büyütün = 0.4 0,7.
- BG11 ~ 2 ml BG11 ve tekrar süspansiyon ile bir kez hücreleri yıkayın.
- Bir tüpe konsantre edildi hücreleri 0.8 ml ilave edilir. Daha sonra% 80 filtreyle sterilize gliserol, 0.2 ml ekleyin.
- İsteğe bağlı: başka bir tüpe Yoğunlaşan hücreler, 0.93 ml ilave edilir. Bu tüp DMSO 0.07 ml ilave edilir.
UYARI: DMSO toksik ve uygun koruma ile ele alınmalıdır. - -80 ° C 'de her iki tüpü saklayın. suşları tüpü çıkarın ve künt bir diş ile bazı hücreleri kazımak canlandırmak içinantibiyotikler olmadan bir agar plaka üzerine almak. Normal olarak steril bir döngü kullanarak dışarı Streak.

Şekil 1: işaretlenmiş ve işaretlenmemiş tırnakları, Synechocystis örneğin cpcC1 ve cpcC2 üretimi için plazmid yapımı (B) cpcC1 ve cpcC2 ve bitişik genlerin bulunduğu Synechocystis genomunun (A) 'bölgesi.. mutant Silinecek genomun bölge siyah olduğunu vurguladı. PCR ile amplifiye edilmiş genom (C) Yer. 5 '(mavi renkte gösterilir) yan bölgesi ve 3' (kırmızı ile gösterilmiştir) kuşatıcı bölge pUC19 klonlama için kısıtlama endonükleaz siteleri ile amplifiye edilir. komşu bölge 5 '(ya da 3'), pUC19 + 3 '(ya da 5 içinde pUC19 üzerinden kesilir ve sokulur9; plazmid B. (D) pUM24 gelen NPT1 / sacB kaset Plazmid A. oluşturmak için bölgelere komşu 5 've 3' arasında Bam HI sindirimi yoluyla çıkarıldı ve sokulur oluşturmak için bölge plazmid yan) bir büyük görmek için tıklayınız Bu rakamın sürümü.
{kind=link}
Sonuçlar
Plazmid tasarımı hem de işaretlenmiş ve işaretlenmemiş mutantların başarılı üretimi için büyük önem taşımaktadır. Şekil 1 plazmid bir örnek verir ve oda Synechocystis genlerinin cpcC1 ve cpcC2 13 bir iptal mutantının üretimi için kullanılır. Her iki durumda da, 5 've 3' komşu bölgeleri yaklaşık 900-1,000 bp. başarılı trialed en küçük yaklaşık 500 bp'lik bir olmasına rağmen azaltılm...
Tartışmalar
işaretsiz mutantların nesil en kritik adımlar şunlardır: 1) dikkatli plazmid tasarımı sadece hedeflenen bölge değişmiş sağlamak; 2) numuneler sukroz üzerine, özellikle kültür aksenik kalmasını sağlamak; 3) ile kaplanmış, ilk 24 saat sonra ağarı artı antibiyotik ilave edilir antibiyotikler, eksik BG11 agar plakaları üzerine işaretlenir mutant üretimi için hücrelerin transforme; 4) kültür BG11 artı sakaroz agar plakaları üzerinde kaplama öncesinde 4 tam gün mutantlar işaretlenmiş: 5)...
Açıklamalar
The authors declare that they have no competing financial interests.
Teşekkürler
Biz Çevre Hizmetleri Birliği Eğitim Vakfı'nın mali destek için Cambridge SynBio fonda Sentetik Biyoloji ve Sosyal Adalet ve Güçlendirilmesi, Hindistan Hükümeti Bakanlığı'na müteşekkiriz.
Malzemeler
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
Referanslar
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis?. Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
Yeniden Basımlar ve İzinler
Bu JoVE makalesinin metnini veya resimlerini yeniden kullanma izni talebi
Izin talebiThis article has been published
Video Coming Soon
JoVE Hakkında
Telif Hakkı © 2020 MyJove Corporation. Tüm hakları saklıdır