
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
تحليل Transcriptomic من
In This Article
Summary
ظهرت المجرة وDAVID كأدوات الشعبية التي تسمح للمحققين دون تدريب المعلوماتية الحيوية لتحليل وتفسير البيانات RNA يليها. نحن تصف بروتوكول للC. ايليجانس الباحثين لإجراء التجارب، والوصول RNA يليها ومعالجة بيانات باستخدام جالاكسي والحصول على المعلومات البيولوجية ذات مغزى من القوائم الجين باستخدام DAVID.
Abstract
أحدثت ثورة في الجيل القادم التسلسل (خ ع) تقنيات طبيعة التحقيق البيولوجي. ومن بين هؤلاء، برزت RNA تسلسل (RNA-تسلسل) كأداة قوية لتحليل الجينات التعبير ورسم الخرائط Transcriptome على. ومع ذلك، والتعامل مع قواعد البيانات-RNA يليها يتطلب خبرة حسابية معقدة وتحديات الملازمة للباحثين علم الأحياء. تم تخفيف هذه العقبة من خلال المشروع الوصول المفتوح غالاكسي التي تسمح للمستخدمين دون المهارات المعلوماتية الحيوية لتحليل البيانات RNA يليها، وقاعدة بيانات عن الشرح، والتصور، والمتكاملة ديسكفري (DAVID)، وجين علم الوجود (GO) جناح تحليل المصطلح الذي يساعد استخلاص المعنى البيولوجي من مجموعات البيانات الكبيرة. ومع ذلك، لأول مرة للمستخدمين الهواة والمعلوماتية الحيوية "، والتعلم الذاتي والتعريف مع هذه المنصات يمكن أن يكون وشاقة تستغرق وقتا طويلا. نحن تصف سير العمل واضحة من شأنها أن تساعد C. ايليجانس الباحثين لعزل دودة RNA، إجراء تجربة-RNA يليهاوتحليل البيانات باستخدام غالاكسي وDAVID المنصات. يوفر هذا البروتوكول تعليمات تدريجية لاستخدام مختلف وحدات غالاكسي للوصول إلى البيانات الخام NGS والشيكات مراقبة الجودة، والمحاذاة، وتحليل التعبير الجيني التفاضلية، وتوجيه المستخدم مع المعلمات في كل خطوة لإنشاء قائمة الجينات التي يمكن فرزهم للتخصيب دروس الجينات أو العمليات البيولوجية باستخدام DAVID. وبشكل عام، فإننا نتوقع أن هذه المادة سوف توفر معلومات إلى C. ايليجانس الباحثون بإجراء تجارب RNA يليها لأول مرة وكذلك المستخدمين بشكل متكرر تشغيل عدد قليل من العينات.
Introduction
التسلسل الأول من الجينوم البشري، قام باستخدام طريقة dideoxynucleotide التسلسل فريد سانجر، واستغرق 10 سنوات، وتكلف ما يقدر بنحو 3 مليارات $ 1 و 2. ومع ذلك، الجيل التالي من التسلسل جعلت (خ ع) التكنولوجيا في ما يزيد قليلا على عقد من الزمان منذ إنشائها من الممكن لتسلسل الجينوم البشري بأكمله في غضون أسبوعين والولايات المتحدة 1000 $. أدوات NGS الجديدة التي تسمح بسرعة جمع تسلسل البيانات المتزايدة مع كفاءة لا يصدق، جنبا إلى جنب مع تخفيضات حادة في التكاليف، هي ثورة البيولوجيا الحديثة بطرق لا يمكن تصورها كما هي مشاريع تسلسل الجينوم أصبحت بسرعة شائعا. وبالإضافة إلى ذلك، قد حفزت هذه التطورات التقدم في العديد من المجالات الأخرى مثل تحليل الجينات التعبير من خلال RNA-التسلسل (RNA-يليها)، ودراسة التعديلات جينية على نطاق الجينوم، وتفاعلات الحمض النووي والبروتينات، والكشف عن التنوع الميكروبي في المضيف البشري. NGS القائم على RNA سيجعلت ف على وجه الخصوص من الممكن تحديد وترنسكربيتوم خريطة شاملة مع الدقة والحساسية، وحلت محل تكنولوجيا متطورة مثل طريقة الاختيار لتحديد ملامح التعبير. في حين تكنولوجيا متطورة استخدمت على نطاق واسع، ويقتصر من قبل اعتمادها على المصفوفات القائمة من قبل مع المعلومات الجينية المعروفة، وعيوب أخرى مثل التهجين عبر ومجموعة محدودة من التغييرات التعبير التي يمكن قياسها بشكل موثوق. RNA وما يليها، من ناحية أخرى، يمكن أن تستخدم لكشف كل النصوص المعروفة وغير المعروفة في حين أن إنتاج منخفضة الضوضاء الخلفية نظرا لطبيعة لا لبس فيها رسم خرائط الحمض النووي. RNA-تسلسل جنبا إلى جنب مع أدوات الوراثية كثيرة عرضت من قبل الكائنات نموذج مثل الخميرة والذباب والديدان والأسماك والفئران، وقد خدم، كأساس للعديد من الاكتشافات الطبية الحيوية الهامة الأخيرة. ومع ذلك، تبقى تحديات كبيرة التي تجعل NGS قابلة للوصول إلى المجتمع العلمي الأوسع، بما في ذلك القيود المفروضة على التخزين والتجهيز، والأهم من ذلك كله، م تحليل المعلوماتية الحيوية eaningful كميات كبيرة من البيانات التسلسل.
خلقت التقدم السريع في تكنولوجيا التسلسل وتراكم البيانات الأسي حاجة كبيرة لمنصات الحسابية التي من شأنها أن تسمح للباحثين للوصول وتحليل وفهم هذه المعلومات. وكانت النظم الأولى تعتمد بشكل كبير على المعرفة برمجة الكمبيوتر، في حين المتصفحات الجينوم مثل NCBI التي سمحت غير المبرمجين للوصول إلى وتصور البيانات لا تسمح تحليلات متطورة. منصة، والوصول المفتوح على شبكة الإنترنت، غالاكسي ( https://galaxyproject.org/ )، وقد شغل هذا الفراغ وثبت أن خط أنابيب قيمة تمكن الباحثون لمعالجة البيانات NGS وتنفيذ مجموعة بسيطة-لمجمع تحليل المعلوماتية الحيوية. وقد أنشأت غالاكسي في البداية، وحافظت، من خلال مختبرات انطون Nekrutenko (جامعة ولاية بنسلفانيا)، وجيمس تايلور (جامعة جونز هوبكنز)و "> 3. غالاكسي يقدم مجموعة واسعة من المهام الحسابية مما يجعلها" محطة واحدة "لتلبية الاحتياجات المعلوماتية الحيوية لا تعد ولا تحصى، بما في ذلك جميع الخطوات المتبعة في دراسة الحمض النووي الريبي يليها. Itallows للمستخدمين تنفيذ معالجة البيانات إما على خوادمها أو محليا على أجهزتهم الخاصة. البيانات وسير العمل يمكن استنساخها وتبادلها. البرامج التعليمية عبر الإنترنت، والمساعدة القسم، ويكي الصفحات ( https://wiki.galaxyproject.org/Support ) مخصصة لمشروع غالاكسي توفر الدعم المستمر. ومع ذلك، لأول مرة للمستخدمين، وخاصة أولئك الذين ليس لهم تدريب المعلوماتية الحيوية، يمكن أن تظهر في خط أنابيب شاقة وعملية التعلم الذاتي والتعريف يمكن أن يكون مضيعة للوقت. وبالإضافة إلى ذلك، درس النظام البيولوجي، وتفاصيل التجربة وطرق استخدامها، والأثر القرارات التحليلية في عدة خطوات، وهذه يمكن أن يكون من الصعب التنقل دون توجيه.
وعموما RN يتكون A-تسلسل غالاكسي سير العمل في تحميل البيانات والتأكد من جودتها تليها تحليل باستخدام سهرة جناح 4، 5، 6، 7، 8، 9، وهو الجماعية لمختلف الأدوات اللازمة لمراحل مختلفة من تحليل البيانات RNA يليها 10، 11، 12، 13، 14. تتكون التجربة-RNA يليها نموذجية من الجزء التجريبي (إعداد العينات، والعزلة مرنا وكدنا] إعداد مكتبة)، وNGS وتحليل المعلوماتية الحيوية البيانات. لمحة عامة عن هذه الأقسام، والخطوات المتبعة في خط أنابيب غالاكسي، وتظهر في الشكل 1.
3fig1.jpg "/>
الشكل (1): نظرة عامة على سير عمل RNA-تسلسل. التوضيح من الخطوات التجريبية والحسابية تشارك في تجربة-RNA يليها مقارنة ملامح الجينات التعبير من سلالتين دودة (A و B، وخطوط برتقالية وخضراء والسهام، على التوالي). وتظهر وحدات مختلفة من غالاكسي المستخدمة في مربعات مع الخطوة المقابلة في بروتوكول لدينا هو مبين في الحمراء. تتم كتابة مخرجات العمليات المختلفة في الرمادي مع تنسيقات الملفات هو مبين في الزرقاء. الرجاء انقر هنا لمشاهدة نسخة أكبر من هذا الرقم.
{kind=link}
الأداة الأولى في جناح سهرة هو برنامج المواءمة يسمى 'Tophat. ينهار مدخلات NGS يقرأ إلى أجزاء أصغر ومن ثم خرائط لهم الجينوم المرجعية. وتضمن هذه عملية من خطوتين الذي يقرأ تغطي المناطق intronic تتضاعف الذي يمكن أن يكون الأمر خلاف ذلك دي محاذاةsrupted أو تحتسب وتعيين تفويتها. وهذا يزيد من التغطية ويسهل التعرف على تقاطعات لصق جديدة. وتفيد التقارير الانتاج Tophat كما ملفين، ملف BED (مع معلومات حول تقاطعات لصق التي تشمل موقع الجيني) وملف BAM (مع تفاصيل رسم الخرائط من كل قراءة). بعد ذلك، يتم محاذاة الملف BAM ضد الجينوم مرجعية لتقدير وفرة من النصوص الفردية داخل كل عينة باستخدام أداة لاحقة في جناح سهرة بعنوان "أزرار". أزرار وظائف عن طريق مسح المحاذاة إلى تقرير شظايا النص بالطول أو "transfrags" التي تشمل جميع المتغيرات لصق المحتملة في إدخال البيانات عن كل جينة. وبناء على هذا، فإنه يقوم بإنشاء "Transcriptome على" (تجميع كل النصوص ولدت في الجينات لكل الجينات) لكل عينة يجري التسلسل. ثم انهارت هذه التجميعات أزرار أكمام أو دمجها معا جنبا إلى جنب مع إعادةference الجينوم لإنتاج ملف الشرح واحد لتحليل التفاضلية المصب باستخدام أداة المقبلة، "Cuffmerge. وأخيرا، فإن "Cuffdiff" التعبير الجيني أداة التدابير التفاضلية بين العينات عن طريق مقارنة النتائج TopHat كل من العينات إلى ملف الإخراج Cuffmerge النهائي (الشكل 1). أزرار أكمام تستخدم FPKM / RPKM (شظايا / يقرأ لكل كيلو قاعدة من نسخة لكل مليون معين يقرأ) القيم أن يقدم فرة نسخة. وتعكس هذه القيم تطبيع البيانات NGS الخام لعمق (متوسط عدد القراءات من عينة أن محاذاة إلى الجينوم مرجعية) وطول الجين (الجينات لها أطوال مختلفة، لذلك لها التهم ليتم تطبيع للطول الجيني لمقارنة مستويات بين الجينات). FPKM وRPKM لا تختلف في جوهرها مع RPKM تستخدم لنهاية واحدة RNA-تسلسل حيث يتوافق مع كل قراءة لجزء واحد، في حين يستخدم FPKM لإقران نهاية RNA-تسلسل، كما يفسر حقيقة أن اثنين من يقرأ يمكن أن تتوافق مع نفس الجزء. وفي النهاية، فإن نتائج هذه التحليلات هي قائمة من الجينات وأعرب تفاضلي بين الظروف و / أو السلالات التي تم اختبارها.
بمجرد الانتهاء من مسيرته الناجحة غالاكسي ويتم إنشاء "قائمة الجينات، فإن الخطوة المنطقية المقبلة تتطلب المزيد المعلوماتية الحيوية تحليل للاستدلال على المعرفة ذات مغزى من قواعد البيانات. ظهرت العديد من حزم البرامج لتلبية هذه الحاجة، بما في ذلك المتاحة للجمهور حزم الحسابية على شبكة الإنترنت مثل DAVID (قاعدة بيانات عن الشرح، والتصور واكتشاف المتكاملة) 15. DAVID يسهل تعيين المعنى البيولوجي للقوائم الجينات كبيرة من الدراسات الإنتاجية العالية من خلال مقارنة قائمة الجينات التي تم تحميلها على المعرفة البيولوجية المتكاملة والكشف عن شروح البيولوجية المرتبطة قائمة الجينات. ويلي ذلك تحليل والإثراء، أي من الاختبارات لبيئة تطوير متكاملةntify إذا كان زائدا أي عملية أو الجينات الطبقة البيولوجية في قائمة الجينات (ق) بطريقة ذات دلالة إحصائية. فقد أصبح خيارا شعبيا بسبب مزيج من اسعة ومتكاملة قاعدة المعرفة والخوارزميات التحليلية القوية التي تمكن الباحثين للكشف عن مواضيع البيولوجية التخصيب داخل الجينوم المشتقة 'القوائم الجين "10 و 16. وتشمل مزايا إضافية قدرتها على معالجة قوائم الجينات التي تم إنشاؤها على أي منصة التسلسل واجهة عالية سهل الاستعمال.
الديدان الخيطية انواع معينة ايليجانس هي نموذج نظام وراثي، معروف جيدا لمزاياها العديدة مثل صغر حجمها، وجسم شفاف، خطة هيئة بسيطة، وسهولة الثقافة وقابليته الكبيرة للتشريح الجيني والجزيئي. الديدان لها الجينوم صغيرة وبسيطة ومشروحة جيدا أن تشمل ما يصل الى 40٪ من الجينات الحفظ مع homologs البشرية المعروفة 17. في الواقع، C. ايليجانسكان متعددة الخلايا الأولى التي كان التسلسل تماما 18 الجينوم، واحدة من الأنواع الأولى حيث تم استخدام الحمض النووي الريبي تسلسل إلى خريطة Transcriptome على الكائن الحي 19 و 20. دراسات دودة في وقت مبكر وشملت التجارب مع أساليب مختلفة لالتقاط عالية الإنتاجية RNA، وإعداد مكتبة والتسلسل وكذلك خطوط الأنابيب المعلوماتية الحيوية التي ساهمت في النهوض التكنولوجيا 21 و 22. في السنوات الأخيرة، أصبح التجريب أساس RNA-تسلسل في الديدان شائعا. ولكن، لعلماء الأحياء دودة التقليدية للتحديات التي تفرضها التحليل الحسابي لبيانات RNA بعدها لا تزال تشكل عائقا رئيسيا لأكبر وأفضل استخدام لهذه التقنية.
في هذه المقالة، نحن تصف بروتوكول لاستخدام منصة غالاكسي لتحليل البيانات RNA يليها الإنتاجية العالية المتولدة من C. ايليجانس. بالنسبة للعديد من أول مرة وهيئة السلع التموينية صغيرةالمستخدمين جنيه، والطريقة الأكثر وضوحا فعالة من حيث التكلفة لإجراء تجربة-RNA يليها هو عزل الحمض النووي الريبي في المختبر والاستفادة منشأة NGS التجاري (أو في المنزل) لإعداد مكتبات كدنا] تسلسل وNGS نفسها. وبالتالي، لدينا لأول مرة بالتفصيل الخطوات المتبعة في عزلة، الكمي ونوعية تقييم C. ايليجانس عينات الحمض النووي الريبي لRNA-تسلسل. بعد ذلك، نحن نقدم إرشادات خطوة بخطوة لاستخدام واجهة غالاكسي لتحليل البيانات NGS، بدءا من الاختبارات لاجراء فحوص مراقبة الجودة بعد التسلسل تليها المحاذاة، والتجمع، والفارق الكمي في التعبير الجيني. وبالإضافة إلى ذلك، أدرجنا الاتجاهات للتدقيق في قوائم الجينات الناتجة عن غالاكسي للدراسات البيولوجية تخصيب باستخدام DAVID. كخطوة نهائية في سير العمل، ونحن نقدم تعليمات لتحميل البيانات RNA يليها الدخول إلى ملقمات العامة مثل تسلسل مقروءة الأرشيف (SRA) على NCBI ( HTTP: // ثww.ncbi.nlm.nih.gov/sra) لجعلها في متناول مجانا للمجتمع العلمي. وبشكل عام، فإننا نتوقع أن هذه المادة سوف توفر معلومات شاملة وكافية لعلماء الأحياء دودة إجراء تجارب RNA يليها لأول مرة وكذلك المستخدمين بشكل متكرر تشغيل عدد قليل من العينات.
Access restricted. Please log in or start a trial to view this content.
Protocol
1. عزل الحمض النووي الريبي
- تدابير وقائية
- مسح أسفل كامل العمل السطح والأدوات والماصات باستخدام رذاذ ريبونوكلياز المتاحة تجاريا للقضاء على أي RNases الحاضر.
- ارتداء القفازات في جميع الأوقات، وتغيير بشكل منتظم بأخرى جديدة خلال خطوات مختلفة من البروتوكول.
- استخدام النصائح مرشح الوحيد والحفاظ على جميع العينات على الجليد قدر الإمكان لتجنب تدهور الحمض النووي الريبي.
ملاحظة: من أجل الحصول على أفضل البيانات من منصات NGS، فمن الأهمية بمكان أن تبدأ مع RNA عالية الجودة. تختلف العزلة RNA وإعداد طرق اعتمادا على أصل عينة، طريقة التسلسل ومحقق تفضيل. عدة مجموعات المتاحة تجاريا يمكن استخدامها لهذا الغرض أو يمكن أيضا أن تكون معزولة RNA باستخدام طريقة الفينول كلوروفورم القياسي لاستخراج الحمض النووي الريبي. مع أي منهجية، ينبغي اتباع التدابير الوقائية المذكورة أعلاه في جميع مراحل عملية للحد من التلوث وOBTعين عينات الحمض النووي الريبي البكر.
- حصاد الديدان
- مزامنة السكان الدودة عن طريق العلاج تبيض الهيبوكلوريت 23 للحصول على 1،000-1،500 C. الديدان ايليجانس الكبار في العمر كعينة لكل سلالة.
- غسل الديدان من لوحات باستخدام M9 حل العازلة وتدور في 325 x ج على طاولة قمة الطرد المركزي لمدة 30 ثانية. نضح من المنطقة العازلة M9 تاركا وراءه بيليه من الديدان. كرر هذه الخطوة ما لا يقل عن ثلاث مرات للقضاء على المرحل البكتيرية.
- لبيليه دودة، إضافة ~ 500 ميكرولتر من تحلل العازلة (في حالة استخدام عدة التجارية) أو Trizol (حل أحادي طوري من الفينول وثيوسيانات غوانيدين، وإذا الفينول: تم إجراء استخراج الكلوروفورم وصفها في 1.3.3) لتعطيل الأنسجة دودة ، إلغاء RNases واستقرار الأحماض النووية.
ملاحظة: بروتوكول يمكن أن يكون مؤقتا هنا فلاش تجميد العينات في النيتروجين السائل تليها تخزين في -80 ° C.
- عزل الحمض النووي الريبي
- عينات الدودة يصوتن في 45٪ السعة في دورات من 20 ثانية. "ON" و40 ق. "OFF" (8-12 دورة في سلالة). الاحتفاظ بعينات على الجليد في جميع الأوقات.
ملاحظة: تأكد من أن التحقيق sonicator مغمورة في المخزن المؤقت ويتم الاحتفاظ بها على مستوى ثابت طوال الوقت. تجنب مزبد من العينة والتنظيف التحقيق بدقة في الفترات الفاصلة بين العينات. قد تختلف دورات صوتنة تبعا لنوع من sonicator المستخدمة. فمن المستحسن أن الظروف صوتنة يتم أولا الأمثل على عينة الاختبار قبل بدء التجربة. - في حالة استخدام عدة متاحة تجاريا، والمضي قدما مع عزل RNA وفقا للبروتوكول المحدد. لعزل الحمض النووي الريبي باستخدام طريقة الفينول كلوروفورم، قم بالخطوات التالية.
- sonicated الطرد المركزي العينات في 16000 x ج لمدة 10 دقيقة. في 4 درجات مئوية.
- نقل طاف في أنبوب microfuge 1.5 مل ريبونوكلياز خالية وإضافة 100 ميكرولتر من كلوروفورم (1/5 عشر حجم RNA / DNA العزلة كاشف).
الحذر: الكلوروفورم السامة. للحد من التعرض، وتجنب استنشاق، والعمل في غطاء الكيميائية عند التعامل مع هذه المادة. - دوامة العينات بدقة ل30 - 60 ق. والسماح للعينات الجلوس في درجة حرارة الغرفة لمدة 3 دقائق.
- أجهزة الطرد المركزي في 11750 x ج لمدة 15 دقيقة. في 4 درجات مئوية. نقل سوى الطبقة العليا المائية لريبونوكلياز خالية microfuge أنبوب رعاية جديدة لا لنضح واجهة بيضاء تحتوي على الحمض النووي. كرر الخطوات من 1.3.4 خلال 1.3.6.
- إضافة 250 ميكرولتر (70٪ من المرحلة المائية أو 1/2 RNA / DNA العزلة حجم الكاشف) من 2-بروبانول وعكس الأنبوب إلى المزيج. دعونا أنابيب الجلوس في درجة حرارة الغرفة لمدة 10 دقيقة أو ترك بين عشية وضحاها في -80 ° C.
- عينات الطرد المركزي في 11750 x ج لمدة 10 دقيقة. في 4 درجات مئوية. صب وطاف بعناية فائقة، تاركا وراءه قليل ميكرولتر في أسفل الأنبوب بحيث لا تشعر بالانزعاج بيليه.
- غسل بيليه مع 500 ميكرولتر من 75٪ من الإيثانول (مصنوعة باستخدام ريبونوكلياز خالية من المياه) وتدور باستمرار في 16000 x ج لمدة 5 دقائق. ار 4 درجات مئوية.
- إزالة أكبر قدر من طاف ممكن من دون إزعاج بيليه. الهواء الجاف بيليه في غطاء محرك السيارة لبضع دقائق.
- إضافة 30 ميكرولتر من ريبونوكلياز خالية من المياه ومساعدة حل بيليه RNA عن طريق التسخين لمدة 10 دقيقة. في 60 ° C.
- والتحقق من جودة RNA وكمية باستخدام bioanalyzer.
ملاحظة: Bioanalyzer يولد R NA I ntegrity N بني مصفر (رين) كمقياس لجودة RNA. وRIN لا يقل عن 8 هو العتبة الموصى بها لعينات الحمض النووي الريبي يليها (أعلى هي أحسن). كمية RNA والجودة يمكن أيضا أن يتم التحقق طيفيا ولكن ينبغي أيضا أن يتبعها تقييم البصري للسلامة الحمض النووي الريبي. للقيام بذلك، قم بتشغيل العينات على هلام الاغاروز 1.2٪ لفترة كافية للحصول على فصل مناسبة لل28S و 18S العصابات RNA الريباسي. وجود شريطين مميزة (1.75 كيلوبايت ل18S الريباسي و 3.5 كيلوبايت ل28S الريباسي في حالة C. ايليجانس) هو مقياس مقبول من الجودة RNA. - استخدام ~ 100 نانوغرام / ميكرولتر RNA لشيص إلى البائع منشأة / NGS لإعداد مكتبات التسلسل.
يجب أن يتم شحن عينات الحمض النووي الريبي على الجليد الجاف لمزود الخدمة التسلسل: ملاحظة. معظم مقدمي بإجراء اختبار الحمض النووي الريبي لمراقبة الجودة مستقلة قبل إعداد مكتبة.
- عينات الدودة يصوتن في 45٪ السعة في دورات من 20 ثانية. "ON" و40 ق. "OFF" (8-12 دورة في سلالة). الاحتفاظ بعينات على الجليد في جميع الأوقات.
2. RNA-تسلسل تحليل البيانات
- تحميل الخام بيانات التسلسل
- تحميل تسلسل البيانات fastq الخام مضغوط المشفرة في شكل fastq.gz من مزود NGS باستخدام "بروتوكول نقل الملفات" (FTP).

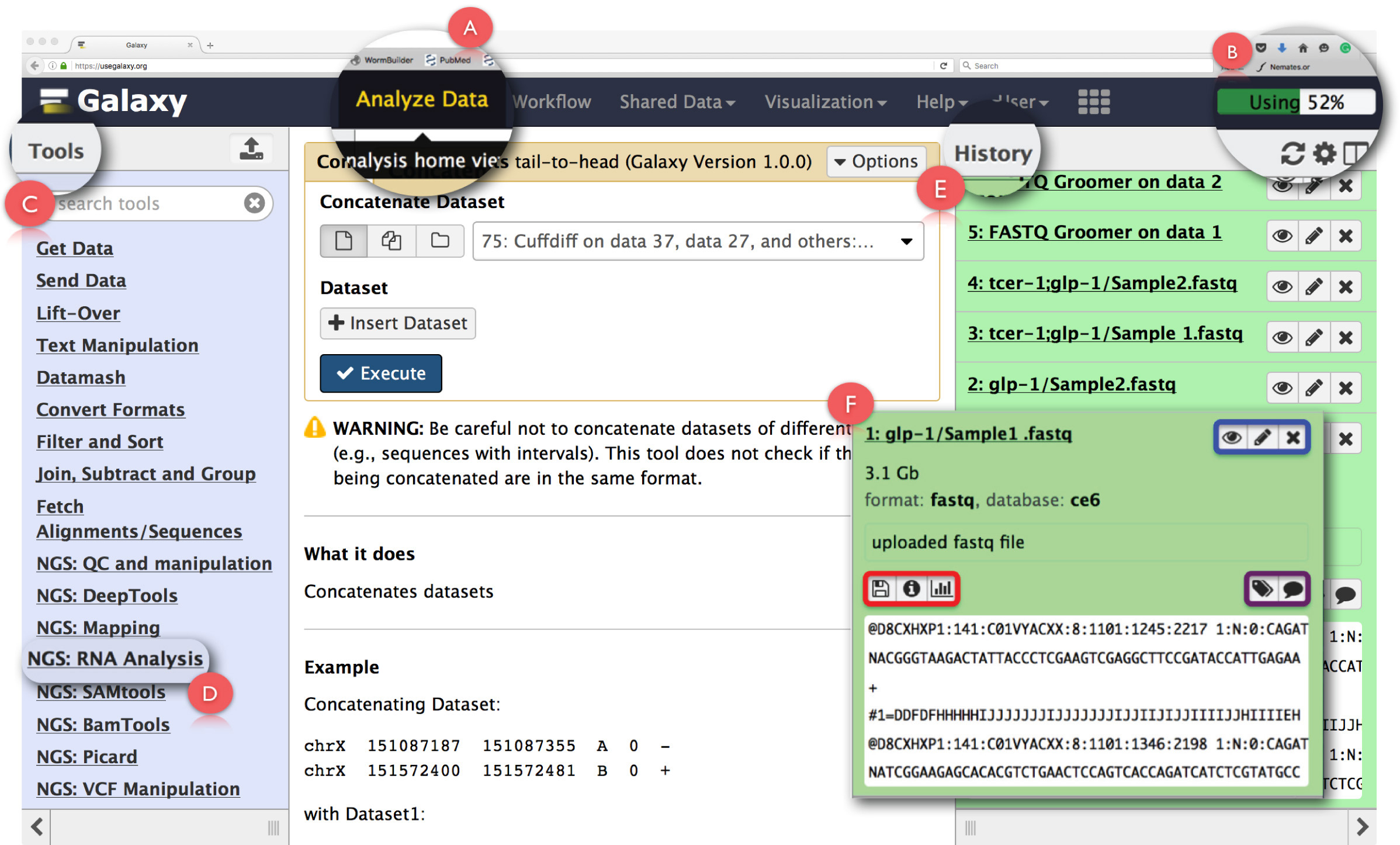
الشكل 2: تخطيط واجهة لوحة وظائف أساسية RNA يليها العضو غالاكسي. يتم توسيع الملامح الرئيسية الصفحة وسلط الضوء. (A) يسلط الضوء على وظيفة "تحليل البيانات" في رأس صفحة ويب تستخدم للوصول تحليل الرئيسية عرض. (B) هو "شريط التقدم" الذي يشير إلى الفضاء على الخادم غالاكسي التي تستخدمها العملية. (C) هو "القسم أدوات" تسرد كل الأدوات التي يمكن تشغيلها على واجهة غالاكسي. (D) يظهر 'NGS: تحليل الحمض النووي الريبي "قسم أداة تستخدم لتحليل الحمض النووي الريبي يليها. (E) يصور لوحة "التاريخ" الذي يسرد كافة الملفات التي تم إنشاؤها باستخدام غالاكسي. (F) يدل على سبيل المثال من مربع الحوار الذي يفتح عند النقر على أي ملف في قسم التاريخ. خلال (F)، المربع الأزرق يسلط الضوء على الرموز التي يمكن استخدامها لعرض وeditthe سمات أو حذف بيانات، مربع الأرجواني يسلط الضوء على الرموز التي يمكن استخدامها ل'تحرير' علامات بيانات أو الشرح، ويشير إلى مربع أحمر الرموز لتحميل البيانات، وعرض تفاصيل مهمة أداء أو إعادة تشغيل العملية. الرجاء انقر هنا لمشاهدة نسخة أكبر من هذا الرقم.
{kind=link}
- الشروع في العمل مع غالاكسي
يمكن تشغيل غالاكسي في الخادم العام المجاني باستخدام منصة على شبكة الإنترنت توفير إمكانية الحصول السحابية والتخزين مجانية محدودة: ملاحظة. كما يمكن تحميلها وتشغيلها محليا على جهاز المستخدم أو مجموعات الحسابية التي تستضيفها المؤسسات ولكن التجهيز المحلي، قد تكون مقيدة بحدود تخزين البيانات والقيود قوة المعالجة من أجهزة المستخدمين. التفاصيل حول تحميل وتركيب يمكن الحصول عليه من https://wiki.galaxyproject.org/Admin/GetGalaxy . في هذا البروتوكول وصفنا استخدام على شبكة الإنترنت من خط الانابيب غالاكسي.- بعد البيانات NGS تحميل وتخزين على جهاز المستخدم، والوصول إلى غالاكسي فيlaxy.org/ "الهدف =" _blank "> https://usegalaxy.org/.
- تسجيل حساب المستخدم عن طريق النقر على "المستخدم" في رأس الصفحة، تسجيل الدخول وتبدأ من خلال التعرف على لوحة واجهة المستخدم.
ملاحظة: من المستحسن أن المستخدمين لأول مرة الاستفادة من البرنامج التعليمي "ابدأ من هنا" المقدمة على الصفحة الرئيسية للحصول على إطلاع مع مجموعة تصل الأساسي للمجرة ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) . - انقر على "تحليل البيانات" (الشكل 2A) في لوحة رأس للوصول إلى "تحليل الرئيسية عرض" الذي هو أيضا شاشة بدء التشغيل على غالاكسي.
ملاحظة: رأس يضم أيضا وصلات الأخرى التي يمكن أن ينظر إليه من قبل تحوم مؤشر الماوس فوقها التفاصيل. في الزاوية اليمنى العليا من رأس بها بار التقدم الذي يراقب الفضاء تستخدم للمهام (الشكل 2B). - Cلعق على 'NGS: تحليل الحمض النووي الريبي "مهمة في" قائمة أدوات "على اللوحة اليسرى (الشكل 2C) للوصول إلى جميع الأدوات اللازمة لتحليل البيانات RNA وما يليها.
ملاحظة: "أدوات القائمة 'كتالوجات جميع العمليات التي غالاكسي العروض. يتم تقسيم هذه القائمة على أساس المهام والنقر على أي واحد سوف تفتح قائمة بجميع الأدوات اللازمة لإنجاز هذه المهمة. - إنشاء التاريخ تحليل جديد من خلال النقر على رمز الترس في الجزء العلوي من "التاريخ" لوحة على اليمين (الشكل 2E). اختيار 'إنشاء جديد "خيار من القائمة المنبثقة. يعطي هذا "التاريخ" اسم مناسب للتعرف على التحليل.
ملاحظة: تظهر لوحة و"التاريخ" كافة الملفات التي تم تحميلها لتحليلها وكذلك جميع ملفات الإخراج التي يتم إنشاؤها عن طريق تشغيل المهام على غالاكسي. النقر على اسم الملف في هذه اللوحة تفتح مربع حوار مع معلومات مفصلة حول المهمة أنجزتومقتطف من مجموعة البيانات (الشكل 2F). الرموز في هذا الإطار تمكن المستخدم من 'الرأي'، 'تحرير سمات' أو 'حذف' مجموعة البيانات (الشكل 2F، أبرزت باللون الأزرق). بالإضافة إلى ذلك، يمكن للمستخدم أيضا 'تحرير' علامات بيانات أو الشرح (الشكل 2F، سلط الضوء في اللون الأرجواني)، "تنزيل" البيانات "عرض تفاصيل" المهمة، "اعادة" المهمة أو حتى "تصور" مجموعة البيانات من هذا مربع الحوار (الشكل 2F، أبرز باللون الأحمر). - انقر على وظيفة "تحميل الملف" تحت "إحضار بيانات" في سياق "ToolsMenu" لتحميل الملفات fastq الخام.
ملاحظة: النقر على هذا أو أي أداة أخرى تفتح وصفا موجزا لهذه العملية، واختبار نفسه في وسط لوحة "واجهة تحليل. المقسم هذا الفريق معا"أدوات" من اللوحة اليسرى و "إدخال الملفات" من لوحة الحق "التاريخ" (الشكل 2E). هنا، يتم اختيار الملفات المدخلات من "التاريخ" وغيرها من المعالم محددة لتشغيل مهمة معينة. يتم حفظ بيانات الناتج الناتجة من كل اختبار مرة أخرى في "التاريخ". وشملت مع اختبار في لوحة "تحليل واجهة" هي تفسيرات لجميع المعلمات المتاحة لتشغيل أداة معينة جنبا إلى جنب مع قائمة مفصلة لجميع ملفات الإخراج الأداة يولد. - بعد فتح المهمة في "واجهة تحليل"، انقر على 'اختيار ملف محلي "أو" اختر ملف بروتوكول نقل الملفات (تحميل أسرع)، انتقل إلى المجلد الذي يحتوي على ملفات تسلسل واختر ورقة العمل المناسب وسيتم تحميلها.
- تسمح غالاكسي ل"لصناعة السيارات في كشف" والتي تم تحميلها نوع الملف (الإعداد الافتراضي). اختر 'C. ايلفندق Egans 'في القائمة المنسدلة للجينوم.
- انقر على "ابدأ" لبدء تحميل البيانات. مرة واحدة يتم تحميل الملف، فإنه سيتم حفظها في لوحة "التاريخ"، ويمكن الوصول إليها من هناك.
- إذا يتم إنتاج ملفات البيانات التسلسل متعددة لعينة واحدة، الجمع بينهما باستخدام "سلسل" أداة. للقيام بذلك، وفتح خيار "معالجة النصوص" في "قائمة أدوات".
- انقر على أداة "سلسل"، اختر الملفات التي تحتاج إلى أن تكون مجتمعة من مربع القائمة المنسدلة في منتصف "واجهة تحليل" وانقر على 'تنفيذ'.
ملاحظة: يتم إنشاء ملفات الإخراج المنتجة باستخدام هذه المهمة في شكل fastq. برنامج رسم الخرائط لديه الحد من 16،000،000 تسلسل لكل ملف fastq وعندما يتم التوصل إلى هذا الحد يتم إنشاء ملف fastq جديد للتسلسل المتبقية. و"، وهناك حاجة سلسل أداة في مثل هذه الحالات على الجمع بين مجموعات البيانات. - تحويل شكل ملفات fastq تحميلها على شكل fastqsanger اللازمة لغالاكسي تحليل الحمض النووي الريبي بعدها عن طريق استخدام 'مربية fastq "أداة وجدت تحت" NGS: مراقبة الجودة والتلاعب "قسم (انظر ملف إضافي).
- اختيار مجموعة البيانات fastq المناسب ضمن "ملف لالعريس 'الخيار وتشغيل الأداة باستخدام المعلمات الافتراضية.
يتم إنشاء ملفات الإخراج المنتجة باستخدام هذه المهمة في شكل fastqsanger: ملاحظة.
- الاختبارات fastqsanger بيانات الجودة مراقبة
- تحقق من نوعية fastqsanger تحميلها يقرأ باستخدام أداة "FastQC 'الواقعة تحت" NGS: QC والتلاعب "في قائمة" أدوات ".
- اختيار ملف بيانات fastqsanger اعدادهم من القائمة المنسدلة ل 'شورر قراءة البيانات من المكتبة الحالية "وتشغيل الأداة باستخدام المعلمات الافتراضية.
ملاحظة: إيلاء اهتمام خاص لنوعية يقرأ وجود أي تسلسل محول. وعادة ما يتم إزالة محولات كجزء من مرحلة ما بعد المعالجة بيانات RNA يليها من قبل مقدمي NGS ولكن في بعض الحالات، قد تركت وراءها. لشرح معايير الجودة الذهاب إلى http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - تحقق مع مزود NGS وإذا محولات موجودة، وتقليم لهم باستخدام أداة "كليب 'من' NGS: مراقبة الجودة والتلاعب" القائمة المهمة.
يتم إنشاء ملفات الإخراج المنتجة باستخدام هذه المهمة في شكل النص الخام وكذلك في HTML والتي يمكن فتحها في أي متصفح ويب: ملاحظة.
- تحليل البيانات مع جناح سهرة
- قبعة عالية
- تحميل أحدث إصدار من C. ايليجانس FASTA إشارة الجينوم وGTF (تنسيق نقل الجينات) الملفات من ملف تحميل "كما هو موضح أعلاه في 2.2.6.
- فتح 'NGS: تحليل الحمض النووي الريبي "قسم وانقر على أداة" TopHat "لتعيين تسلسل يقرأ لجينوم إشارة تحميلها.
- اختر الإجابة المناسبة من القائمة المنسدلة على السؤال "هل هذه نهاية واحدة أو البيانات المقترنة نهاية؟
- اختيار ملف fastq المناسب.
- اختر "استخدام الجينوم من التاريخ" في القائمة المنسدلة المقبلة واختيار الجينوم إشارة بتحميلها في الخطوة 2.4.1.1.
- اختر 'افتراضي' للمعلمات أخرى وانقر على 'تنفيذ'.
ملاحظة: من بين ملفات الإخراج المنتجة باستخدام هذه المهمة، يتم استخدام ملف "الفعالية مقبول" لخطوات لاحقة.
- أزرار أكمام وCuffmerge
- حدد "صفعة"أداة في" الروابط NGS: قسم تحليل الحمض النووي الريبي "لتجميع النصوص، وتقدر وفرة واختبار للتعبير التفاضلية.
- في القائمة المنسدلة الأولى، اختر تعيين "يضرب مقبول (تنسيق BAM) 'ملف تم الحصول عليها من تحليل TopHat.
- في القائمة المنسدلة الثانية، تعيين الشرح الإشارة إلى ملف GTF بتحميلها في الخطوة 2.4.1.1.
- حدد "نعم" لخيار "تنفيذ تصحيح التحيز 'وتشغيل المهمة باستخدام الإعدادات الافتراضية لجميع المعلمات الأخرى.
ملاحظة: من بين ملفات الإخراج المنتجة باستخدام هذه المهمة، يتم استخدام الملف 'مقبول النصوص "لخطوات لاحقة. - أداة فتح 'Cuffmerge' في 'NGS: تحليل الحمض النووي الريبي "لدمج" تجميعها النصوص "المنتجة لجميع العينات-RNA يليها.
ملاحظة: المربع الأول في الأداة بملء النفس وقوائم جميع ملفات GTF التي تنتجها أزرار أكمام. - حدد الملف 'تجميعها النصوص "لجميع السلالات / الظروف المختبرة، بما في ذلك مكررات البيولوجية من نفس السلالة / حالة (انظر المناقشة لمكررات البيولوجية).
- اختر 'نعم' ل 'استخدام المرجعي الشرح "واختيار ملف GTF بتحميلها في الخطوة 2.4.1.1.
- في الإطار التالي، ومرة أخرى اختر "نعم" لخيار "استخدام سيكوينس داتا 'واختيار كاملة ملف الجينوم FASTA بتحميلها في الخطوة 2.4.1.1.
- الحفاظ على معايير أخرى كما الافتراضي، انقر فوق "تنفيذ".
ملاحظة: Cuffmerge بإنشاء ملف الانتاج GTF واحد.
- Cuffdiff
- انتقل إلى أداة "Cuffdiff 'في' NGS: تحليل الحمض النووي الريبي" القسم. في القائمة "النصوص"، حدد ملف الإخراج المدمجة من Cuffmerge.
- ضع الكلمة المناسبةالشروط 1 و 2 مع أسماء سلالتين / حالة.
ملاحظة: Cuffdiff يمكن أن تؤدي مقارنات بين أكثر من سلالتين أو شروط، فضلا عن تجارب دوام. ببساطة استخدام خيار "اضافة شروط جديدة" لإضافة كل سلالات جديدة / حالة، حسب الحاجة. - لكل سلالة / حالة، تحت عنوان "يعيد" فرد حدد ملفات الإخراج "الفعالية مقبول" من TopHat التي تتوافق مع مكررات البيولوجية المختلفة من تلك السلالة / حالة. اضغط باستمرار على المفتاح 'كمد'، في حالة استخدام كمبيوتر ماكنتوش، ومفتاح 'السيطرة'، في حالة استخدام جهاز كمبيوتر، لتحديد ملفات متعددة.
- ترك جميع الخيارات الأخرى كمعلمات الافتراضية. انقر على "تنفيذ" لتشغيل المهمة.
ملاحظة: Cuffdiff يولد العديد من ملفات الإخراج في شكل جداول كما قراءات النهائية للتحليل الحمض النووي الريبي يليها. تشمل هذه الملفات مع تتبع FPKM عن نصوص أو الجينات (جنبا إلى جنبقيم FPKM النصوص تقاسم هوية الجينات)، النصوص الأساسية وتسلسل الترميز. جميع ملفات البيانات التي تم إنشاؤها يمكن أن ينظر في أي طلب البيانات ويحتوي سمات مشابهة مثل اسم الجينات، موضع، أضعاف التغيير (في نطاق log2) فضلا عن بيانات إحصائية عن المقارنات بين السلالات / الشروط، بما في ذلك قيمة احتمالية والقيم ف. يمكن فرز البيانات في هذه الملفات بناء على الدلالة الإحصائية للفروق أو أضعاف تغيير في التعبير الجيني (حجم واتجاه التغيير، كما هو الحال في حتى- أو أسفل-الجينات تنظيم) والتلاعب بها وفقا لمتطلبات المستخدمين. إذا كانت هناك حاجة التحويل بين معرفات الجينات المختلفة (على سبيل المثال، Wormbase ID الجينات مقابل عدد كوزميد)، الأدوات المتاحة على Biomart ( http://www.biomart.org/ ) يمكن استخدامها.
- قبعة عالية
3. جين علم الوجود (GO) تحليل الأجل باستخدام DAVID
- وصول DAVID من موقع حttps: //david.ncifcrf.gov/. انقر على "ابدأ التحليل" في رأس صفحة ويب. في "الخطوة 1، نسخ ولصق قائمة من الجينات التي تم الحصول عليها من غالاكسي في مربع A. في" الخطوة 2 "، حدد" Wormbase ID جين "كمعرف للجينات الإدخال.
ملاحظة: تعترف DAVID فئات الشرح الأكثر متاحة للجمهور، بحيث يمكن أيضا معرفات الجينات الأخرى (مثل ID الجينات Entrez أو رمز الجين) يمكن استخدامها. - في "الخطوة 3"، اختر "قائمة جين" (الجينات ليتم تحليلها) تحت عنوان "قائمة نوع 'ثم اضغط على" إرسال قائمة "رمز.
ملاحظة: "معالج تحليل، تفتح لسرد كافة الأدوات DAVID الارتباطات التشعبية التي يمكن تشغيلها على لائحة الجينات التي تم تحميلها (الشكل 3). انقر على هذه الروابط للوصول إلى وحدات المقابلة ذات الصلة وفقا لمتطلبات المستخدم. لتحديد الأدوات المناسبة لمهمة معينة، انقر على 'أي أدوات DAVID للاستخدام؟ "وصلة على ' . الصفحة معالج تحليل. انقر على رابط 'ابدأ التحليل "في رأس للعودة إلى الصفحة الرئيسية لل" معالج تحليل "في أي وقت أثناء التحليل.

الشكل (3): تخطيط من DAVID تحليل معالج صفحة ويب وأمثلة من المخرجات العملية. يسرد "معالج تحليل" على شبكة الإنترنت واجهة المستخدم والأدوات المستخدمة لتحليل الجينات قائمة محملة لتخصيب اليورانيوم على أساس معايير مختلفة. النقر على هذه الأدوات تقارير تحليل البيانات في صفحة ويب جديدة. وترد أمثلة لتقارير جداول الناتجة عن "جين التصنيف الوظيفي '،' الوظيفي الرسم البياني الشرح" و "تجميع الشرح الوظيفي 'كما إدراجات (السهام).> الرجاء انقر هنا لمشاهدة نسخة أكبر من هذا الرقم.
- وظيفية أداة الشرح 1: ظيفية الشرح تجميع
- انقر على وحدة "الوظيفي الشرح تجميع" للذهاب إلى صفحة ملخص. حافظ على فئات الشرح الافتراضي وانقر على "تجميع الشرح الوظيفي" لتوليد مجموعات من حيث الشرح مماثلة من قبل تقدمهم في تخصيب اليورانيوم في المرتبة.
- انقر على اسم ارتباط تشعبي كل فصل دراسي لقراءة التفاصيل حول هذا الموضوع و "RT" (المصطلحات ذات الصلة) لقائمة مصطلحات أخرى مشابهة تتعلق الفئة.
- انقر على شريط الأرجواني لسرد الجينات المرتبطة بالمصطلح و'G' أحمر لسرد كافة الجينات المرتبطة مع جميع المصطلحات ضمن كتلة.
- انقر على أيقونة خضراء لمشاهدة عرض ثنائي الأبعاد من جميع الجينات وشروط في كتلة.
ملاحظة: قائمة الأعمدة الثلاثة الماضية نتائج التحليلية والإحصائية لكلمصطلح. نتائج هذا وكل تحليلات أخرى يمكن تحميلها في شكل. TXT من خلال النقر على رابط "تحميل الملف".
- وظيفية أداة الشرح 2: الوظيفي الشرح الرسم البياني
- عودة إلى صفحة ملخص وانقر على "مخطط الشرح الوظيفي" لتحديد الشروط البيولوجية زائدة التمثيل إلى حد كبير (مثل النسخ النشاط عامل أو النشاط كيناز) المرتبطة قائمة الجينات.
- انقر على اسم المدى للحصول على معلومات أكثر تفصيلا و "RT" (المصطلحات ذات الصلة) إلى قائمة المصطلحات الأخرى ذات الصلة.
- انقر على شريط الأرجواني لسرد كافة الجينات المرتبطة المقابلة الفئة الفردية.
ملاحظة: تسرد العمودين الأخيرين نتائج الاختبارات الإحصائية 'لكل فئة.
- أداة الشرح وظيفية 3: الوظيفي الجدول الشرح
- عودة إلى صفحة ملخص وانقر على 'Functioنال الجدول الشرح "لرؤية قائمة بجميع شروح المرتبطة الجينات على القائمة دون أي حسابات الإحصائية.
ملاحظة: هذه الأداة يمكن أن تكون مفيدة لتحليل الجينات بواسطة الجينات من قائمة أو لننظر، جينات معينة مثيرة للاهتمام للغاية.
- عودة إلى صفحة ملخص وانقر على 'Functioنال الجدول الشرح "لرؤية قائمة بجميع شروح المرتبطة الجينات على القائمة دون أي حسابات الإحصائية.
- الجينات الوظيفية أداة تصنيف
- العودة إلى معالج تحليل "وانقر على وحدة" جين التصنيف الوظيفي "للفصل بين قائمة الجينات مساهمة في المجموعات ذات الصلة وظيفيا من الجينات في المرتبة وفقا لها" نقاط التخصيب "، وهو مقياس لتخصيب العام للمجموعة الجينات في القائمة.
- انقر على اسم المدى للحصول على معلومات أكثر تفصيلا و "RG" للكشف عن الجينات ذات الصلة وظيفيا من مجموعة الجينات
- انقر على 'T' أحمر (تقارير المدى) إلى قائمة الأحياء المرتبطة به ورمز أخضر لمشاهدة عرض ثنائي الأبعاد من جميع الجينات وشروط.
- الجينات اسمعارض دفعة
- العودة إلى معالج تحليل "وانقر على 'اسم جين عارض دفعة" لترجمة "Wormbase جين معرفات" إلى أسماء الجين يناظرها. (WBGene00022855 = حدات التخفيض المعتمد المؤقتة-1).
- انقر على اسم الجين للحصول على مزيد من المعلومات الجينات المحددة.
- انقر على "RG" الارتباط (الجينات ذات الصلة) بجانب كل جين للكشف عن الجينات وتوقع أن تكون مرتبطة وظيفيا لهذا الجين من الفائدة.
4. تحميل البيانات RAW على NCBI تسلسل مقروءة الأرشيف (SRA)
- الوصول إلى صفحة ويب SRA في تسجيل الدخول إلى رابط NCBI "أو تسجيل حساب جديد.
- انقر على 'Bioproject.
- انقر على 'تقديم' تحت 'استخدام Bioproject' العنوان على اليسار.
- حدد الخيار "تقديم الجديد". تحديث التفاصيل من مقدم. تواصل من خلال علامات التبويب السبعة المتبقية، وملء في تفاصيل التجربة والبيانات التي يتم تحميلها. انقر على "إرسال" عند اكتماله.
ملاحظة: في علامة التبويب الخامس "العينة البيولوجية"، وترك فتحة ل"العينة البيولوجية" فارغة. - تحديث الصفحة الناتجة عن طريق النقر على رابط 'بلدي التقديمات. وسيتم إدراج البيانات المقدمة مع عدد تقديمها المخصصة، وصف موجز ومركز تحميل.
- انقر على 'العينة البيولوجية "في الجزء العلوي من هذه الصفحة، في" بدء تقديم الجديد "مربع وإنشاء" تقديم الجديد ". إرسال طلبات منفصلة لكل عينة.
- كما هو الحال مع "Bioproject" في 4.4، تحديث تفاصيل مقدم والاستمرار من خلال ما تبقى من علامات التبويب ملء في تفاصيل كل علامة تبويب. وبمجرد الانتهاء مراجعة وانقر فوق "إرسال".
- انتقل إلى HTTP: //www.ncbi.nlm.nih.goت / حساب الاحتياطي الخاص لإنشاء النهائي "تسلسل مقروءة الأرشيف (SRA)" الاستسلام.
- انقر على 'تسجيل الدخول إلى حساب الاحتياطي الخاص "تحت عنوان" الشروع في العمل ".
- في الصفحة التالية انقر على رابط 'NCBI PDA. سيتم فتح رابط 'تفضيلات تحديث' حتى. أكمل النموذج وانقر على 'تفضيلات حفظ'.
- في الصفحة التي تظهر، انقر على 'إنشاء الجديدة تقديم "الارتباط. أدخل اسم مناسب تحت "اسم مستعار" ثم انقر على "حفظ". سيتم إنشاء الجدول مع ID تقديم وغيرها من التفاصيل.
- انقر على "تجربة جديدة" وتسجيل مكتبة تسلسل فريد واحد على الأقل لكل "العينة البيولوجية.
- تعيين وربط إنشاؤها مسبقا "BioProject" وID تقديم ل"العينة البيولوجية. سيتم إنشاء A 'تجربة جديدة.
- انقر على "تشغيل جديد" في أسفل الصفحةبعد أن تم إجراء تجربة SRA والتعرف على ملفات البيانات التي تحتاج إلى أن تكون مرتبطة بها.
- حساب مجموع MD5 كل ملف البيانات. للقيام بذلك على محطة ماكنتوش، انتقل إلى تطبيقات / المرافق / الطرفي. في المحطة، ونوع في "MD5" (بدون علامات التنصيص) تليها مسافة. سحب وإسقاط الملفات التي تحتاج إلى تحميلها إلى محطة من مكتشف وانقر على 'أدخل'.
- سوف محطة بإرجاع مبلغ MD5 أبجدية. أدخل هذا كجزء من عملية التقديم لتحميل الملف. استخدام اسم المستخدم وكلمة المرور التي يوفرها نظام لتحميل الملفات باستخدام FTP.
Access restricted. Please log in or start a trial to view this content.
النتائج
في C. elegans، يكون القضاء على الخلايا الجذعية سلالة الجرثومية (GSCs) يمتد عمر، ويعزز مرونة الإجهاد، ويرفع الدهون في الجسم 24 و 28. فقدان GSCs، سواء الناجمة عن الليزر الاجتثاث أو عن طريق الطفرات مثل GLP-1، يسبب تمديد عمر ?...
Access restricted. Please log in or start a trial to view this content.
Discussion
أهمية التسلسل منصة غالاكسي في علم الأحياء الحديث
أصبح مشروع غالاكسي دورا أساسيا في مساعدة علماء الأحياء دون تدريب المعلوماتية الحيوية لمعالجة وتحليل البيانات تسلسل عالية الإنتاجية بطريقة سريعة وفعالة. كانت تعتبر مهمة شاقة، جعلت...
Access restricted. Please log in or start a trial to view this content.
Disclosures
الكتاب ليس لديهم ما يكشف.
Acknowledgements
فإن الكتاب أود أن أعرب عن امتناني لمختبرات والمجموعات والأفراد الذين طوروا غالاكسي وDAVID، وبالتالي جعل NGS الوصول إليها على نطاق واسع للمجتمع العلمي. واعترف المساعدة والمشورة التي يقدمها زملاؤه في جامعة بيتسبرغ خلال التدريب المعلوماتية الحيوية لدينا. وأيد هذا العمل من قبل المؤسسة الطبية الباحث إليسون الجديد في الشيخوخة جائزة (AG-NS-0879-12) ومنحة من المعاهد الوطنية للصحة (R01AG051659) إلى AG.
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
References
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22(2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085(2011).
- Consortium, C. eS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383(2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, Web Server issue W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639(2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788(2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158(2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved