
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Транскриптомный Анализ
В этой статье
Резюме
Galaxy и DAVID появились в качестве популярных инструментов, которые позволяют следователям без подготовки биоинформатики для анализа и интерпретации РНК-Seq данных. Мы опишем протокол для C. Элеганс исследователей проводить РНК-Seq экспериментов, доступ и обрабатывать набор данных с помощью Galaxy и получить значимую биологическую информацию из списков генов с использованием DAVID.
Аннотация
Следующее поколение секвенирования (NGS) технологии революционизировали природу биологических исследований. Из них РНК Секвенирование (Секвенирование РНК) возникла как мощный инструмент для анализа экспрессии генов и картирования транскриптом. Однако, обработка РНК-Seq наборов данных требует сложной вычислительной опыт и создает присущие проблемы для биологии исследователей. Это узкое место было смягчено Galaxy проекта открытого доступа, что позволяет пользователям без навыков биоинформатики для анализа РНК-Seq данных, а также базы данных для аннотации, визуализации и Integrated Discovery (DAVID), ген Онтология (ГО) термин свита анализ, который помогает получить биологическое значение из больших массивов данных. Тем не менее, для начинающих пользователей и любителей биоинформатики, самообучения и ознакомлении с этими платформами может быть отнимающим много времени и сложной. Опишем простой рабочий процесс , который поможет C. Элеганс исследователи изолировать червь РНК, проводят РНК-Seq эксперименти анализировать данные с помощью Galaxy и DAVID платформ. Этот протокол обеспечивает пошаговые инструкции по использованию различных Galaxy модулей для доступа необработанных данных NGS, проверки контроля качества, выравнивание и анализ экспрессии дифференциального гена, направляя пользователя с параметрами на каждом этапе, чтобы создать список генов, которые могут быть подвергнуты скринингу для обогащения классы генов или биологические процессы с использованием Давида. В целом, мы ожидаем , что эта статья будет предоставлять информацию C. Элеганс исследователей , проводящим РНК-Seq экспериментов впервые, а также частых пользователей , работающих с небольшим количеством образцов.
Введение
Первое секвенирование генома человека, осуществляется с использованием методы дидеоксинуклеотидного-секвенирование Фреда Сэнгера, потребовалось 10 лет, а стоимость , по оценкам US $ 3 млрд 1, 2. Однако, в чуть более десяти лет с момента своего создания следующего поколения секвенирования технологии (NGS) позволило секвенировать весь геном человека в течение двух недель и за $ 1000 долларов США. Новые инструменты NGS, которые позволяют постоянно увеличивающиеся скорости сбора данных секвенирования-с невероятной эффективностью, наряду с резким сокращением стоимости, реконструируют современную биологию в немыслимых способах, как секвенирование генома проекты быстро становятся обычными явлением. Кроме того, эти события оцинкованных прогресса во многих других областях, таких как анализ экспрессии генов с помощью РНК-секвенировании (РНК-Seq), изучения генома эпигенетических модификаций, ДНК-белковых взаимодействий, и скрининг на микробное разнообразие в человеческих хостах. НГС на основе РНК-Seд, в частности, позволило выявить и карты Транскриптом Комплексным точности и чувствительности, а также заменила технологию микрочипов в качестве метода выбора для выражения профилирования. Хотя технология микрочипов широко используется, она ограничена его опорой на уже существующих массивов с известной геномной информации, а также другие недостатки, такие как кросс-гибридизации и ограниченного диапазона изменений экспрессии, которые могут быть надежно измерить. РНК-сло, с другой стороны, может быть использованы для обнаружения известных и неизвестных транскриптов при производстве низкого фонового шума из-за его однозначное отображение ДНК природы. РНК-Seq, вместе с многочисленными генетическими инструментами, предлагаемые модельными организмами, такие как дрожжи, мухами, черви, рыбы и мышей, послужили основу для многих важных недавних биомедицинских открытий. Тем не менее, остаются значительные проблемы, которые делают NGS недоступными для широкого научного сообщества, в том числе ограничения, хранения, обработки, и больше всего, м eaningful анализ биоинформатики больших объемов данных секвенирования.
Быстрое развитие технологий секвенирования и накопления экспоненциальной данных создали большую потребность в вычислительных платформах, что позволит исследователям получить доступ, анализировать и понимать эту информацию. Ранние системы были в значительной степени зависят от компьютерного программирования знаний, в то время как геномные браузеры, такие как NCBI, что позволило не программистам доступ и визуализировать данные не позволяют сложные анализы. Платформа веб-, открытого доступа, Galaxy ( https://galaxyproject.org/ ), заполнил эту пустоту и доказали свою ценность трубопровода , что позволяет исследователям для обработки данных NGS и выполнять целый спектр простых в комплексе биоинформатики анализ. Галактика была первоначально создана и поддерживается, лабораториями Антона Некрутенко (Penn State University) и Джеймс Тейлор (Университет Джона Хопкинса)е "> 3. Галактика предлагает широкий спектр вычислительных задач , что делает его„одной остановки магазин“для бесчисленных нужд биоинформатики, включая все этапы , участвующих в исследовании РНК-Seq. Itallows пользователям выполнять обработку данных либо на своих серверах или локально на своих машинах. Данные и рабочие процессы могут быть воспроизведены и совместно. Интерактивные руководства, раздел справки, и вики-страницы ( https://wiki.galaxyproject.org/Support ) , посвященный проекту Galaxy обеспечивают постоянную поддержку. Тем не менее, для начинающих пользователей, особенно тех, без обучения биоинформатики, трубопровод может оказаться сложными и процесс самообучения и ознакомлению может занять много времени. Кроме того, биологическая система изучена, и особенность эксперимента и используемые методов, влияние аналитические решения на несколько шагов, и они могут быть трудно ориентироваться без инструкции.
Общий Р.Н. А-Seq Галактики Рабочий процесс состоит из загрузки данных и проверки качества с последующим анализом с помощью Tuxedo Suite 4, 5, 6, 7, 8, 9, которая является коллективным различных инструментов , необходимых для различных этапов анализа данных 10 РНК-Seq, 11, 12, 13, 14. Типичная Секвенирование РНК эксперимент состоит из экспериментальной части (подготовки образца, изоляции мРНК и кДНК библиотеки препарата), то НГС и анализ биоинформатики данных. Обзор этих секций, а также шаги , участвующих в трубопроводе Galaxy, показаны на рисунке 1.
3fig1.jpg»/>
Рисунок 1: Обзор РНК-Seq рабочий процесс. Иллюстрация экспериментальных и вычислительных стадий, участвующих в РНК-Seq эксперимент, чтобы сравнить ген-профили экспрессии двух червячных штаммов (А и В, оранжевый и зеленый линиями и стрелками, соответственно). Различные модули используются Галактики показаны в коробках с соответствующим шагом в нашем протоколе, указанном в красном цвете. Выходы различных операций записываются в сером цвете с форматами файлов, показанных синим цветом. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
Первый инструмент в смокинге Suite , это программа выравнивания называется "Tophat. Он расщепляет входной NGS читает на более мелкие фрагменты, а затем отображает их на референсный геном. Этот двухэтапный процесс обеспечивает то, что читает охватывающих интронных регионов, выравнивание может быть иначе диsrupted или пропущенный учитываются и отображаются. Это увеличивает охват и облегчает идентификацию нового сплайсинга. Tophat выход сообщаются в виде двух файлов, кровать файлы (с информацией о сплайсинге , которые включают в себя геномное местоположение) и БАМ файл (с подробной информацией отображения каждого чтения). Затем файл BAM выровнен относительно эталонного генома оценить обилие индивидуальных транскриптов в пределах каждого образца с помощью последующего инструмента в смокинге люкс под названием «Запонки». Запонки функции путем сканирования выравнивания , чтобы сообщить полнометражных фрагменты транскриптов или «transfrags» , которые охватывают все возможные варианты сплайсинга во входных данных для каждого гена. Исходя из этого, он генерирует «» транскрипта (сборку всех транскриптов, полученные на ген для каждого гена) для каждого образца быть секвенированы. Эти Запонки сборки затем разрушилась или объединены вместе вместе с реFerence геном для создания одного файла аннотаций для нисходящего дифференциального анализа с использованием следующего инструмента, «Cuffmerge». Наконец, экспрессия гена инструмент измеряет дифференциальное в «» Cuffdiff между образцами путем сравнения Tophat выходов каждого из образцов до конечного выходного файла Cuffmerge (рисунок 1). Запонки используют FPKM / RPKM (Фрагменты / Считывает За килобазу транскрипта на миллион отображенных прочтений) значения , чтобы сообщить транскрипты содержаний. Эти значения отражают нормализацию необработанных данных NGS для глубины (среднего числа считывает из образца, который выравнивать к опорному геном) и длина гена (гены имеют разную длину, так что отсчеты должны быть нормализованы по длине гена, чтобы сравнить уровни между генами). FPKM и RPKM, по существу, то же самое с RPKM используется для одностороннего РНК-Seq, где каждый чтения соответствует одному фрагменту, в то время, FPKM используется дляпарноконцевое РНК-Seq, как это объясняет тот факт, что два читает могут соответствовать одному и тому же фрагменту. В конечном счете, результаты этих анализов приведен список генов, выраженных дифференциально между условиями и / или штаммами.
После успешного запуска Galaxy завершен и «список генов» генерируется, следующий логический шаг требует больше биоинформатики анализа выводить значимые знания из массивов данных. Многие программные пакеты появились , чтобы удовлетворить эту потребность, в том числе публично доступные веб-вычислительные пакетов , такие как DAVID (базы данных для аннотаций, визуализации и интегрированного открытия) 15. DAVID облегчает назначение биологического смысла для больших списков генов из исследований с высокими пропускной способностью, сравнивая закачанный список генов в его комплексных биологических базе знаний и выявлении биологических аннотаций, связанные со списком генов. После этого следует обогатительному анализ, то есть тесты на язьntify, если любой биологический процесс или ген класс перепредставлен в списке гена (ов) в статистически значимом образе. Он стал популярным выбором из - за сочетанием широкого, комплексной базы знаний и мощных аналитических алгоритмы , которые позволяют исследователям обнаружить биологические темы , обогащенные в геномика производных «списки генов» 10, 16. Дополнительные преимущества включают его способность обрабатывать списки генов, созданные на любой платформе секвенирования и очень удобный интерфейс.
Нематоды Caenorhabditis Элеганс является генетическая модель системы, хорошо известна своими многочисленными преимуществами , такими , как малый размер, прозрачный корпус, простой план тела, легкость культуры и большой аменабельностью генетической и молекулярной диссекции. Черви имеют небольшой, простой и хорошо аннотированный геном , который включает до 40% консервативных генов с известными человеческими гомологами 17. Действительно, C. Элегансбыл первым метазоа чей геном был полностью секвенирован 18, и один из первых видов , где Секвенирование РНК была использованы для отображения транскрипта организма 19, 20. В начале червячные исследованиях участвовали экспериментирование с различными методами с высокой пропускной способностью захвата РНК, подготовка библиотеки и последовательности, а также биоинформатики трубопроводов , которые внесли свой вклад в развитие технологии 21, 22. В последние годы, РНК-Seq на основе экспериментирования у червей стало обычным явлением. Но для традиционных червячных биологов проблема, связанная с компьютерным анализом РНК-Seq данных остается основным препятствием для более широкого и эффективного использования техники.
В этой статье мы опишем протокол для использования платформы Galaxy для анализа данных РНК-Seq высокой пропускной способности, полученные от С. Элеганс. Для многих, впервые и малого АССпользователи ле, наиболее экономически эффективным и простым способом провести РНК-Seq эксперимент является выделение РНК в лаборатории и использовать коммерческую (или в доме) NGS средство для получения кДНК библиотек секвенирования и самой NGS. Таким образом, мы первым подробно шаги , вовлеченные в изоляции, количественное и качественное оценка C. Элеганс образцов РНК для РНК-Seq. Далее, мы обеспечиваем шаг за шагом инструкции по использованию интерфейса Galaxy для анализа данных NGS, начиная с тестами для проверки контроля качества после секвенирования с последующим выравниванием, сборкой и дифференциальной количественной оценкой экспрессии гена. Кроме того, мы включили направления внимательно изучить списки генов, полученные из Галактики для изучения биологического обогащения с использованием DAVID. В качестве последнего шага в рабочем процессе, мы предоставляем инструкции для загрузки РНК-Seq данных на публичных серверах , таких как последовательность чтения архива (SRA) на NCBI ( HTTP: // шww.ncbi.nlm.nih.gov/sra) , чтобы сделать его свободно доступным для научного сообщества. В целом, мы ожидаем, что эта статья будет оказывать всестороннюю и достаточную информацию для червячных биологов, осуществляющих РНК-Seq экспериментов впервые, а также частых пользователей, работающих с небольшим количеством образцов.
Access restricted. Please log in or start a trial to view this content.
протокол
Выделение 1. РНК
- Меры предосторожности
- Вытирают все рабочую поверхность, инструменты и пипетку с использованием коммерчески доступными РНКазами спрей для устранения любых РНКазов присутствуют.
- Надевайте перчатки во все времена, регулярно меняя их свежими в течение различных этапов протокола.
- Используйте только советы фильтра и сохранить все образцы на льду как можно больше, чтобы избежать деградации РНК.
ПРИМЕЧАНИЕ: Для получения наилучших данных от NGS платформ, очень важно начать с высоким качеством РНК. Выделение РНК и подготовка методы различаются в зависимости от образца происхождения, метода секвенирования и исследователем предпочтений. Несколько коммерчески доступных наборов могут быть использованы для этой цели, или РНК могут быть также выделены с использованием стандартного метода фенол-хлороформ экстракции РНК. При любой методике, предупредительные меры, перечисленные выше, должны соблюдаться на протяжении всего процесса, чтобы свести к минимуму загрязнение и ОБТAin образцы нетронутой РНК.
- Сбор Worms
- Синхронизировать популяцию червя гипохлорита отбеливающей обработкой 23 для получения 1000-1500 подобранных по возрасту C. Элеганс взрослых червей на штамм.
- Промыть червь от пластины с использованием буферного раствора M9 и спины при 325 мкг на стол центрифуги в течение 30 секунд. Аспирата из буфера M9 оставляя за таблеткой червей. Повторите этот шаг, по крайней мере, три раза, чтобы исключить бактериальный унос.
- К осадку червячного, добавить ~ 500 мкл буфер для лизиса (при использовании коммерческого набора) или Trizol (моно-фазной раствор фенола и гуанидинизотиоцианата, если фенол: хлороформ экстракция описан в разделе 1.3.3 предпринимается), чтобы разрушить ткани червя , деактивировать РНКазы и стабилизации нуклеиновых кислот.
Примечание: Этот протокол может быть приостановлен здесь вспышкой замораживания образцов в жидком азоте с последующим хранением при -80 ° С.
- Выделение РНК
- Соникатные образцы червей на 45% амплитуды в циклах 20 с. 'ON' и 40 сек. 'OFF' (8-12 циклов в деформации). Храните образцы на льду во все времена.
Примечание: Убедитесь, что Sonicator зонд погружали в буфер и поддерживается на постоянном уровне в течение всего. Избегайте вспенивания образца и тщательно очистить зонд в промежутке между образцами. циклы обработки ультразвука могут варьироваться в зависимости от типа используемого для обработки ультразвука. Рекомендуется, чтобы условия обработки ультразвуком сначала оптимизированы на тестовом образце перед началом эксперимента. - При использовании коммерчески доступного набора, продолжения выделения РНК в соответствии с предписанным протоколом. Для выделения РНК с использованием способа фенол-хлороформ, выполняют следующие действия.
- Центрифуга образцы обрабатывали ультразвуком при 16000 х г в течение 10 мин. при 4 ° С
- Передача супернатант в 1,5 мл РНКазы микроцентрифужную пробирку и добавляют 100 мкл хлороформа (1/5 го объема реагента для выделения РНК / ДНК).
предосторожность: Хлороформ является токсичным. Для того, чтобы свести к минимуму воздействие и избегать вдыхания, работать в химической капотом при обращении с этим веществом. - Vortex образцы тщательно в течение 30 - 60 с. и пусть образцы сидеть при комнатной температуре в течение 3 мин.
- Центрифуга при 11750 х г в течение 15 мин. при 4 ° С. Передача только верхний водный слой к новому РНКазов микрофужных трубок, стараясь не аспирата белого интерфейса ДНК-содержащей. Повторите шаги 1.3.4 через 1.3.6.
- Добавьте 250 мкл (70% водной фазы или 1/2 РНК / ДНК, выделение объема реагента) 2-пропанола и инвертировать трубку для смешивания. Пусть трубки сидят при комнатной температуре в течение 10 мин или оставить в течение ночи при -80 ° С.
- Центрифуга образцов при 11750 х г в течение 10 мин. при 4 ° С. Слейте супернатант очень осторожно, оставляя за собой несколько мкл на дне пробирки так, чтобы осадок не нарушается.
- Промыть осадок 500 мкл 75% -ного этанола (сделанный с использованием РНКазы свободной воды) и спином вниз при 16000 х г в течение 5 мин.т 4 ° С.
- Удалить столько супернатант насколько это возможно, не нарушая гранул. Воздух сухой осадок в шкафу в течение нескольких минут.
- Добавьте 30 мкл РНКазы свободной воды и помогают растворить РНК гранул при нагревании в течение 10 мин. при 60 ° C.
- Проверьте качество РНК и количество с помощью Bioanalyzer.
Примечание: Bioanalyzer генерирует R НС I ntegrity N умбру (RIN) в качестве меры качества РНК. RIN, по меньшей мере, 8 рекомендуемый порог для РНК-Seq образцов (выше, тем лучше). количество и качество РНК также могут быть проверены спектрофотометрически, но и должны сопровождаться визуальной оценки целостности РНК. Чтобы сделать это, запустить образцы на агарозном геле 1,2% достаточно долго, чтобы получить подходящий разделение 28s и 18s рибосомальной РНК полос. Наличие двух отдельных полос (1,75 кб для 18S рРНК и 3,5 кб для 28s рРНК в случае C. Элеганс) является приемлемой мерой качества РНК. - Используйте ~ 100 нг / мкл РНК шир податель / NGS объект для подготовки библиотек секвенирования.
Примечание: образцы РНК должны быть отправлены на сухой лед к поставщику услуг секвенирования. Большинство провайдеров провести независимую проверку РНК контроля качества перед приготовлением библиотеки.
- Соникатные образцы червей на 45% амплитуды в циклах 20 с. 'ON' и 40 сек. 'OFF' (8-12 циклов в деформации). Храните образцы на льду во все времена.
2. Секвенирование РНК Анализ данных
- Загрузка сырья секвенирования данных
- Загрузить сжатые данные секвенирования сырого fastq закодированных в формате fastq.gz от поставщика NGS , используя «протокол передачи файлов (FTP»).

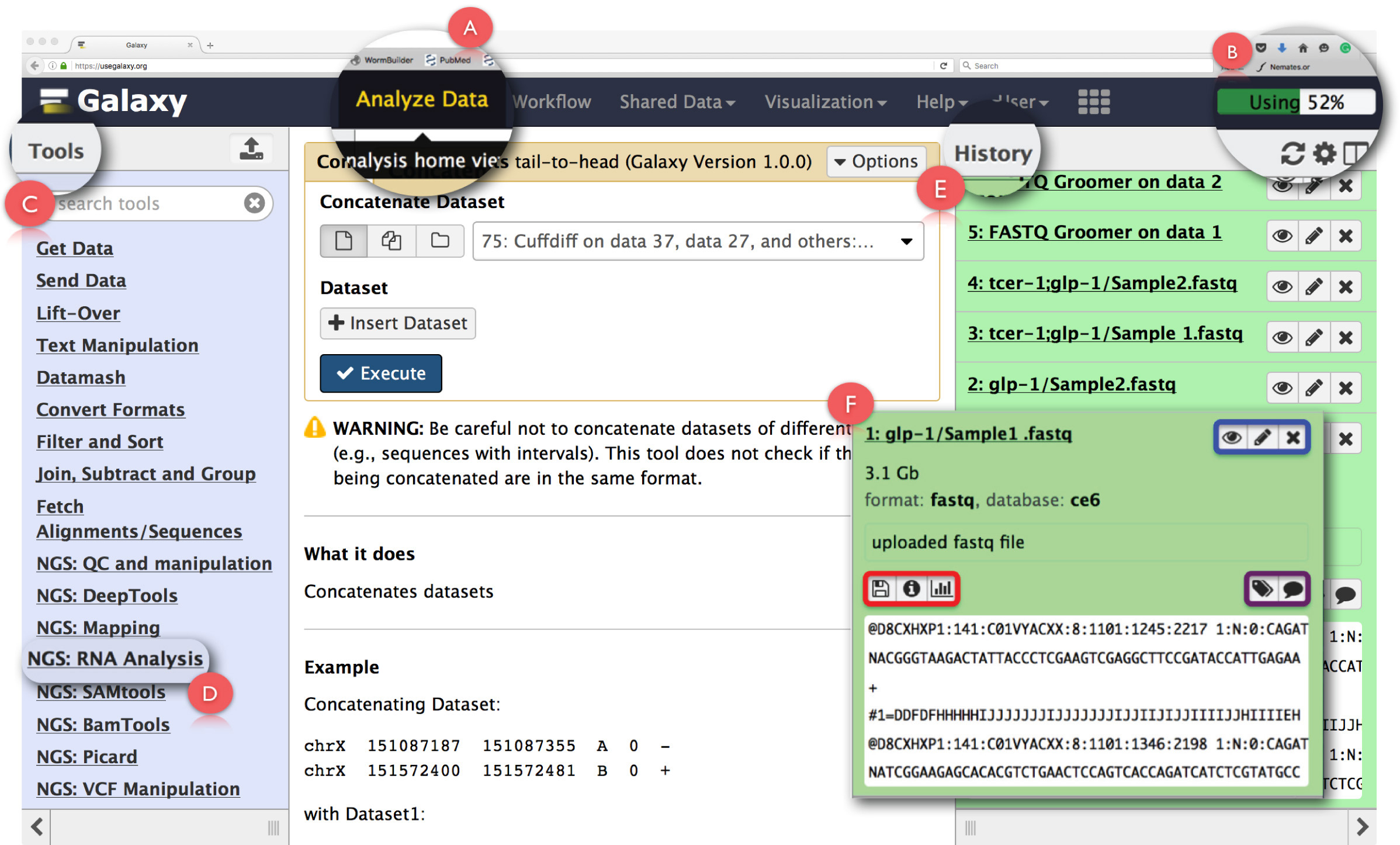
Рисунок 2: Компоновка пользователя Galaxy панели интерфейса и основные РНК-Seq функции. Основные особенности страниц расширены и подсвечиваются. (A) указывает на функцию «Анализ данных» в заголовке веб - страницы , используемый для доступа Анализ Главная Просмотреть. (В) является «Прогресс бар» , что указывает на пространство на сервере Galaxy используемой операции. (C) является «Инструменты Раздел» , в котором перечислены все инструменты , которые могут быть запущены на интерфейсе Galaxy. (D) , показывает «NGS: РНК анализа» раздела инструмент , используемый для РНК-Seq анализа. (E) изображает панель «История» , в котором перечислены все файлы , созданные с помощью Galaxy. (F) показан пример диалогового окна , которое открывается при нажатии на любой файл в разделе Истории. В (F), синяя коробка подчеркивает значки , которые могут быть использованы для просмотра, editthe атрибутов или удалить набор данных, фиолетовое окно подчеркивает значки , которые могут быть использованы для «редактирования» наборов данных тегов или аннотации, а красный прямоугольник показывает иконку для загрузки данных, просматривать детали задания выполняются или повторите операцию. Пожалуйста , нажмите здесь , чтобы посмотреть увеличенную версию этой фигуры.
{kind=link}
- Начало работы с Galaxy
Примечание: Galaxy может быть запущен на бесплатном общедоступном сервере, используя веб-платформу, обеспечивающую доступ к облаку и бесплатный ограниченное хранение. Кроме того, можно загрузить и запустить локально на компьютере пользователя или вычислительных кластеров, проводимых учреждениями, но локальной обработки, могут быть ограничены пределами данных для хранения и ограничения мощности обработки пользовательских машин. Сведения о загрузке и установке можно получить по адресу https://wiki.galaxyproject.org/Admin/GetGalaxy . В этом протоколе мы опишем веб-использование трубопровода Galaxy.- После загрузки и сохранения данных NGS на компьютере пользователя, Galaxy доступа вlaxy.org/»целевых = "_blank"> https://usegalaxy.org/.
- Зарегистрировать учетную запись пользователя, нажав на «User» в заголовке страницы, логин и начать с ознакомления с панелью пользовательского интерфейса.
Примечание: Рекомендуется , чтобы неопытные пользователи используют «Начать здесь» учебник предоставляется на главной странице , чтобы ознакомиться с базовой установкой до Галактики ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) , - Нажмите на «Анализ данных» (Рисунок 2A) в панели заголовка , чтобы получить доступ к «Анализ Home View» , который также начальный экран на Galaxy.
Примечание: В заголовке также имеются другие ссылки, чьи детали можно увидеть при наведении указателя мыши на них. Верхний правый угол заголовка имеет индикатор , который контролирует пространство , используемое для выполнения задач (рис 2В). - Слизать на «NGS: РНК анализ» задача в «меню Tools» на левой панели (рис 2С) , чтобы получить доступ все инструменты , необходимые для анализа данных РНК-сл.
Примечание: «Меню Инструменты» каталоги всех операций , что Galaxy предложения. Это меню разделено на основе задач и нажать на любом из откроет список всех инструментов, необходимых для выполнения этой задачи. - Создать новую историю анализа, нажав на значок шестеренки в верхней части панели «История» справа (рис 2E). Выберите «Создать новый» вариант из всплывающего меню. Дайте эту «историю» подходящее имя для идентификации анализа.
Примечание: панель «История» показывает все файлы, загруженные для анализа, а также все выходные файлы, которые создаются путем запуска задач по Галактике. Щелчок по имени файла в этой панели открывает диалоговое окно с подробной информацией о задаче, выполняемойи фрагмент набора данных (рис 2F). Иконки в этом окне позволяют пользователю «зрения», «редактировать атрибуты» или «удалить» набор данных (рис 2F, выделены синим цветом). Кроме того, пользователь может также «редактировать» набор данных тегов или аннотацию (рис 2F, выделены фиолетовым цветом), «загрузить» в данных, «показать детали» от задачи, «перезапустить» задачу или даже «визуализировать» набор данных из этого диалоговое окно (рис 2F, выделены красным цветом). - Нажмите функцию «Загрузить Файл» в разделе «Получить данные» в «ToolsMenu» , чтобы загрузить исходные файлы fastq.
ПРИМЕЧАНИЕ: При нажатии на этот или любой другой инструмент открывает краткое описание работы, а сам тест, в средней панели «Analysis интерфейса». Эта панель шнурки вместе«Инструменты» из левой панели и «Input Files» из панели справа «History» (рис 2Е). Здесь входные файлы из «Историй» выбраны и другие параметры , определенные для выполнения данной задачи. Результирующий выходной набор данных из каждого теста сохраняется обратно в «Истории». В комплекте с тестом на панели "Анализ Interface" является объяснение всех доступных параметров для запуска данного инструмента вместе с подробным списком всех выходных файлов генерирует инструмент. - После открытия задачи в «Analysis Interface», нажмите на кнопку «Выбрать локальный файл» или «Выберите FTP File» (быстрее загрузка), перейдите в папку , содержащую файлы секвенирования и выбрать подходящий набор данных для загрузки.
- Разрешить Гэлакси «Автоопределение» Загруженный файл типа (настройка по умолчанию). Выбрать 'C. эльEgans 'в раскрывающемся меню для генома.
- Нажмите на «Start» , чтобы начать загрузку данных. После того , как файл будет загружен, он будет сохранен в панели «История» и может быть доступен оттуда.
- Если файлы данных множественного секвенирования производятся для одного образца, объединить их с помощью инструмента «Concatenate». Чтобы сделать это, откройте опцию «Text Manipulation» в "меню Tools.
- Нажмите на инструмент «Соединить», выберите файлы , которые должны быть объединены с раскрывающимся вниз в середине «Анализ интерфейса» и нажмите кнопку «Выполнить».
Примечание: Выходные файлы , созданные с помощью этой задачи генерируется в формате fastq. Программа отображения имеет предел 16000000 последовательностей в fastq файл и при достижении этого предела нового файл fastq генерируется для остальных последовательностей. '; Объединить»инструмент необходим в таких случаях объединить наборы данных. - Преобразование загруженных файлов формата fastq в требуемый формат fastqsanger для Galaxy РНК-Seq анализа с помощью «fastq грумер» инструмент найден под «NGS: QC и манипуляция» раздел (см дополнительный файл).
- Выберите подходящий fastq набор данных под «File жениху» опции и запустить инструмент с использованием параметров по умолчанию.
Примечание: Выходные файлы , созданные с помощью этой задачи генерируются в формате fastqsanger.
- Тесты fastqsanger данных контроля качества
- Проверьте качество загружаемой fastqsanger считывает с помощью инструмента «FastQC» , расположенного под «NGS: QC и манипуляция» в меню «Инструменты».
- Выберите ухоженную fastqsanger файл данных из выпадающего меню для "Шорт читать данные из текущей библиотеки»и запустите инструмент с использованием параметров по умолчанию.
Примечание: Обратите особое внимание на качество чтений и наличие каких-либо адаптеры последовательностей. Адаптеры обычно удаляются как часть РНК-Seq обработки данных после поставщиками NGS, но в некоторых случаях, может быть оставлен позади. Для объяснения стандартов качества перейти к http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - Проверьте с поставщиком NGS и если адаптеры присутствуют, обрезать их с помощью инструмента «Клип» от «NGS: QC и манипуляция» меню задач.
Примечание: Выходные файлы , созданные с помощью этой задачи создаются в сыром текстовом формате, так и в HTML , который можно открыть на любом веб - браузере.
- Анализ данных с помощью Tuxedo Люкс
- TopHat
- Скачать последнюю версию С. Элеганс референсный геном FASTA и ГТФ (Gene Transfer Format) файлы Загрузка файла» , как описано выше в 2.2.6.
- Откройте «NGS: РНК анализ» раздел и нажмите на «TopHat» инструмент для отображения секвенирования читает загруженном референсный геном.
- Выберите подходящий ответ из выпадающего меню на вопрос "Является ли это одним концом или парноконцевое данные?
- Выберите подходящий файл fastq.
- Выберите «Использовать геном из истории» в следующем выпадающем меню и выбрать ссылочный геном , загруженный на шаге 2.4.1.1.
- Выберите « По умолчанию» для остальных параметров и нажмите кнопку «Выполнить».
Примечание: Среди выходных файлов , произведенных с помощью этой задачи, файле «ПРИНИМАЮТСЯ Hits» используется для последующих шагов.
- Запонки и Cuffmerge
- Выберите «Каффаинструмент в „Links NGS: раздел РНК анализа“ , чтобы собрать стенограммы, оценить их численность и тест для дифференциальной экспрессии.
- В первом раскрывающемся меню выберите отображенные «Принятые хиты (формат BAM)» файл , полученные из анализа TopHat.
- Во втором раскрывающемся меню, установите опорный аннотацию к УПГ файл , загруженный на шаге 2.4.1.1.
- Выберите «Да» для параметра «Выполнение коррекции смещения» и выполнить задачу , используя настройки по умолчанию для всех остальных параметров.
Примечание: Среди выходных файлов , произведенных с помощью этой задачи, файл «ПРИНИМАЮТСЯ транскриптов» используется для последующих шагов. - Инструмент Open «Cuffmerge» в «NGS: РНК - анализ» , чтобы объединить «» в собранном виде транскрипты производится для всех РНК-Seq образцов.
Примечание: Первое поле в инструмент самостоятельно заполнит и списки всех GTF файлы , созданные Запонки. - Выберите «Сборные транскрипты» файл для всех штаммов / тестируемых условий, в том числе биологических повторностей одного и то же штамм / состоянии (см обсуждения для биологических повторностей).
- Выберите «Да» для «использования ссылочной Аннотации» и выберите GTF файл , загруженный на шаге 2.4.1.1.
- В следующем окне, снова выберите «Да» для опции «Использовать Секенс Дата» и выбрать весь файл геном Fasta загруженный на шаге 2.4.1.1.
- Сохраняя другие параметры по умолчанию, нажмите кнопку «Выполнить».
Примечание: Cuffmerge генерирует один GTF выходного файла.
- Cuffdiff
- Перейдите к инструменту «Cuffdiff» в «NGS: РНК анализа» раздел. В меню «транскрипты», выберите слитый выходной файл из Cuffmerge.
- меткаусловия 1 и 2 с именами двух штаммов / состояние.
Примечание: Cuffdiff может выполнять сравнение между более двух штаммами или условиями , а также экспериментами , конечно времени. Просто используйте опцию «Добавить новые условия» для добавления каждого новых штаммов / состояния, по мере необходимости. - Для каждого штамма / условия, при «повторности» выбрать отдельные выходные файлов «принимаются хитами» из Tophat , которые соответствуют различным биологическим повторам этого штамма / состояния. Удерживая нажатой клавишу «CMD», если с помощью компьютера Macintosh, и клавишу «Ctrl», если с помощью компьютера, чтобы выбрать несколько файлов.
- Оставьте все остальные параметры в качестве параметров по умолчанию. Нажмите кнопку «Выполнить» , чтобы выполнить задачу.
Примечание: Cuffdiff генерирует множество выходных файлов в табличном формате в качестве конечного отсчета РНК-Seq анализа. Они включают в себя файлы с FPKM слежения за транскриптов генов (комбинированныеЗначения FPKM транскриптов, разделяющих идентичность генов), первичных транскриптов и кодирующих последовательностей. Все файлы данные, генерируемые можно просматривать на любом приложении электронной таблицы и содержат одинаковые атрибуты, такие как название гена, локус, изменение раза (в log2 масштабе), а также статистические данные о сравнениях между штаммами / условиями, в том числе р д значения и значение. Данные в этих файлах могут быть отсортированы на основе статистической значимости различий или сложить изменение в экспрессии генов (величина и направление изменения, как и в вверх или понижающего регулируемые гены) и манипулировать ими в соответствии с требованиями пользователей. Если преобразование между различными идентификаторами генов необходимо (например, ген , Wormbase ID по сравнению с числом космиды), инструменты , доступные на Biomart ( http://www.biomart.org/ ) могут быть использованы.
- TopHat
3. Джин Онтология (ГО) Term Анализ с использованием DAVID
- Доступ DAVID с сайта чTTPS: //david.ncifcrf.gov/. Нажмите на «Start Analysis» в заголовке страницы. В «Шаг 1», копировать и вставить список генов, полученных из Галактики в коробке А. В «Шаг 2», выберите «Wormbase Gene ID» в качестве идентификатора для генов ввода.
Примечание: Дэвид распознает большинство общедоступных категории аннотаций, так что другие идентификаторы генов (например, гена Entrez ID или символ гена) также могут быть использованы. - В «Шаг 3», выберите «List Gene» (гены , которые будут проанализированы) под «Тип списка» , а затем нажмите на «Submit List» значок.
Примечание: «Анализ Wizard», откроется в список всех гиперссылка инструменты DAVID , которые могут быть запущены на загруженном список генов (рис 3). Нажмите на эти ссылки для доступа к соответствующим соответствующие модули согласно требованию пользователя. Для того, чтобы определить инструменты , подходящие для данной задачи, нажмите на кнопку "Какие инструменты DAVID использовать? «Ссылка на» ; Страница Мастер анализа». Нажмите на ссылку «Начать анализ» в заголовке , чтобы вернуться на главную страницу «Мастер анализа» в любой момент во время анализа.

Рисунок 3: Макет DAVID анализа мастера веб - страницы и примеры работы выходов. Пользовательский веб-интерфейс «Мастер анализа» перечисляет средства , используемые для анализа загруженного списка генов для обогащения на основе различных параметров. При нажатии на этих инструментах сообщают анализируемые данные в новой веб-странице. Примеры отчетов , генерируемых из табличных «Gene функциональной классификации», «Функциональной диаграммы Реферата» и «функциональной аннотации Кластеризации» показаны как вставки (стрелка).> Пожалуйста, нажмите здесь, чтобы посмотреть увеличенную версию этой фигуры.
- Функциональная Аннотация Инструмент 1: Функциональная Аннотация Кластеризация
- Нажмите на модуле «функциональная аннотация кластеризации» , чтобы перейти к странице сводки. Держите категории аннотаций по умолчанию и нажмите на кнопку «функциональная аннотация кластеризации» для создания кластеров сходных терминов аннотаций , ранжированных по их обогащению счета.
- Нажмите на гиперссылке имени каждого термина , чтобы прочитать подробную информацию об этом и «RT» (связанные термины) в список других аналогичных терминов , относящихся к данной категории.
- Нажмите на фиолетовой панели, чтобы список генов, связанные с термином и красным «G», чтобы перечислить все гены, связанные со всеми условиями в пределах кластера.
- Нажмите на зеленый значок, чтобы увидеть двухмерное представление всех генов и терминов в кластере.
Примечание: список Последние три колонки аналитические и статистические результаты для каждогосрок. Результаты этого и все другие аналитики могут быть загружены в формате .txt, нажав «Скачать файл» ссылку.
- Функциональная Аннотация Инструмент 2: Функциональная диаграмма Аннотация
- Возврат к странице сводки и нажмите на кнопку «Функциональная диаграмма аннотаций» определить существенно сверхпредставленных биологические термины (например , фактор активности транскрипции или киназа активности) , связанных со списком генов.
- Нажмите на термин имя , чтобы получить более подробную информацию и «RT» (связанные термины) в список других связанных с ним терминов.
- Нажмите на фиолетовой панели, чтобы перечислить все связанные с ним гены, соответствующие отдельные категории.
Примечание: список Последние две колонков результатов статистических-тесты для каждой категории.
- Функциональная Аннотация Инструмент 3: Функциональная Аннотация Таблица
- Возврат к странице сводки и нажмите на кнопку "FunctioNAL Аннотация Таблица ' , чтобы увидеть список всех аннотаций , связанных с генами на список без каких - либо статистических расчетов.
Примечание: Этот инструмент может быть полезен для генов по-ген анализа списка или посмотреть на конкретных, весьма интересные генах.
- Возврат к странице сводки и нажмите на кнопку "FunctioNAL Аннотация Таблица ' , чтобы увидеть список всех аннотаций , связанных с генами на список без каких - либо статистических расчетов.
- Gene Функциональная классификация инструментов
- Возвращение в «Мастере анализа» и нажмите на модуль «Gene функциональной классификации» для разделения списка ввода генов в функционально связанных групп генов , ранжированных в соответствии с их «Обогащение Score», мера общего обогащения группы генов в списке.
- Нажмите на термин имя , чтобы получить более подробную информацию и «RG» , чтобы выявить функционально связанных генов группы генов
- Нажмите на красном «T» (термин отчеты) в список ассоциированной биологии и зеленый значок, чтобы увидеть двухмерное представление всех генов и терминов.
- Gene имяПакетный просмотра
- Возвращение в «Мастере анализа» и нажмите на кнопку «имя-Gene Batch просмотра» , чтобы перевести «Wormbase Gene идентификаторов» в их соответствующих имена генов. (WBGene00022855 = вС-1).
- Нажмите на название гена, чтобы получить более подробную информацию генов конкретного.
- Нажмите на «RG» (родственные гены) рядом с каждым из гена , чтобы выявить гены , по прогнозам, будет функционально связан с геномом.
4. Загрузка RAW данные на NCBI последовательность чтения архива (SRA)
- Перейти на веб - страницу SRA на Войти в ссылке NCBI»или зарегистрировать новую учетную запись.
- Нажмите на кнопку «Bioproject».
- Нажмите «Представление» под «Использование Bioproject» заголовка слева.
- Выберите опцию «New Подчинение». Обновление деталь подателя. Продолжить через оставшиеся семь вкладок, Заполняя детали эксперимента и данные загружаются. Нажмите кнопку «Отправить» , когда завершена.
Примечание: На вкладке «пятой биопробы», оставьте слот для «биопробы» пустым. - Обновить результирующую страницу, нажав на ссылку "Мои ресурсы . Представленные данные будут перечислены с присвоенным номером представления, кратким описанием и статусом загрузки.
- Нажмите на кнопку «биопробы» в верхней части этой страницы, в «начать новую подачу» коробки и создать «новое представление». Отправить отдельные представления для каждого образца.
- Как и в случае с «Bioproject» в 4.4, обновить данные заявителя и продолжить через остальные вкладки заполнения в деталях каждой вкладки. После завершения обзора и нажмите кнопку «Отправить».
- Перейдите к HTTP: //www.ncbi.nlm.nih.gov / SRA для создания окончательного «Последовательность чтения архива (SRA)» представление.
- Нажмите на ссылку «Вход в SRA» в разделе «Начало работы».
- На следующей странице нажмите на ссылку «NCBI КПК». Ссылка «обновить настройки» откроется. Заполните форму и нажмите «Сохранить настройки».
- На открывшейся странице нажмите на ссылку «Создать новое представление» ссылку. Введите подходящее имя в разделе «Псевдоним» и нажмите «Сохранить». Таблица с идентификатором представления и другими деталями будет создана.
- Нажмите на «Новый эксперимент» и зарегистрировать по крайней мере одну уникальную библиотеку секвенирования для каждого «биопробы».
- Назначают и связать ранее созданный «BioProject» и «» биопробы представления идентификаторов. Будет создан «Новый эксперимент».
- Нажмите на кнопку «New Run» в нижней части страницыпосле того, как эксперимент SRA был сделан и идентифицировать файлы данных, которые должны быть связаны с ним.
- Вычислить сумму MD5 для каждого файла данных. Для этого на терминале MacIntosh, перейдите к Applications / Utilities / Terminal. В терминале, введите «md5» (без кавычек) с последующим пробелом. Перетащите файлы , которые должны быть загружены в терминал с искателем и нажмите «Enter».
- Терминал будет возвращать буквенно-цифровую сумму MD5. Введите это как часть процесса подачи для загрузки файла. Используйте имя пользователя и пароль, предоставленные системой для загрузки файлов с помощью FTP.
Access restricted. Please log in or start a trial to view this content.
Результаты
В С. Элеганс, устранение зародышевых стволовых клеток (GSCs) продлевает жизнь, повышает устойчивость стресс и поднимает жир тела 24, 28. Потеря GSCs, либо вызванные лазерной абляции или путем мутаций , таких как GLP-1, вызывает увеличени?...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
Значение Галактики секвенирования платформы в современной биологии
Проект Galaxy стал важную роль в содействии биологов без обучения биоинформатики для обработки и анализа данных секвенирования с высокой пропускной способностью быстро и эффективно. После того, как счита?...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторы не имеют ничего раскрывать.
Благодарности
Авторы хотели бы выразить свою благодарность лаборатории, групп и отдельных лиц, которые разработали Galaxy и DAVID, и, таким образом, сделали NGS широко доступной для научного сообщества. Помощь и рекомендации, представленные коллегами из Университета Питтсбурга во время нашего обучения биоинформатики признается. Эта работа была поддержана Эллисон Medical Foundation Нью Scholar в Старении награды (AG-NS-0879-12) и грант от Национального института здоровья (R01AG051659) на AG.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
Ссылки
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22(2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085(2011).
- Consortium, C. eS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383(2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, Web Server issue W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639(2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788(2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158(2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены