
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Análise transcriptomic de
Neste Artigo
Resumo
Galaxy e David surgiram como ferramentas populares que permitem que os investigadores sem formação bioinformática para analisar e interpretar dados de RNA-Seq. Descreve-se um protocolo para C. elegans investigadores para executar ARN-Seq experiências, o acesso e processar o conjunto de dados usando Galaxy e obter informação biológica significativa a partir das listas de genes utilizando DAVID.
Resumo
Próxima geração seqüenciamento (NGS) tecnologias têm revolucionado a natureza da investigação biológica. Destes, Sequenciação de ARN (ARN-SEQ) emergiu como uma ferramenta poderosa para a análise de expressão genética e mapeamento transcriptoma. No entanto, a manipulação de dados de RNA-Seq requer experiência computacional sofisticada e coloca desafios inerentes para pesquisadores de biologia. Este gargalo tem sido mitigada pelo projeto acesso aberto Galaxy que permite que usuários sem habilidades de bioinformática para analisar os dados de RNA-Seq, eo banco de dados para anotação, visualização e Integrated Discovery (David), um Gene Ontology (GO) Suíte análise termo que ajuda derivar significado biológico a partir de grandes conjuntos de dados. No entanto, para usuários iniciantes e amadores bioinformática, auto-aprendizagem e familiarização com essas plataformas podem ser demorado e difícil. Nós descrevemos um fluxo de trabalho simples que irá ajudá C. elegans pesquisadores para isolar RNA verme, realizar um experimento RNA-Seqe analisar os dados usando plataformas Galaxy e David. Este protocolo fornece instruções passo a passo para a utilização dos vários módulos Galaxy para aceder aos dados em bruto NGS, verificações de controlo de qualidade, o alinhamento, e análise de expressão diferencial de genes, orientando o utilizador com os parâmetros em cada passo para gerar uma lista de genes que podem ser rastreados para o enriquecimento de as classes de genes ou processos biológicos utilizando Davi. No geral, prevemos que este artigo irá fornecer informações para C. elegans pesquisadores realizam experimentos de RNA-Seq, pela primeira vez, bem como usuários freqüentes executando um pequeno número de amostras.
Introdução
A primeira sequenciação do genoma humano, realizada usando o método didesoxinucle�ido-seqüenciamento de Fred Sanger, levou 10 anos e um custo estimado de US $ 3 bilhões 1, 2. No entanto, em pouco mais de uma década desde a sua criação, Next-Generation Sequencing tecnologia (NGS) tornou possível sequenciar todo o genoma humano dentro de duas semanas e para US $ 1.000. Novos instrumentos NGS que permitem velocidades de recolha de sequenciamento de dados cada vez maiores com incrível eficiência, juntamente com reduções acentuadas nos custos, estão revolucionando a biologia moderna de maneiras inimagináveis como projetos de sequenciação do genoma estão rapidamente se tornando comuns. Além disso, estes desenvolvimentos tenham galvanizado progresso em muitas outras áreas, tais como a análise de expressão genética através de RNA-Sequencing (ARN-SEQ), estudo de modificações epigenética do genoma, as interacções ADN-proteína, e rastreio para a diversidade microbiana em hospedeiros humanos. NGS à base de ARN-Seq, em particular, tornou possível identificar e transcriptomes mapa abrangente com precisão e sensibilidade, e substituiu tecnologia de microarrays como o método de escolha para a criação de perfis de expressão. Embora a tecnologia de microarray tem sido amplamente utilizado, que é limitada pela sua dependência em matrizes pré-existentes com informação genómica conhecida, e outros inconvenientes, tais como hibridação cruzada e gama restrita de mudanças de expressão que pode ser medida com fiabilidade. RNA-seq, por outro lado, podem ser utilizados para detectar transcritos conhecidos e desconhecidos, enquanto a produção de baixo nível de ruído de fundo devido à sua natureza não ambígua de mapeamento de ADN. RNA-Seq, em conjunto com as várias ferramentas genéticas oferecidos por organismos modelo tais como leveduras, moscas, vermes, peixes e ratos, tem servido como base para muitas recentes descobertas importantes biomédicas. No entanto, subsistem desafios significativos que fazem NGS inacessíveis à comunidade científica em geral, incluindo as limitações de armazenamento, processamento e acima de tudo, m análise bioinformática eaningful de grandes volumes de dados de sequenciação.
Os rápidos avanços em tecnologias de sequenciamento e acumulação exponencial de dados criaram uma grande necessidade de plataformas computacionais que permitirão que os pesquisadores para acessar, analisar e compreender esta informação. Os primeiros sistemas foram fortemente dependente do conhecimento de programação de computadores, enquanto que, os navegadores do genoma como NCBI que permitiram não-programadores para acessar e visualizar dados não permitiu análises sofisticadas. A plataforma, a abertura de acesso baseado na web, Galaxy ( https://galaxyproject.org/ ), preencheu este vazio e provou ser um oleoduto valioso que permite aos pesquisadores para processar dados NGS e realizar um espectro de simples de complexo análises de bioinformática. Galaxy foi inicialmente estabelecida, e é mantido, pelos laboratórios de Anton Nekrutenko (Penn State University) e James Taylor (Johns Hopkins University)f "> 3. Galaxy oferece uma ampla gama de tarefas computacionais tornando-se um 'balcão único' para as necessidades de bioinformática inúmeras, incluindo todas as etapas envolvidas em um estudo RNA-Seq. Itallows usuários para executar o processamento de dados, quer em seus servidores ou localmente em suas próprias máquinas. os dados e fluxos de trabalho pode ser reproduzido e compartilhado. os tutoriais online, seção de ajuda, e um wiki-página ( https://wiki.galaxyproject.org/Support ) dedicada ao Projeto Galaxy fornecer apoio consistente. no entanto, para usuários de primeira viagem, especialmente aqueles com nenhum treinamento bioinformática, o gasoduto pode parecer intimidante e do processo de auto-aprendizagem e familiarização pode ser demorado. Além disso, o sistema biológico estudado, e especificidades da experiência e métodos utilizados, o impacto as decisões de análise em vários passos, e estes podem ser difícil de navegar sem instrução.
O RN geral A-Seq Galaxy fluxo de trabalho consiste de carregamento de dados e de verificação de qualidade, seguido por análise usando o smoking Suite 4, 5, 6, 7, 8, 9, que é um colectivo de várias ferramentas necessárias para diferentes fases de análise de dados de RNA-Seq 10, 11, 12, 13, 14. Uma experiência típica de ARN-Seq consiste na parte experimental (preparação da amostra, de isolamento de ARNm e ADNc preparação biblioteca), o NGS e a análise de dados bioinformática. Uma visão geral destas secções, e os passos envolvidos na calha Galaxy, são mostrados na Figura 1.
3fig1.jpg"/>
Figura 1: Vista geral de uma ARN-Seq fluxo de trabalho. Ilustração dos passos experimentais e computacionais envolvidos numa experiência de RNA-Seq para comparar os perfis de duas estirpes de vermes (A e B, as linhas de laranja e verdes e setas, respectivamente) de expressão genética. Os diferentes módulos de Galaxy utilizados são apresentadas em caixas com o passo correspondente no nosso protocolo indicado no vermelho. As saídas de várias operações são escritos em cinza com os formatos de arquivo mostrados em azul. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
A primeira ferramenta no Tuxedo Suite é um programa de alinhamento chamado 'Tophat'. Ele quebra a entrada NGS lê em fragmentos menores e, em seguida, mapeia-los para um genoma de referência. Este processo de dois passos garante que lê abrangendo regiões intrónicas cujo alinhamento pode ser de outra forma disrupted ou não atendidas são contabilizados e mapeada. Isto aumenta a cobertura e facilita a identificação de novas junções de processamento alternativo. Saída Tophat é relatado como dois arquivos, um arquivo de BED (com informações sobre junções de emenda que incluem localização genômica) e um arquivo BAM (com detalhes de mapeamento de cada leitura). Em seguida, o arquivo BAM está alinhada contra um genoma de referência para estimar a abundância de transcritos individuais dentro de cada amostra, utilizando a ferramenta posterior na Suite Tuxedo chamado 'Abotoaduras'. Botão de punho funções por digitalizar o alinhamento para relatar fragmentos transcrito de comprimento completo ou 'transfrags' que abrangem todas as possíveis variantes de processamento nos dados de entrada para cada gene. Com base nisso, que gera um 'transcriptoma' (montagem de todos os transcritos gerados por cada gene para gene) para cada amostra a ser sequenciada. Estes conjuntos Abotoaduras são então recolhido ou fundidos em conjunto, juntamente com a reference genoma para produzir um arquivo de anotação única para análise diferencial a jusante usando a ferramenta seguinte, 'Cuffmerge'. Finalmente, a expressão do gene ferramenta medidas diferencial da 'Cuffdiff' entre amostras por comparação das saídas TOPHAT de cada uma das amostras para o ficheiro de saída Cuffmerge final (Figura 1). Abotoaduras usa FPKM / RPKM (Fragmentos / leituras por kilobase de transcrição por milhão mapeados lê) valores para relatar abundâncias transcrição. Estes valores reflectem a normalização dos dados NGS matérias para a profundidade (número médio de leituras a partir de uma amostra que se alinham para o genoma de referência) e o comprimento do gene (genes têm diferentes comprimentos, de modo a contagem tem de ser normalizado para o comprimento de um gene para comparar os níveis entre os genes). FPKM e RPKM são essencialmente o mesmo com RPKM a ser utilizado para um único final de ARN-Seq onde cada leitura corresponde a um único fragmento, enquanto que, é utilizada para FPKM-Fim emparelhado ARN-Seq, uma vez que representa o facto de que duas leituras pode corresponder ao mesmo fragmento. Em última análise, o resultado destas análises é uma lista de genes diferencialmente expressos entre as condições e / ou estirpes testadas.
Uma vez uma temporada de sucesso Galaxy está concluído e uma 'lista gene' é gerado, o próximo passo lógico requer mais bioinformática analisa deduzir conhecimento significativo dos conjuntos de dados. Muitos pacotes de software surgiram para atender a essa necessidade, incluindo pacotes computacionais baseados na web publicamente disponíveis, como David (o banco de dados para anotação, visualização e descoberta Integrado) 15. DAVID facilita a atribuição de significado biológico para listas de genes grandes a partir de estudos de alta produtividade através da comparação da lista de genes carregado para a sua base de conhecimento biológico integrado e revelando as anotações biológicos associados com a lista de genes. Isto é seguido por Análise de Enriquecimento, ou seja, testes para identify se qualquer processo ou classe de genes biológica é sobre-representados na lista (s) de genes de uma forma estatisticamente significativa. Tornou-se uma escolha popular por causa de uma combinação de uma ampla, base de conhecimento integrada e algoritmos de análise poderosas que permitem aos pesquisadores detectar temas biológicos enriquecido dentro genômica derivado 'listas de genes' 10, 16. Outras vantagens incluem sua capacidade de processar listas de genes criadas em qualquer plataforma sequenciamento e uma interface altamente user-friendly.
O nemátodo Caenorhabditis elegans é um sistema modelo genético, bem conhecidos pelas suas muitas vantagens, tais como tamanho pequeno, o corpo transparente, o plano de corpo simples, facilidade de cultura e excelente receptividade ao dissecção genética e molecular. Vermes têm uma pequena, simples e bem-anotada genoma que inclui até 40% de genes conservados com homólogos humanos conhecidos 17. De fato, C. elegansfoi o primeiro metazoários cujo genoma foi completamente sequenciado 18, e uma das primeiras espécies onde ARN-Seq foi usada para mapear transcriptoma de um organismo 19, 20. Estudos de vermes cedo envolvido experimentação com diferentes métodos de captura-ARN de alto rendimento, a preparação da biblioteca e a sequenciação assim como condutas de bioinformática que contribuíram para o avanço da tecnologia de 21, 22. Nos últimos anos, a experimentação baseada em RNA-Seq em vermes tem se tornado comum. Mas, para os biólogos vermes tradicionais os desafios colocados pela análise computacional de dados de RNA-Seq continuam a ser um grande obstáculo para uma maior e melhor utilização da técnica.
Neste artigo, descreve-se um protocolo para a utilização da plataforma Galaxy para analisar os dados de RNA-Seq high-throughput gerados a partir de C. elegans. Para muitos pela primeira vez e pequeno-scale utilizadores, a forma mais eficaz e económica e simples de realizar uma experiência de RNA-Seq é isolar RNA no laboratório e utilizar uma (ou em casa) facilidade NGS comercial para a preparação de bibliotecas de cDNA e sequenciação do próprio NGS. Assim, temos primeiro detalhada das etapas envolvidas no isolamento, quantificação e qualidade de avaliação de C. elegans amostras de ARN por ARN-Seq. Em seguida, nós fornecemos passo-a-passo para o uso da interface Galaxy para análises dos dados NGS, começando com testes de pós-sequenciação de verificações de controlo de qualidade, seguido por alinhamento, montagem, e a quantificação diferencial da expressão do gene. Além disso, nós incluímos as direções para controlar a lista de genes resultantes da Galaxy para estudos de enriquecimento biológicos utilizando DAVID. Como etapa final no fluxo de trabalho, nós fornecemos instruções para fazer upload de dados de RNA-Seq para servidores públicos, como a Sequência Leia Archive (SRA) no NCBI ( http: // www.ncbi.nlm.nih.gov/sra) para torná-lo livremente acessível à comunidade científica. No geral, prevemos que este artigo irá fornecer informações completas e suficientes para biólogos vermes que realizam experimentos de RNA-Seq, pela primeira vez, bem como usuários freqüentes executando um pequeno número de amostras.
Access restricted. Please log in or start a trial to view this content.
Protocolo
Isolamento de ARN 1.
- Medidas de precaução
- Limpe as inteiras de trabalho de superfícies, instrumentos e pipetas usando um pulverizador de RNase comercialmente disponível para eliminar quaisquer RNases presente.
- Usar luvas em todos os momentos, regularmente mudá-las por outras novas durante as diferentes etapas do protocolo.
- Utilizar apenas pontas de filtro e manter todas as amostras sobre gelo tanto quanto possível para evitar a degradação de ARN.
NOTA: Para obter os melhores dados a partir de plataformas NGS, é fundamental começar com RNA de alta qualidade. métodos de isolamento de RNA e preparação de variar dependendo da origem da amostra, método de sequenciação e investigador preferência. Vários kits disponíveis comercialmente podem ser utilizados para este propósito ou ARN também pode ser isolada utilizando um método de fenol-clorofórmio padrão de extracção de ARN. Com qualquer metodologia, as medidas de precaução listadas acima deve ser seguido ao longo do processo para minimizar a contaminação e obtain amostras de ARN intocada.
- colheita worms
- Sincronizar a população sem-fim por meio de tratamento de branqueamento de hipoclorito de 23 para se obter 1,000-1,500 pareados por idade C. elegans vermes adultos por estirpe.
- Lavam-se os vermes fora das placas utilizando uma solução tampão M9 e centrifugação a 325 xg numa centrifugadora de bancada durante 30 s. Aspirar para fora o tampão M9 deixando para trás um sedimento de vermes. Repetir esta etapa, pelo menos três vezes para eliminar transição bacteriana.
- Para o sedimento sem-fim, adicionar ~ 500 uL de tampão de lise (se utilizando um kit comercial) ou Trizol (uma solução de mono-fásica de fenol e isotiocianato de guanidina; se extracção com fenol: clorofórmio descrito em 1.3.3 é levada a cabo) para romper tecidos vermes , desactivar RNases e estabilizar os ácidos nucleicos.
NOTA: O protocolo pode ser pausado aqui pelo flash congelamento as amostras em azoto luido seguido de armazenagem a -80 ° C.
- Isolamento de ARN
- Sonicar amostras de vermes em 45% da amplitude em ciclos de 20 s. 'ON' e 40 s. 'OFF' (8-12 ciclos por estirpe). Manter as amostras em gelo em todos os momentos.
NOTA: Verifique se a sonda de ultra-sons é imerso no buffer e é mantido a um nível constante por toda parte. Evitar a formação de espuma da amostra e limpar a sonda completamente em-entre amostras. ciclos de sonicação pode variar dependendo do tipo de ultra-sons utilizado. Recomenda-se que as condições de sonicação são optimizados primeiro em uma amostra de teste antes de iniciar uma experiência. - Se usando um kit disponível comercialmente, prosseguir com Isolamento de ARN de acordo com o protocolo prescrito. Para o isolamento de ARN utilizando um método de fenol-clorofórmio, executar as seguintes etapas.
- Centrífuga sonicada amostras a 16.000 xg durante 10 min. a 4 ° C.
- Transferir o sobrenadante para um tubo de microcentrífuga de 1,5 ml sem RNase e adicionar 100 mL de clorofórmio (1/5 do volume de reagente de isolamento de ARN / ADN).
Cuidado: Clorofórmio é tóxico. Para minimizar a exposição e evitar a inalação, trabalhar em uma capa química durante o manuseamento desta substância. - Vortex as amostras cuidadosamente durante 30 - 60 s. e deixar que as amostras de sentar à temperatura ambiente durante 3 min.
- Centrifugar a 11750 xg durante 15 min. a 4 ° C. Transferir apenas a camada aquosa superior a um novo isento de ARNase tubo de microcentrífuga tomando cuidado para não aspirar o interface de branco contendo ADN. Repita os passos 1.3.4 através 1.3.6.
- Adicionar 250 mL (70% de fase aquosa ou 1/2 de ARN / ADN isolamento volume de reagente) de 2-propanol e inverter o tubo de misturar. Deixe tubos repousar à temperatura ambiente durante 10 min ou deixar durante a noite a -80 ° C.
- Centrifugar as amostras a 11.750 xg durante 10 min. a 4 ° C. Decantar o sobrenadante cuidadosamente, deixando para trás um pouco uL na parte inferior do tubo, de modo que a pastilha não é perturbado.
- Lavar sedimento com 500 ul de 75% de etanol (feita usando água isenta de RNase) e girar para baixo a 16.000 xg durante 5 min. umat 4 ° C.
- Remover o máximo possível sobrenadante sem perturbar o sedimento. Ar secar o sedimento em um capuz durante alguns minutos.
- Adicionar 30 mL de água sem RNase e ajudar a dissolver o sedimento de ARN por aquecimento durante 10 min. a 60 ° C.
- Verifique a qualidade RNA e quantidade usando um Bioanalyzer.
NOTA: Bioanalyzer gera um R ND I ntegrity N úmero (NIR) como uma medida de qualidade de ARN. Um RIN de, pelo menos, 8 é o limiar recomendado para amostras de ARN-SEQ (maior é melhor). quantidade de RNA e qualidade também pode ser verificada por espectrofotometria, mas também deve ser seguido por avaliação visual da integridade do RNA. Para fazer isso, executar as amostras sobre um gel de agarose a 1,2% o tempo suficiente para se obter uma separação adequada das bandas 28S e 18S de ARN ribossomal. A presença de duas bandas distintas (1,75 kb para o rRNA 18S e 3,5 kb para rRNA 28S, no caso de C. elegans) constitui uma medida aceitável de qualidade de ARN. - Uso ~ 100 ng / pL de ARN para ship para o fornecedor de instalação / NGS para preparação de bibliotecas de sequenciação.
NOTA: As amostras de RNA devem ser enviadas em gelo seco para o prestador de serviços de seqüenciamento. A maioria dos provedores de realizar um teste de RNA de controle de qualidade independente antes da preparação da biblioteca.
- Sonicar amostras de vermes em 45% da amplitude em ciclos de 20 s. 'ON' e 40 s. 'OFF' (8-12 ciclos por estirpe). Manter as amostras em gelo em todos os momentos.
2. ARN-Seq Análise de Dados
- Download do Raw Seqüenciamento de Dados
- Faça o download dos dados de sequenciamento fastq cru comprimido codificados no formato fastq.gz do provedor NGS usando um "protocolo de transferência de arquivos" (ftp).

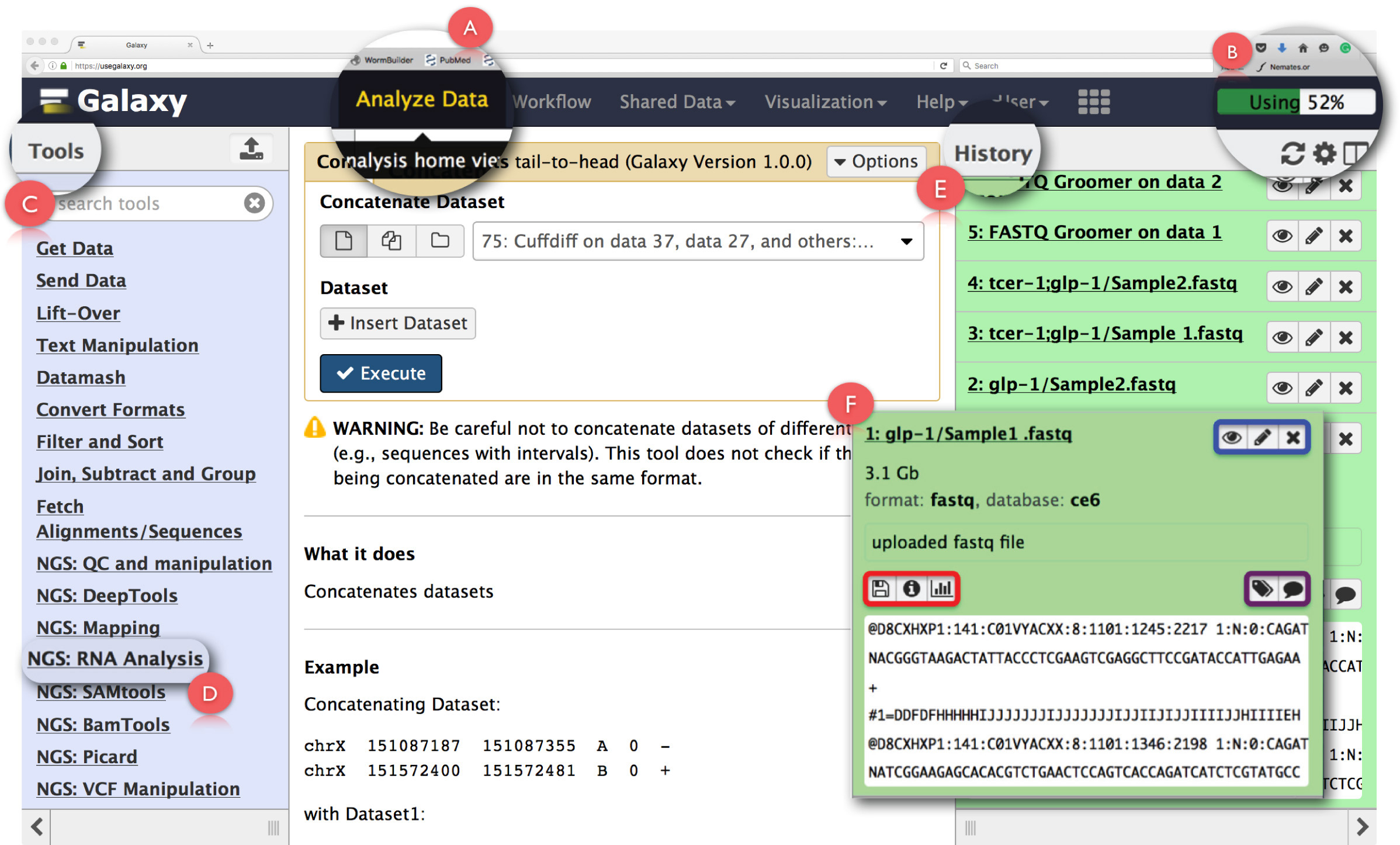
Figura 2: Esquema do Galaxy Painel de interface e as principais funções de ARN-Seq. As principais características da página são expandidas e destacou. (A) realça a função 'Analise dados' no cabeçalho da página da web utilizado para acesso Análise Home View. (B) é o 'Progresso bar', que indica o espaço no servidor Galaxy utilizado pela operação. (C) é o 'Tools Section' que lista todas as ferramentas que podem ser executados na interface do Galaxy. (D) mostra os 'NGS: Análise de ARN a secção ferramenta utilizada para a análise de RNA-Seq. (E) mostra o painel 'História' que lista todos os arquivos gerados usando Galaxy. (F) mostra um exemplo da caixa de diálogo que se abre ao clicar em qualquer arquivo na seção Histórico. Within (F), a caixa azul destaca ícones que podem ser usados para ver, editthe atributos ou excluir o conjunto de dados, a caixa roxa destaca ícones que podem ser usados para 'editar' as tags conjunto de dados ou anotação, e, a caixa vermelha indica ícones para baixar os dados, ver detalhes da tarefa executada ou executar novamente a operação. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Introdução ao Galaxy
NOTA: Galaxy pode ser executado em um servidor público gratuito usando uma plataforma baseada na web fornecendo acesso em nuvem e armazenamento limitado livre. Ele também pode ser baixado e executado localmente na máquina do usuário ou aglomerados computacionais hospedados por instituições mas o processamento local, pode ser restringida por limites de armazenamento de dados e limitações de potência de processamento de máquinas dos usuários. Detalhes sobre o download e instalação pode ser acessado no https://wiki.galaxyproject.org/Admin/GetGalaxy . Neste protocolo, descrevemos o uso baseado na Web do gasoduto Galaxy.- Depois de baixar e armazenar os dados NGS na máquina do usuário, acesso Galaxy emlaxy.org/" target = "_blank"> https://usegalaxy.org/.
- Registrar uma conta de usuário clicando em 'Usuário' no cabeçalho da página, login e começar a familiarizar-se com o painel de interface do usuário.
NOTA: É recomendável que os usuários primeira vez utilizar o tutorial 'Comece aqui' fornecido na página inicial para se familiarizar com a configuração básica do Galaxy ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) . - Clique em 'Analisar dados' (Figura 2A) no painel de cabeçalho para aceder ao 'Análise Início Ver', que também é a tela de inicialização no Galaxy.
NOTA: O cabeçalho também abriga outros links cujos detalhes podem ser vistos ao passar o ponteiro do mouse sobre eles. O canto superior direita do cabeçalho tem uma barra de progresso que monitora o espaço utilizado pelas tarefas (Figura 2B). - Clamber 'NGS: Análise RNA' tarefa no 'Menu Ferramentas' no painel esquerdo (Figura 2C) para acessar todas as ferramentas necessárias para análise de dados de RNA-seq.
NOTA: O 'Menu Ferramentas' cataloga todas as operações que oferece Galaxy. Este menu é dividido com base em tarefas e clicando em qualquer um vai abrir uma lista de todas as ferramentas necessárias para realizar essa tarefa. - Criar nova história análise clicando no ícone de engrenagem na parte superior do painel 'Histórico' à direita (Figura 2E). Escolha 'Criar Novo' opção no menu pop-up. Dê este 'História' um nome adequado para identificar a análise.
NOTA: O painel de 'Histórico' mostra todos os arquivos enviados para análise, bem como todos os arquivos de saída que são gerados pela execução de tarefas no Galaxy. Ao clicar em um nome de arquivo neste painel abre-se uma caixa de diálogo com informações detalhadas sobre a tarefa executadae um fragmento do conjunto de dados (Figura 2F). Ícones neste caixa permitir que o usuário 'view', 'editar os atributos' ou 'delete' o conjunto de dados (Figura 2F, destacada em azul). Além disso, o usuário pode também 'editar' tags conjunto de dados ou anotação (Figura 2F, em destaque na roxo), 'download' os dados, 'ver detalhes' da tarefa, 'reprise' a tarefa ou mesmo 'visualizar' o conjunto de dados a partir deste caixa de diálogo (Figura 2F, destacado em vermelho). - Clique na função 'Carregar Arquivo' em 'Obter dados' no 'menu Ferramentas' para fazer upload de arquivos fastq matérias.
NOTA: Ao clicar sobre este ou qualquer outra ferramenta abre uma breve descrição da operação, eo próprio teste, no painel do meio 'Análise de Interface'. Este painel laços juntos o'Ferramentas' no painel da esquerda e os 'Arquivos de Entrada' no painel da direita 'História' (Figura 2E). Aqui, arquivos de entrada de 'História' são selecionados e outros parâmetros definidos para executar uma determinada tarefa. O conjunto de dados de saída resultante de cada teste é salvo de volta em 'História'. Incluído com o teste no painel 'Análise Interface "são explicações para todos os parâmetros disponíveis para a execução de uma determinada ferramenta, juntamente com uma lista detalhada de todos os arquivos de saída da ferramenta gera. - Depois que a tarefa é aberta no 'Análise de Interface', clique em 'Escolher arquivo local' ou 'Escolher arquivo FTP' (upload mais rápido), navegue até a pasta que contém os arquivos de sequenciamento e selecione o conjunto de dados apropriado para ser carregado.
- Permitir Galaxy para 'Auto-detectar' a (configuração padrão) carregado tipo de arquivo. Selecione 'C. elEgans "no menu suspenso para o genoma.
- Clique em 'Start' para iniciar upload de dados. Uma vez que o arquivo é carregado, ele será salvo no painel 'História' e pode ser acessado a partir de lá.
- Se arquivos de dados múltiplos de sequenciamento são produzidos para uma única amostra, combiná-los usando a ferramenta 'Concatenate'. Para fazer isso, abra a opção 'Manipulação de texto' no 'Menu Ferramentas'.
- Clique na ferramenta 'Concatenate', escolher os arquivos que precisam ser combinados a partir da caixa drop-down no meio da 'interface Analysis' e clique em 'Executar'.
NOTA: Os arquivos de saída produzidos utilizando esta tarefa são gerados no formato fastq. O programa de mapeamento tem um limite de 16.000.000 sequências por arquivo fastq e quando esse limite for atingido um novo ficheiro fastq é gerada para as sequências restantes. a '; Concatenate ferramenta' é necessária em tais casos para combinar os conjuntos de dados. - Converter os arquivos no formato fastq enviados para o formato fastqsanger necessário para Galaxy análise de RNA-Seq usando a função 'fastq groomer' encontrado em dos NGS: QC e manipulação 'seção (ver arquivo suplementar).
- Escolha o conjunto de dados fastq apropriada sob o 'Arquivo para Groom' opção e executar a ferramenta usando parâmetros padrão.
NOTA: Os arquivos de saída produzidos utilizando esta tarefa são gerados no formato fastqsanger.
- testes fastqsanger Data Quality-Control
- Verifique a qualidade do fastqsanger carregado lê usando a ferramenta 'FastQC' localizado em 'NGS: QC e manipulação' no menu 'Ferramentas'.
- Escolha o arquivo de dados fastqsanger preparado a partir do menu drop-down para 'Short ler dados a partir da biblioteca atual e executar a ferramenta usando parâmetros padrão.
NOTA: Preste atenção especial para a qualidade da leitura e presença de quaisquer sequências de adaptador. Os adaptadores são geralmente removidos como parte do processamento de dados de RNA-Seq pós por prestadores de NGS mas em alguns casos, pode ser deixado para trás. Para uma explicação de padrões de qualidade ir para http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - Verifique com o provedor de NGS e se adaptadores estão presentes, apará-los usando a ferramenta 'grampo' do 'NGS: QC e manipulação' menu de tarefas.
NOTA: Os arquivos de saída produzidos utilizando esta tarefa são gerados no formato txt matéria, bem como em HTML que pode ser aberto em qualquer navegador web.
- Análise de dados com Tuxedo Suíte
- Cartola
- Baixar a última versão do C. elegans fasta genoma de referência e arquivos de upload do arquivo gtf (Gene Transferência Format)', como descrito acima, em 2.2.6.
- Abra as 'NGS: Análise RNA' seção e clique na ferramenta 'TopHat' para mapear o sequenciamento lê o genoma de referência baixado.
- Seleccione a resposta adequada no menu suspenso à pergunta "É este single-end ou dados-end emparelhado?
- Escolha o arquivo fastq apropriado.
- Selecione 'Use um genoma da história' no próximo menu suspenso e escolher genoma de referência baixou na etapa 2.4.1.1.
- Selecione 'Default' para os outros parâmetros e clique em 'Executar'.
NOTA: Entre os arquivos de saída produzidos utilizando esta tarefa, arquivo dos hits aceites »é utilizado para as etapas subseqüentes.
- Abotoaduras e Cuffmerge
- Selecione o 'Cuffferramenta nas 'ligações NGS: Seção de Análise RNA' para montar as transcrições, estimar sua abundância e teste para expressão diferencial.
- No primeiro menu suspenso, escolha os 'hits aceites (formato BAM)' mapeados arquivo obtidos através da análise TopHat.
- No segundo menu suspenso, definir anotação da referência para o arquivo gtf baixou na etapa 2.4.1.1.
- Selecione 'Sim' para a opção 'Executar a correção viés' e execute a tarefa usando as configurações padrão para todos os outros parâmetros.
NOTA: Entre os arquivos de saída produzidos utilizando esta tarefa, o arquivo 'aceites Transcrições' é usado para as etapas subseqüentes. - Ferramenta Open 'Cuffmerge' no 'NGS: Análise RNA' para mesclar os 'Transcrições montados' produzidos para todas as amostras de RNA-Seq.
NOTA: A primeira caixa na ferramenta de auto-povoa e listas de todo o arquivos gtf produzidos por abotoaduras. - Selecione o arquivo 'montado Transcrições' para todas as estirpes / condições testadas, incluindo réplicas biológicas da mesma cepa / condição (ver discussão de repetições biológicas).
- Selecione 'Sim' para 'Use anotação da referência' e escolha o arquivo gtf baixou na etapa 2.4.1.1.
- Na caixa a seguir, selecione novamente 'Sim' para a opção 'Dados Use Sequence' e escolha o arquivo inteiro genoma fasta baixou na etapa 2.4.1.1.
- Manter os outros parâmetros como padrão, clique em 'Executar'.
NOTA: Cuffmerge gera um único arquivo de saída GTF.
- Cuffdiff
- Navegue até a ferramenta 'Cuffdiff' no 'NGS: Análise RNA' seção. No menu 'Transcrições', selecione o arquivo de saída resultante da fusão de Cuffmerge.
- Rótuloas condições 1 e 2 com os nomes de duas estirpes / condição.
NOTA: Cuffdiff pode realizar comparações entre mais de duas estirpes ou condições, bem como experimentos curso de tempo. Basta usar a opção 'Adicionar novas condições' para adicionar cada novo cepas / condição, conforme necessário. - Para cada estirpe / condição, sob indivíduo seleccione 'réplicas' ficheiros de saída 'hits aceites' de TopHat que correspondem às diferentes réplicas biológicas daquela estirpe / condição. Mantenha pressionada a tecla 'cmd', se estiver usando um computador Macintosh, e tecla 'ctrl', se estiver usando um PC, para selecionar vários arquivos.
- Deixe todas as outras opções como parâmetros padrão. Clique em 'Executar' para executar a tarefa.
NOTA: Cuffdiff gera vários arquivos de saída em formato tabular como a leitura final da análise de RNA-Seq. Estes incluem arquivos com rastreamento FPKM para transcrições, genes (combinadosFPKM valores de transcritos que partilham uma identidade de genes), as transcrições primárias e as sequências de codificação. Todos os ficheiros de dados gerados podem ser vistos em qualquer aplicação folha de cálculo e conter atributos semelhantes, tais como o nome do gene, lus, dobrar as alterações (em escala log 2), bem como dados estatísticos sobre as comparações entre estirpes / condições, incluindo o valor de p e valores de q. Os dados nestes ficheiros podem ser classificados com base na significância estatística das diferenças ou dobrar mudança na expressão do gene (magnitude e direcção da mudança, como em cima ou para baixo- genes regulados) e manipulados de acordo com os requisitos dos utilizadores. Se for necessária a conversão entre diferentes identificadores de genes (por exemplo, Wormbase ID gene versus número cosmídeo), ferramentas disponíveis no Biomart ( http://www.biomart.org/ ) pode ser utilizado.
- Cartola
3. Gene Ontology (GO) Análise Termo usando DAVID
- Acesso DAVID a partir do site https: //david.ncifcrf.gov/. Clique em 'Iniciar análise' no cabeçalho da página. Em 'Passo 1', copiar e colar a lista de genes obtidos a partir de Galaxy na caixa A. No 'Passo 2', seleccione 'Wormbase Gene ID' como o identificador para os genes introduzidos.
NOTA: DAVID reconhece categorias de anotação mais acessíveis ao público, para que outros identificadores de genes (como ID gene Entrez ou símbolo do gene) também pode ser usado. - Em 'Passo 3', escolha 'Lista Gene' (genes a serem analisados) em 'Tipo de lista' e, em seguida, clique no botão 'Enviar List' ícone.
NOTA: 'Assistente de análise', vai abrir para listar todas as ferramentas DAVID hiperlinks que podem ser executados na lista gene carregado (Figura 3). Clique nos links para acessar os módulos correspondentes relevantes como pela exigência do usuário. Para identificar as ferramentas apropriadas para uma determinada tarefa, clique em 'Quais as ferramentas DAVID de usar? 'Link na' ; Página Análise Wizard'. Clique no link 'Iniciar análise' no cabeçalho para retornar à home page do 'Assistente de Análise' em qualquer ponto durante a análise.

Figura 3: Esquema do DAVID Análise Assistente página da web e exemplos de funcionamento saídas. User-interface web do 'Assistente de Análise' lista as ferramentas utilizadas para analisar lista gene carregado para o enriquecimento com base em vários parâmetros. Clicando sobre essas ferramentas reporta os dados analisados em uma nova página web. Exemplos dos relatórios tabulares gerados a partir de 'Gene Classificação Funcional', 'Guia de anotação funcional' e 'clustering anotação funcional' são mostrados como inserir (setas).> Por favor clique aqui para ver uma versão maior desta figura.
- Ferramenta de anotação funcional 1: Funcional Clustering Anotação
- Clique no módulo 'Anotação Clustering funcional' para ir para a página de resumo. Mantenha as categorias de anotação padrão e clique em 'Clustering anotação funcional' para gerar conjuntos de termos de anotação semelhantes classificados por sua pontuação de enriquecimento.
- Clique no nome hiperlinks de cada termo para ler detalhes sobre ele e 'RT' (termos relacionados) para listar outros termos semelhantes relacionados com a categoria.
- Clique na barra de roxo para listar os genes associados com um termo e o 'G' vermelho para listar todos os genes associados com todos os termos dentro de um cluster.
- Clique no ícone verde para ver uma visão bidimensional de todos os genes e os termos em um cluster.
NOTA: As três últimas colunas listar os resultados analíticos e estatísticos para cadaprazo. Os resultados para este e todos os outros analytics pode ser baixado em formato .txt, clicando no link 'Download File'.
- Ferramenta de anotação funcional 2: anotação funcional Gráfico
- Voltar para a página de resumo e clique sobre 'Gráfico anotação funcional' para identificar termos biológicos significativamente sobre-representados (por exemplo, de actividade do factor de transcrição ou a actividade da quinase), associadas com a lista de genes.
- Clique no nome prazo para obter informações mais detalhadas e 'RT' (termos relacionados) para listar outros termos relacionados.
- Clique na barra de roxo para listar todos os genes associados da categoria individual correspondente.
Nota: Relacione As duas últimas colunas resultados as estatísticas-testes para cada categoria.
- Ferramenta de anotação funcional 3: Funcional Tabela Anotação
- Voltar para a página de resumo e clique em 'Functional Tabela anotação 'para ver uma lista de todas as anotações associadas com os genes de uma lista sem quaisquer cálculos estatísticos.
NOTA: Esta ferramenta pode ser útil para a análise de gene-por-gene de uma lista ou de olhar para genes específicos e altamente interessantes.
- Voltar para a página de resumo e clique em 'Functional Tabela anotação 'para ver uma lista de todas as anotações associadas com os genes de uma lista sem quaisquer cálculos estatísticos.
- Gene Ferramenta de Classificação Funcional
- Voltar ao 'Assistente de análise' e clique em módulo 'gene funcional Classification' para segregar a lista gene de entrada em grupos relacionados com o funcionalmente de genes classificados como por seu 'Índice de Enriquecimento', uma medida de enriquecimento global do grupo gene na lista.
- Clique no nome prazo para obter informações mais detalhadas e 'RG' para revelar genes funcionalmente relacionados do grupo gene
- Clique em 'T' vermelho (relatórios prazo) para listar biologia associado eo ícone verde para ver uma visão bidimensional de todos os genes e termos.
- Gene-nameVisualizador de lote
- Voltar ao 'Assistente de análise' e clique em 'Gene-Nome do lote Visualizador' traduzir 'Wormbase Gene IDs' em seus nomes de genes correspondentes. (WBGene00022855 = tRCE-1).
- Clique no nome gene para obter mais informações específicas do gene.
- Clique sobre o 'RG' (genes relacionados) ligação ao lado de cada gene para revelar os genes previstos como sendo relacionadas funcionalmente com o gene de interesse.
4. Carregamento de RAW Dados para o NCBI sequência lida Arquivo (SRA)
- Acesse o site SRA em Entrar no link de NCBI' ou registar uma nova conta.
- Clique em 'Bioproject'.
- Clique em 'Submission' sob o 'Usando Bioproject' cabeçalho do lado esquerdo.
- Selecione a opção 'New Submission'. Detalhes da actualização do apresentador. Continuar através dos restantes sete abas, Preenchendo os detalhes da experiência e dados a ser carregado. Clique em 'Enviar' quando concluído.
NOTA: Na quinta guia 'amostra biolica', deixar o slot para 'amostra biolica' vazio. - Atualize a página resultante clicando no link 'Minhas inscrições'. Os dados apresentados serão listados com um número de apresentação atribuído, breve descrição e status upload.
- Clique em 'amostra biolica' no topo desta página, na caixa 'iniciar uma nova submissão' e criar uma 'nova submissão'. Enviar submissões separadas para cada amostra.
- Tal como no caso com 'Bioproject' em 4,4, actualizar os detalhes do apresentador e continuar através do resto dos separadores de enchimento nos detalhes de cada guia. Uma vez concluída a revisão e clique em 'Enviar'.
- Navegue até http: //www.ncbi.nlm.nih.gov / SRA para criar o 'Sequence Leia Archive (SRA)' submissão final.
- Clique em 'Login para SRA' em 'Getting Started'.
- Na próxima página, clique no link 'NCBI PDA'. Um link 'Preferências update' vai abrir. Preencha o formulário e clique em 'Salvar preferências'.
- Na página resultante, clique no botão 'Create New Submission' link. Digite um nome adequado sob 'Alias' e clique em 'Salvar'. Uma tabela com o ID submissão e outros detalhes serão criados.
- Clique em 'New Experiment' e registrar pelo menos uma biblioteca de sequenciamento único para cada 'amostra biolica'.
- Designar e vincular o criado anteriormente 'Bioproject' e 'amostra biolica' ID da submissão do. A 'New Experiment' será criado.
- Clique em 'New Run' na parte inferior da páginaApós o experimento SRA foi feito e identificar os arquivos de dados que precisam ser ligadas a ele.
- Calcular a soma MD5 de cada arquivo de dados. Para fazer isso em um terminal MacIntosh, navegue até Aplicativos / Utilitários / Terminal. No terminal, tipo em 'MD5' (sem as aspas), seguido por um espaço. Arraste e solte os arquivos que precisam ser carregados no terminal do Finder e clique 'Enter'.
- Terminal irá retornar uma soma MD5 alfanumérico. Digite isso como parte do processo de submissão para o upload de arquivos. Use o nome de usuário e senha fornecidos pelo sistema para fazer upload de arquivos utilizando FTP.
Access restricted. Please log in or start a trial to view this content.
Resultados
Em C. elegans, a eliminação das células estaminais da linha germinal (GSCS) prolonga a vida, aumenta a capacidade de resistência de stress, e eleva a gordura corporal 24, 28. Perda de GSCS, quer provocada por laser de ablação ou por mutações, tais como GLP-1, faz com que a vida útil de extensão através da activação de uma rede de 29 factores de transcrição. Um destes factor...
Access restricted. Please log in or start a trial to view this content.
Discussão
Importância do sequenciamento Platform Galaxy em Biologia Moderna
O Projeto Galaxy tornou-se fundamental para ajudar os biólogos sem formação bioinformática para processar e analisar dados de sequenciamento de alto rendimento de uma forma rápida e eficiente. Uma vez considerado uma tarefa hercúlea, esta plataforma disponível publicamente fez correr algoritmos de bioinformática complexos para analisar os dados NGS um processo simples, confiável e fácil. Além de hospedar uma ampla gam...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Os autores gostariam de expressar a sua gratidão aos laboratórios, grupos e indivíduos que desenvolveram Galaxy e David, e, portanto, feitas NGS amplamente acessível para a comunidade científica. A ajuda e conselhos fornecidos por colegas da Universidade de Pittsburgh durante a nossa formação bioinformática é reconhecido. Este trabalho foi apoiado por uma Ellison Medical Foundation New Acadêmico em Aging prêmio (AG-NS-0879-12) e uma concessão do National Institutes of Health (R01AG051659) para AG.
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
Referências
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22(2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085(2011).
- Consortium, C. eS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383(2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, Web Server issue W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639(2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788(2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158(2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados