
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
의 Transcriptomic 분석
요약
갤럭시 다윗은 생물 정보학 교육을받지 않은 상태에서 연구자들은 분석하고 RNA-SEQ 데이터를 해석 할 수 있도록 인기있는 도구로 등장했다. C. elegans의 연구원들은 RNA-SEQ 실험, 액세스를 수행 갤럭시를 사용하여 데이터 집합을 처리하고 DAVID를 사용하여 유전자 목록에서 의미있는 생물학적 정보를 얻을 수 있도록 우리는 프로토콜을 설명합니다.
초록
차세대 시퀀싱 (NGS) 기술은 생물 조사의 본질을 혁명을 일으켰다. 이들 중, RNA 시퀀싱 (RNA-SEQ)는 유전자 발현 분석 및 사체 매핑을위한 강력한 도구로 떠오르고있다. 그러나 RNA-SEQ 데이터 세트를 처리하는 것은 복잡한 계산 전문 지식을 필요로하며, 생물학 연구자 고유의 과제를 포즈. 이 병목 현상은 유전자 온톨로지 (GO) 기간 분석 스위트는 데 도움이 생물 정보학 기술이없는 사용자가 RNA-SEQ 데이터를 분석 할 수 있습니다 오픈 액세스 갤럭시 프로젝트 및 주석, 시각화, 및 통합 검색 (DAVID)의 데이터베이스에 의해 완화 된 대규모 데이터 집합으로부터 생물학적 의미를 도출. 그러나 처음 사용자 및 생물 정보학 '아마추어, 이러한 플랫폼과 자기 학습과 친숙위한 시간과 위협이 될 수 있습니다. 우리는 C. elegans의 연구원이 웜 RNA를 분리하는 데 도움이됩니다 간단한 워크 플로우를 설명, RNA를-SEQ 실험을갤럭시와 데이비드 플랫폼을 사용하여 데이터를 분석 할 수 있습니다. 이 프로토콜의 농축을 위해 스크리닝 할 수있는 유전자의리스트를 생성하는 단계마다 파라미터를 사용자에게 원시 NGS 데이터 품질 관리 검사, 정렬, 및 차등 유전자 발현 분석 액세스 안내하는 다양한 갤럭시 모듈을 사용하는 단계적 지침을 제공 유전자 또는 DAVID 클래스를 사용하여 생물학적 과정. 전반적으로, 우리는 C. 샘플의 작은 숫자를 실행하는 처음 RNA-SEQ 실험뿐만 아니라 잦은 사용자를 착수 연구원 엘레 간스에이 문서가 정보를 제공 할 것으로 기대하고 있습니다.
서문
인간 게놈의 첫 번째 순서는, 프레드 생어의 디데 옥시 시퀀싱 방법을 사용하여 수행 10 년했다, 그리고 미국 약 $ 3 억 2 비용. 그러나, 설립 이후 10 년간 작은에서 차세대 시퀀싱 (NGS) 기술은 가능한 한 2 주 이내에 전체 인간 게놈 염기 서열와 $ 1,000 미국을 위해하는했다. 게놈 시퀀싱 프로젝트가 빠르게 보편화되고있다으로 비용 날카로운 감소와 함께 놀라운 효율성을 시퀀싱 데이터 수집의 속도를 계속 증가 허용 새로운 NGS 장비는, 상상할 수없는 방법으로 현대 생물학에 혁명을 일으키고있다. 또한, 이러한 개발은 RNA 시퀀싱 (RNA-SEQ) 게놈 전체의 후생 유전 학적 변형 연구 DNA - 단백질 상호 작용을 통해 유전자 발현 분석과 같은 많은 다른 분야의 진전을 도금 한 인간 호스트 미생물 다양성 스크리닝. RNA-괜찮다 NGS 기반특히 q는 가능 종합적으로 정확성과 민감도와지도의 전 사체를 확인하고했다, 그리고 발현 프로파일 링에 대한 선택의 방법으로 마이크로 어레이 기술을 대체하고있다. 마이크로 어레이 기술은 광범위하게 사용되었지만, 이러한 교차 혼성화하고 신뢰성있게 측정 할 수있는 발현 변화의 제한 범위로 기존의 공지 된 유전자 정보 배열 및 다른 단점에 대한 의존도에 의해 제한된다. RNA-SEQ, 다른 한편으로는, 인해 명확한 DNA 매핑 특성으로 낮은 배경 잡음을 생산하면서 알려진 알려지지 않은 성적을 감지하는 데 사용할 수 있습니다. RNA-SEQ는 효모와 같은 모델 생물에서 제공하는 수많은 유전자 도구, 파리와 함께, 벌레, 물고기와 마우스는 많은 중요한 최근 생물 의학 발견을위한 기반을 역임했다. 그러나, 저장의 한계, 처리, 그리고 무엇보다도, m을 포함하여 폭 넓은 과학 사회에 액세스 할 NGS을 중요한 과제 남아 시퀀싱 대용량 데이터의 eaningful 생물 정보 분석.
시퀀싱 기술과 기하 급수적 인 데이터 축적의 급속한 발전 연구원, 액세스 분석하고이 정보를 이해 할 수 있도록 전산 플랫폼을위한 큰 필요를 만들었습니다. 초기 시스템은 컴퓨터 프로그래밍 지식에 의존했다, 반면, 프로그래머가 아닌 액세스하고 정교한 분석을 허용하지 않은 데이터를 시각화 할 수와 같은 NCBI 같은 게놈 브라우저. 웹 기반 오픈 액세스 플랫폼, 갤럭시 ( https://galaxyproject.org/ ),이 공백을 채워 NGS 데이터를 처리하고의 스펙트럼을 수행 할 수있는 연구를 할 수있는 귀중한 파이프 라인 것으로 입증되었습니다 단순하고 복잡 생물 정보 분석. 갤럭시 안톤 Nekrutenko (펜실베니아 주립 대학)과 제임스 테일러의 실험실에 의해 처음 설립되었으며 유지 (존스 홉킨스 대학)F "> 3. 갤럭시는. 그것에게 RNA-SEQ 연구에 참여한 모든 단계를 포함하여 수많은 생물 정보학 요구에 대한 '원 스톱 쇼핑'을 만드는 연산 작업의 넓은 범위를 제공하는 자사의 서버 또는 하나의 데이터 처리를 수행하는 사용자를 Itallows 로컬 자신의 컴퓨터에. 데이터 및 워크 플로우를 재현하고 공유 할 수 있습니다. 온라인 자습서, 도움말 섹션, 그리고 위키 페이지 ( https://wiki.galaxyproject.org/Support 갤럭시 프로젝트에 전념가) 일관성있는 지원을 제공합니다. 그러나, 처음 사용자를 위해, 특히 어떤 생물 정보학 교육 사람들은, 파이프 라인이 어려운 나타날 수 있으며 자기 학습과 친숙의 과정은 많은 시간이 소요될 수 있습니다. 또한, 생물학적 시스템을 연구하고 실험 및 방법의 세부 사항은 영향을 사용 여러 단계의 분석 의사 결정, 이들은 지시없이 이동하기가 어려울 수 있습니다.
전반적인 RN A-서열 갤럭시 워크 플로우는 데이터 업로드 및 RNA-SEQ 데이터 분석 (10)의 각 단계에 필요한 다양한 도구의 집합 인 턱시도 스위트 4, 5, 6, 7, 8, 9를 이용하여 분석 하였다 품질 검사 이루어져 11, 12, 13, 14. 일반적인 RNA-SEQ 실험 실험 부분 (시료 전처리, mRNA의 단리 된 cDNA 라이브러리 제조)로 구성되어 상기 NGS 및 정보학 데이터 분석. 이러한 섹션, 갤럭시 파이프 라인에 포함 된 단계의 개요는 그림 1에 나타내었다.
3fig1.jpg "/>
그림 1 : RNA-SEQ 워크 플로우의 개요. 두 웜 균주 (각각 A 및 B, 오렌지색 및 녹색 선 및 화살표)의 유전자 발현 양상을 비교하기 위하여 RNA-SEQ 실험에 관련된 실험 및 계산 단계들의 그림. 갤럭시 이용의 다른 모듈이 적색으로 표시 우리의 프로토콜에 대응하는 단계와 박스에 도시된다. 다양한 동작의 출력은 청색에 도시 된 파일 포맷이 회색에 기록된다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
턱시도 스위트의 첫 번째 도구는 'Tophat'라는 맞춤 프로그램입니다. 그것은 NGS 입력이 작은 조각으로 읽은 다음 참조 게놈에 매핑 분해. 이러한 2 단계 공정은 그와 정렬 달리 디 될 수 인트론 영역에 걸쳐 판독 보장srupted 또는를 차지하고 매핑됩니다 놓쳤다. 이것은 커버리지를 증가시키고, 새로운 접합 접합부의 식별을 용이하게한다. Tophat 출력은 두 개의 파일 (게놈 위치를 포함 스플 라이스 접합에 대한 정보) 침대 파일 (각 읽기의 매핑 정보 포함) BAM 파일로보고됩니다. 다음으로, BAM 파일은 '커프스'라는 턱시도 스위트의 다음 도구를 사용하여 각 샘플 내의 개별 성적 증명서의 풍요 로움을 추정 참조 게놈에 정렬됩니다. 전체 길이 사체 조각 또는 모든 유전자에 대한 입력 데이터의 모든 가능한 스플 라이스 변종에 걸쳐 'transfrags'을보고 정렬을 스캔하여 커프스 기능을. 이것에 기초하여, 서열 분석되는 각각의 샘플 (각 유전자에 대한 유전자 당 생성 된 모든 증명서의 조립)는 '사체'를 생성한다. 이 커프스 어셈블리는 붕괴 또는 재와 함께 병합된다ference 게놈 다음 도구 'Cuffmerge'를 사용하여 다운 스트림 차동 분석을위한 하나의 주석 파일을 생성합니다. 마지막으로, 최종 Cuffmerge 출력 파일 (도 1)에 대한 각 시료의 TopHat 출력을 비교하여 샘플 간의 'Cuffdiff'공구 측정 차등 유전자 발현. 커프스 단추가 FPKM / RPKM 사용 증명서의 존재비를보고 값을 (조각을 / 매핑 만 당 사본의 당 킬로 읽고 읽습니다). 유전자 길이 (카운트 수준과 비교하는 유전자의 길이를 정규화 할 필요가 있으므로 유전자가 서로 다른 길이를 갖는 (a 참조 게놈 정렬 샘플로부터 판독 평균 수)이 값은 깊이 원료 NGS 데이터의 정규화를 반영 ) 유전자 간. , FPKM가 사용되는 반면 FPKM RPKM과는 본질적으로 모든 판독 한 조각에 해당하는 단일 엔드 RNA-SEQ 사용되고 RPKM와 동일한 쌍의 엔드 RNA-SEQ 그것은 두 동일한 단편에 해당 할 수 판독 사실을 차지한다. 궁극적으로, 이러한 분석의 결과가 차분 테스트 조건 및 / 또는 균주의 유전자 발현 사이의 목록이다.
성공적인 갤럭시 실행이 완료되고 '유전자 목록'이 생성되면, 다음 논리적 단계는 데이터 세트에서 의미있는 지식을 추론하는 분석 많은 생물 정보학을 필요로한다. 많은 소프트웨어 패키지는 DAVID 15 (주석, 시각화 및 통합 검색을위한 데이터베이스)로 공개적으로 이용 가능한 웹 기반의 전산 패키지를 포함, 이러한 요구에 부응하기 위해 등장했다. DAVID는 집적 생물학적 지식에 업로드 된 유전자의리스트를 비교하여 유전자 목록과 관련된 생물학적 통계를 공개하여 높은 처리량 연구 대규모 유전자 목록에 생물학적 의미를 부여 용이하게한다. 이것은 즉 농축 분석, 뒤에 IDE에 대한 테스트생물학적 방법 또는 유전자 클래스는 통계적으로 유의 한 방식으로 상기 유전자의리스트 (들)에 과대 대표되는 경우 ntify. 이 때문에 폭 넓은 통합 된 지식베이스와 내 풍부한 생물 테마 감지하는 연구자 수 있도록 강력한 분석 알고리즘의 조합으로 인기있는 선택이되었다 게놈을 파생 '유전자 목록'(10), (16). 추가 장점은 시퀀싱 플랫폼과 매우 사용자 친화적 인 인터페이스에 생성 유전자 목록을 처리 할 수있는 능력을 포함한다.
선충 Caenorhabditis 엘레 간스 잘 같은 작은 크기, 투명 몸, 간단한 몸 계획, 문화의 용이성과 유전 및 분자 해부에 큰 복종 할 의무로서 많은 장점 알려진 유전자 모델 시스템입니다. 웜 알려진 인간 동족체 (17) 40 %까지 보존 된 유전자를 포함하는 소형 단순하고 잘 주석 게놈을 갖는다. 실제로, C.는 엘레 간스제 18 게놈 완전히 서열화 된 후생 동물 및 RNA-SEQ 유기체의 전 사체 (19), (20)을 매핑하는 데 사용 된 제 1 화학 종 중 하나였다. 초기 웜 연구는 기술 (21), (22)의 발전에 기여 높은 처리량 RNA 캡처, 도서관 준비와 순서뿐만 아니라 생물 정보학 파이프 라인에 대한 다른 방법으로 실험을하고있었습니다. 최근 몇 년 동안, 웜에서 RNA-SEQ 기반 실험이 보편화되고있다. 그러나, 기존의 웜 생물 학자에 대한 RNA-SEQ 데이터의 전산 분석에 의해 제기 된 문제는 기술의 더 나은 활용을위한 주요 장애물이 남아있다.
이 글에서, 우리는 선충에서 발생하는 높은 처리량 RNA-SEQ 데이터를 분석하는 갤럭시 플랫폼을 사용하기위한 프로토콜을 설명합니다. 많은 처음 작은-SCA를 들어제작 : 사용자는 RNA를-SEQ 실험을 수행 할 수있는 가장 비용 효율적이고 간단한 방법은 실험실에서 RNA를 분리하고 염기 서열의 cDNA 라이브러리의 준비 및 NGS 자체에 대한 상용 (또는 사내) NGS 시설을 활용하는 것입니다. 따라서, 우리가 처음 분리하는 단계를 설명 한 C의 정량 및 품질 평가는 RNA-서열에 대한 RNA 샘플을 엘레 간스. 다음으로 정렬 조립 한 유전자 발현의 차이 정량화 하였다 후 시퀀싱 품질 제어 검사를위한 검사로 시작 NGS 데이터의 분석을위한 갤럭시 인터페이스를 사용하여 단계별로 설명 제공. 또한, 우리는 데이비드를 사용하여 생물 농축 연구를위한 갤럭시로 인한 유전자 목록을 면밀히 검토하는 방향을 포함했다. 워크 플로의 마지막 단계로, 우리는 NCBI에 순서 읽기 아카이브 (SRA) (공공 서버에 RNA-SEQ 데이터를 업로드하기위한 지침을 제공 에 http : // www.ncbi.nlm.nih.gov/sra) 과학계에 자유롭게 접근 할 수 있도록합니다. 전반적으로, 우리는이 문서가 샘플의 작은 숫자를 실행하는 처음 RNA-SEQ 실험뿐만 아니라 잦은 사용자를 착수 웜 생물 학자에 포괄적이고 충분한 정보를 제공 할 것으로 기대하고 있습니다.
Access restricted. Please log in or start a trial to view this content.
프로토콜
1. RNA 분리
- 예방 대책
- 존재하는 RNases을 제거하기 위해 시판 된 RNase 스프레이를 사용하여 전체 작업 표면, 악기 및 피펫을 닦아.
- 정기적으로 프로토콜의 여러 단계 중 신선한 것로 변경, 항상 장갑을 착용 할 것.
- 전용 필터 팁을 사용하여 RNA 저하를 방지하기 위해 최대한 얼음에 모든 샘플을 유지한다.
참고 : NGS 플랫폼에서 최상의 데이터를 얻기 위해서는 고품질의 RNA로 시작하는 것이 중요합니다. RNA 분리 및 제조 방법은 시료의 기원, 시퀀싱 및 연구자 우선 방식에 따라 다양하다. 여러 상용 키트는이 목적을 위해 사용할 수 있습니다 또는 RNA는 RNA 추출의 표준 페놀 - 클로로포름 방법을 사용하여 분리 할 수 있습니다. 하나의 방법론으로, 위에 나열된 예방 조치는 오염 및 OBT를 최소화하기 위해 프로세스 전반에 걸쳐 따라야한다아인 자연 그대로의 RNA 샘플.
- 수확 웜
- 변형 당 1,000-1,500 연령대 C. elegans의 성인 벌레를 얻기 위해 차아 염소산 표백 처리 (23)에 의해 웜 인구를 동기화 할 수 있습니다.
- 30 초 동안 테이블 탑 원심 분리기에 325 XG에 M9 완충액을 사용하여 스핀 플레이트 웜 씻어. 벌레의 펠렛 뒤에 떠나는 M9 버퍼를 대기음. 세균 이월을 제거하기 위해 적어도 세 번이 단계를 반복합니다.
- 웜 펠릿 ~ 500 용해 완충액 μL (경우 상업용 키트를 사용하여) 또는 트리 졸 추가 (페놀 및 구아니딘 이소 티오 시아 네이트의 모노 - phasic 용액을 페놀 경우 : 1.3.3에 기재된 클로로포름 추출이 수행된다) 웜 조직을 방해 , RNases 비활성화 핵산을 안정화.
주 :이 프로토콜은 -80 ℃에서 저장 한 다음 액체 질소에서 시료를 냉동 플래시 의해 여기 일시 정지 될 수있다.
- RNA 분리
- 20 초의 주기로 45 % 진폭으로 초음파 처리 웜 샘플. 'ON'및 40 개의. 'OFF'(주 당 8-12 사이클). 항상 얼음에 샘플을 유지합니다.
주 : 초음파기 탐침이 상기 버퍼에 침지하고, 전체에 일정한 레벨로 유지되는 것을 확인. 샘플의 거품을 피하고 샘플 사이에-철저하게 프로브를 청소합니다. 초음파 사이클 사용 소니 케이의 종류에 따라 달라질 수 있습니다. 초음파 조건이 먼저 실험을 시작하기 전에 테스트 샘플에 최적화되어이 좋습니다. - 시중에서 판매하는 키트를 사용하는 경우, 규정 된 프로토콜에 따라 RNA 분리를 진행합니다. 페놀 - 클로로포름 법을 사용한 RNA의 분리를 위해, 다음 단계를 수행한다.
- 원심 분리는 10 분 동안 16,000 XG에 샘플을 초음파. 4 ℃에서
- 1.5 용액의 RNase가없는 미세 원심 분리 튜브로 상청액을 옮기고 (RNA / DNA 분리 시약의 부피 번째 1/5) 클로로포름 100 μL를 추가한다.
주의: 클로로포름 독성이다. 이 물질을 취급 할 때는 노출을 최소화하고 흡입을 방지하기 위해 화학 후드에서 작동합니다. - 60 초 - 30 철저하게 샘플을 소용돌이. 및 샘플을 3 분 동안 실온에서 앉아 보자.
- 15 분 동안 11,750 XG 원심 분리기. 4 ℃에서. DNA를 함유하는 백색 인터페이스를 흡인하지 새의 RNase가없는 미세 원심 분리 튜브 돌보는에만 상부 수층을 전송. 반복 1.3.6을 통해 1.3.4 단계를 반복합니다.
- 250 μL 2- 프로판올 (1/2 성상 또는 RNA / DNA 분리 시약 용적의 70 %)을 첨가하고 혼합하는 튜브를 반전. 튜브를 실온에서 10 분 동안 앉아 또는 -80 ° C에서 밤새두고 보자.
- 원심 시료를 10 분 동안 11,750 XG에. 4 ℃에서. 펠렛 방해되지 않도록 튜브의 하단에 몇 μL 남겨두고, 매우 신중하게 뜨는을 가만히 따르다.
- (의 RNase없는 물을 사용하여 만든) 75 % 에탄올 500 μL로 펠렛을 씻고 5 분 16,000 XG에 스핀 다운. 에이t의 4 ° C.
- 펠렛을 방해하지 않고 가능한 한 많은 상층 액을 제거합니다. 공기는 몇 분 동안 후드에서 펠렛을 건조.
- 의 RNA 분해 효소가없는 물을 30 μL를 추가하고 10 분 동안 가열하여 RNA 펠렛을 용해 도움이됩니다. 60 ° C에서.
- bioanalyzer를 사용하여 RNA의 질과 양을 확인합니다.
주 : Bioanalyzer은 RNA의 품질의 측정치와 같은 R NA ntegrity I의 N에 엄버 (RIN)를 생성한다. 적어도 8의 RIN는 RNA-SEQ 샘플에 대한 권장 임계 값 (더 낫다)입니다. RNA의 양과 질 또한 분광 확인할 수뿐만 아니라 RNA 무결성의 시각적 평가를 하였다한다. 이를 위해, 28S 및 18S 리보솜 RNA 밴드의 적절한 분리를 얻기 위하여 충분히 긴 1.2 % 아가 로스 겔상에서 샘플을 실행. 두 가지 주파수 대역 (18S rRNA에 대한 1.75의 KB 및 C. 엘레 간스의 경우 28S rRNA의의 3.5 KB)의 존재는 RNA 품질의 허용 척도이다. - 사용 ~ 100 NG /시에 μL RNA시퀀싱 라이브러리의 준비를위한 공급 업체 / NGS 시설에 페이지.
참고 : RNA 샘플 시퀀싱 서비스 제공 업체에 드라이 아이스에 제공해야한다. 대부분의 업체는 라이브러리 준비하기 전에 독립적 인 RNA의 품질 관리 테스트를 실시하고 있습니다.
- 20 초의 주기로 45 % 진폭으로 초음파 처리 웜 샘플. 'ON'및 40 개의. 'OFF'(주 당 8-12 사이클). 항상 얼음에 샘플을 유지합니다.
2. RNA-SEQ 데이터 분석
- 원시 시퀀싱 데이터의 다운로드
- 는 "파일 전송 프로토콜"(FTP)을 사용하여 NGS 제공자로부터 fastq.gz 형식으로 인코딩 된 상기 압축 된 원시 fastq 시퀀싱 데이터 다운로드.

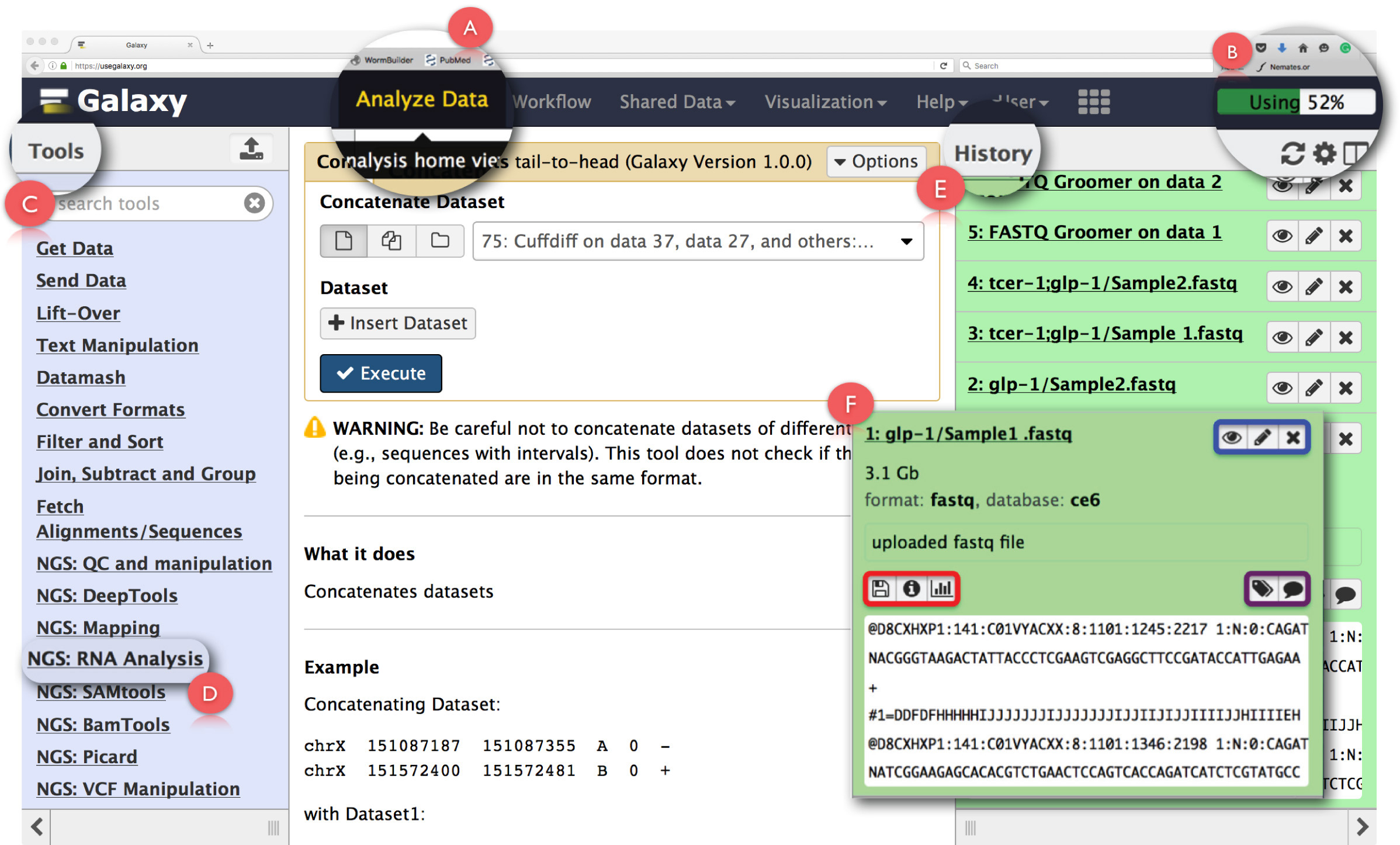
그림 2 : 갤럭시 사용자 인터페이스 패널 및 키 RNA-SEQ 기능의 레이아웃입니다. 페이지의 주요 기능 확장 및 강조 표시됩니다. (A)을 액세스하는 데 사용되는 웹 페이지의 헤더에서의 데이터 분석 '기능을 강조 분석 홈보기. (B)의 조작에 의해 이용 갤럭시 서버의 공간을 나타내는 '진행 바'이다. (C)는 갤럭시 인터페이스에서 실행할 수있는 모든 도구를 나열하는 '도구 섹션'입니다. (D)는 '을 NGS : RNA 분석'도시 툴 영역은 RNA-서열 분석에 사용 하였다. (E)는 갤럭시를 사용하여 생성 된 모든 파일을 나열하는 '역사'패널을 보여줍니다. (F)는 역사 섹션에있는 파일을 클릭하면 열립니다 대화 상자의 예를 보여줍니다. (F) 내에서 파란색 상자를 볼 수 있습니다 아이콘, editthe 특성을 강조하거나, 보라색 상자가 데이터 세트 태그 또는 주석 '편집'에 사용할 수있는 아이콘을 강조하고 데이터 세트를 삭제, 빨간색 상자 아이콘을 나타냅니다 데이터를 다운로드하는 작업의 세부 사항을 볼 수행 또는 작업을 다시 실행하십시오. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
- 갤럭시 시작하기
참고 : 갤럭시 클라우드 액세스 및 제한된 무료 저장 공간을 제공하는 웹 기반의 플랫폼을 사용하여 무료 공용 서버에서 실행할 수 있습니다. 그것은 또한 다운로드 및 사용자의 컴퓨터 또는 기관하지만 현지 처리에 의해 호스팅 계산 클러스터에서 로컬로 실행, 데이터 저장소 제한 및 사용자 시스템의 처리 능력의 한계에 의해 제한 될 수 있습니다. 다운로드 및 설치에 대한 세부 사항은에 액세스 할 수 있습니다 https://wiki.galaxyproject.org/Admin/GetGalaxy . 이 프로토콜에서 우리는 갤럭시 파이프 라인의 웹 기반 사용법을 설명합니다.- 에서 사용자의 컴퓨터, 액세스 갤럭시에 NGS 데이터를 다운로드하여 저장 한 후laxy.org/ "대상 ="_blank "> https://usegalaxy.org/.
- 페이지, 로그인의 헤더에 '사용자'를 클릭하여 사용자 계정을 등록하고 사용자 인터페이스 패널에 익숙해지고 의해 시작합니다.
참고 : 처음 사용자가 홈페이지에서 제공하는 '여기서 시작'튜토리얼은 갤럭시의 기본 셋업과 친숙 얻을 활용하는 것이 좋습니다 ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) . - 또한 갤럭시의 시작 화면 인 '분석 홈보기'를 액세스 할 헤더 패널에서 '분석 자료'(그림 2A)를 클릭합니다.
참고 : 헤더는 그 세부 사항 그들 위에 마우스 포인터를 가져 가면 볼 수있는 다른 링크를 주택. 헤더의 우측 상단의 작업 (도 2b)의 공간 활용을 모니터링 진행 바있다. - 기음에 핥기 'NGS : RNA 분석'왼쪽 패널 (그림 2C)에서 '도구 메뉴'에서 작업 RNA-서열 데이터 분석에 필요한 모든 도구에 액세스 할 수 있습니다.
참고 : '도구 메뉴'모든 작업 갤럭시 제안을 카탈로그. 이 메뉴는 작업과 그 작업을 수행하는 데 필요한 모든 도구의 목록을 열 것 중 하나를 클릭 기준으로 분할됩니다. - 오른쪽 (그림 2E)의 '역사'패널의 상단에있는 톱니 바퀴 아이콘을 클릭하여 새로운 분석 역사를 만듭니다. 선택 팝업 메뉴에서 옵션 '새로 만들기'를. 이 '역사를'분석을 식별 할 수있는 적절한 이름을 지정합니다.
참고 : '역사'패널은 갤럭시에서 작업을 실행하여 생성 된 모든 출력 파일뿐만 아니라 분석에 업로드 된 모든 파일을 보여줍니다. 이 패널에서 파일 이름을 클릭하면 수행 된 작업에 대한 자세한 정보 대화 상자를 엽니 다및 데이터 세트 (그림 2 층)의 조각. 이 상자의 아이콘은 데이터 세트 (그림 2 층은 파란색으로 표시) '속성을 편집'또는 '삭제', '보기'로 사용자를 할 수 있습니다. 또한, 사용자는이에서 데이터 집합을도 또한 '편집'데이터 세트 태그 또는 주석 (그림 2 층은 보라색으로 표시), 작업의 '다운로드'데이터 '세부 정보보기', '다시 실행'작업 또는 '시각화'수 대화 상자 (그림 2 층은 빨간색으로 강조). - 원시 fastq 파일을 업로드하기 위해 '도구 메뉴'에서 '데이터 가져 오기'아래의 '파일 업로드'기능을 클릭합니다.
참고 :이 또는 다른 도구를 클릭하면 중간 '분석 인터페이스'패널의 동작에 대한 간단한 설명, 시험 자체를 연다. 이 패널은 함께 끈왼쪽 패널 오른쪽 '역사'패널에서 '입력 파일'(그림 2E)에서 '도구'. 여기서 '역사'의 입력 파일을 선택하고 다른 매개 변수는 주어진 작업을 실행하도록 정의됩니다. 모든 테스트에서 생성 된 출력 데이터 세트는 '역사'에 다시 저장됩니다. 도구에서 생성 된 모든 출력 파일의 자세한 목록과 함께 주어진 도구를 실행에 사용할 수있는 모든 매개 변수에 대한 설명은 '분석 인터페이스 "패널의 테스트와 함께 포함되어 있습니다. - 작업은 '분석 인터페이스'에서 열리면, '로컬 파일 선택'을 클릭하거나, (빠른 업로드) 'FTP 파일 선택'시퀀싱 파일이 들어있는 폴더로 이동하여 업로드 할 수있는 적절한 데이터 집합을 선택합니다.
- 업로드 된 파일 유형 (기본 설정) '자동 검색'에 갤럭시를 허용합니다. 선택 'C. 엘게놈의 풀다운 메뉴에서 egans '.
- 데이터 업로드를 시작하려면 '시작'을 클릭합니다. 파일이 업로드되면, 그것은 '역사'패널에 저장됩니다 거기에서 액세스 할 수 있습니다.
- 여러 시퀀싱 데이터 파일이 하나의 샘플 용으로 제작하는 경우, 'CONCATENATE'도구를 사용하여 결합합니다. 이렇게하려면 '도구 메뉴'에서 '텍스트 조작'옵션을 엽니 다.
- '분석 인터페이스'의 중간에 드롭 다운 상자에서 결합하고 '실행'을 클릭해야 할 파일을 선택합니다 'CONCATENATE'도구를 클릭합니다.
참고 :이 작업을 사용하여 생성 된 출력 파일이 fastq 형식으로 생성됩니다. 매핑 프로그램은 fastq 파일과 그 한계에 도달하면 새로운 fastq 파일이 남아있는 시퀀스 생성되는 당 16,000,000 시퀀스의 제한이 있습니다. '; 연결하여 '도구는 데이터 집합을 결합하는 등의 경우에 필요하다. - 아래에있는 'fastq 루머'도구를 사용하여 갤럭시 RNA-서열 분석에 필요한 fastqsanger 형식으로 업로드 fastq 형식의 파일을 변환 'NGS를 : 품질 관리 및 조작'섹션을 (추가 파일을 참조하십시오).
- 옵션 '신랑에게'파일에서 해당 fastq 데이터 세트를 선택하고 기본 매개 변수를 사용하여 도구를 실행합니다.
참고 :이 작업을 사용하여 생성 된 출력 파일은 fastqsanger 형식으로 생성됩니다.
- fastqsanger 데이터 품질 관리 테스트
- 아래에있는 'FastQC'도구를 사용하여 읽는 업로드 fastqsanger의 품질을 확인 : '도구'메뉴의 'NGS를 QC 및 조작'.
- 쇼어 '의 드롭 다운 메뉴에서 손질 fastqsanger 데이터 파일을 선택t '는 현재 라이브러리에서 데이터를 읽고 기본 매개 변수를 사용하여 도구를 실행합니다.
참고 : 모든 어댑터 서열하여 읽기의 품질과 존재에 특별한주의를 기울이십시오. 어댑터는 일반적으로 NGS 제공 업체에 의해 게시물 RNA-SEQ 데이터 처리의 일환으로 제거하지만, 일부 경우에, 뒤에 남아있을 수 있습니다. 품질 기준의 설명은 이동 http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - NGS 제공 업체에 문의 및 어댑터가있는 경우, 그들이에서 '클립'도구를 사용하여 트림 'NGS : 품질 관리 및 조작'작업 메뉴를 표시합니다.
참고 :이 작업을 사용하여 생성 된 출력 파일은 원시 TXT 형식뿐만 아니라 모든 웹 브라우저에서 열 수있는 HTML로 생성됩니다.
- 턱시도 스위트 룸과 데이터 분석
- 모자
- 의 최신 버전을 다운로드 C. 엘레 참조 게놈 FASTA 및 2.2.6에서 설명한 바와 같이 GTF (유전자 전달 형식) 파일 업로드에서 파일 '.
- 'RNA 분석 NGS'섹션 및 시퀀싱 다운로드 한 참조 게놈을 읽어 매핑 'TopHat'도구에 클릭을 엽니 다.
- 질문에 대한 드롭 다운 메뉴에서 적절한 답을 선택하는 것은 '이 단일 엔드 또는 쌍 엔드 데이터인가?'
- 적절한 fastq 파일을 선택합니다.
- 선택한 다음 드롭 다운 메뉴에서 '역사에서 게놈 사용'단계 2.4.1.1에서 다운로드 참조 게놈을 선택합니다.
- 다른 매개 변수에 대한 '기본'을 선택하고 '실행'을 클릭합니다.
참고 :이 작업을 사용하여 생성되는 출력 파일 중, '허용 조회수'파일이 후속 단계에 사용됩니다.
- 커프스 단추와 Cuffmerge
- '커프를 선택NGS 링크 '는의 도구': 성적 증명서를 조립하는 RNA 분석 '섹션 발현에 대한 자신의 풍요 로움과 테스트를 추정한다.
- 첫번째 드롭 다운 메뉴에서 TopHat 분석으로부터 얻어진 매핑 '허용 히트 (BAM 포맷)'파일을 선택한다.
- 두번째 드롭 다운 메뉴에서, 단계 2.4.1.1에서 다운로드 한 파일을 참조 GTF 주석 세트.
- '바이어스 보정을 수행'옵션 '예'를 선택하고 다른 모든 매개 변수에 대한 기본 설정을 사용하여 작업을 실행합니다.
참고 :이 작업을 사용하여 생성되는 출력 파일 중, '허용 성적 증명서'파일이 후속 단계에 사용됩니다. - 에서 열기 'Cuffmerge'도구 'NGS : RNA 분석'은 '조립 성적 증명서'생산에 대한 모든 RNA-SEQ 샘플을 병합합니다.
참고 : 도구 자체 채우고 목록의 첫 번째 상자 모든 커프스 단추에 의해 생산 m> GTF 파일. - 같은 변형 / 상태의 생물학적 복제 (생물 복제에 대한 설명을 참조)를 포함하여 테스트 한 모든 균주 / 조건에 대한 '조립 성적 증명서'파일을 선택합니다.
- '사용 참조 주석'에 대한 '예'를 선택하고 단계 2.4.1.1에서 다운로드 한 GTF 파일을 선택합니다.
- 다음 상자에 다시 '사용 세쿠엔스 다타'옵션 '예'를 선택하고 단계 2.4.1.1에서 다운로드 한 전체 게놈 FASTA 파일을 선택합니다.
- 기본값으로 다른 매개 변수를 유지, '실행'을 클릭합니다.
참고 : Cuffmerge은 하나의 GTF 출력 파일을 생성합니다.
- Cuffdiff
- 의 'Cuffdiff'도구로 이동합니다 'NGS : RNA 분석'섹션을. '성적 증명서'메뉴에서 Cuffmerge에서 병합 된 출력 파일을 선택합니다.
- 상표두 균주 / 조건 이름 조건 1 및 2.
참고 : Cuffdiff 두 개 이상의 변종이나 조건뿐만 아니라 시간 경과 실험 사이의 비교를 수행 할 수 있습니다. 간단하게 필요에 따라 각각의 새로운 변종 / 조건을 추가하기 위해 '추가 새로운 조건'옵션을 사용합니다. - '를 복제'선택 개별 균주 / 상태의 다른 생물학적 복제 대응 TopHat으로부터 '허용 조회수'출력 파일 아래의 각 스트레인 / 상태에 대한. 여러 파일을 선택하려면, PC를 사용하는 경우, 'CTRL'키를 Macintosh 컴퓨터를 사용하고, 경우 'cmd를'키를 누르십시오.
- 기본 매개 변수로 다른 모든 옵션을 둡니다. 작업을 실행하는 '실행'을 클릭합니다.
참고 : Cuffdiff는 RNA-SEQ 분석의 최종 판독 등을 표 형식으로 다수의 출력 파일을 생성합니다. 이 결합 성적 증명서, 유전자 FPKM 추적을 파일을 (포함유전자 신원을 공유 사체), 기본 증명서 및 코딩 서열의 FPKM 값. 생성 된 모든 데이터 파일은 P 값 및 Q 값을 포함한 균주 / 조건 간 비교에 (LOG2 규모)의 변화뿐만 아니라, 통계 데이터를 접 스프레드 시트 애플리케이션에서 볼과 같은 유전자 이름 궤적과 유사한 특성을 포함 할 수있다. 이러한 파일의 데이터는 통계적 유의성 차이에 기초하거나 (업 조절 유전자와 같이 크기와 방향의 변화 또는 하향) 유전자 발현 변화를 접어 정렬하고 사용자의 요구에 따라 조작 될 수있다. 다른 유전자 식별자 사이의 변환이 필요한 경우 (예, 코스 미드 Wormbase 번호 대 유전자 ID)는, Biomart (볼 도구 http://www.biomart.org/가 ) 사용될 수있다.
- 모자
3. 유전자 온톨로지 (GO) 기간 분석 DAVID를 사용하여
- 웹 사이트 시간에서 액세스 DAVIDttps : //david.ncifcrf.gov/. 웹 페이지의 헤더에 '분석 시작'을 클릭합니다. '단계 1'에서, 입력 유전자의 식별자로서 '단계 2'선택 'Wormbase 유전자 ID'에 박스로 갤럭시 A.로부터 얻은 유전자의 목록을 복사 및 붙여 넣기.
참고 : 데이비드 대부분의 공개 주석의 범주를 인식하므로 (예 : Entrez의 유전자 ID 또는 유전자 상징으로) 다른 유전자 식별자도 사용할 수 있습니다. - '3 단계'에서, (유전자 분석하기) '목록 유형'에서 '유전자 목록'을 선택하고 '목록 제출'을 아이콘을 클릭합니다.
참고 : '분석 마법사', 업로드 된 유전자 목록에서 실행할 수있는 모든 하이퍼 링크 DAVID 도구 (그림 3)를 나열하는 열립니다. 사용자의 요구 사항에 따라 관련 해당 모듈에 액세스하려면 다음 링크를 클릭하십시오. 어떤 DAVID 도구를 사용하기 '를 클릭, 주어진 작업에 적합한 도구를 확인하려면? '에 링크' ; 분석 마법사 '페이지. 분석 기간 동안 어느 시점에서 '분석 마법사의 홈 페이지로 돌아 헤더에있는'시작 분석 '링크를 클릭하십시오.

그림 3 : DAVID 분석 마법사 웹 페이지 및 운영 출력의 예의 레이아웃입니다. '분석 마법사'웹 사용자 인터페이스는 다양한 매개 변수를 기반으로 농축에 업로드 된 유전자 목록을 분석하는 데 사용되는 도구가 나열되어 있습니다. 이러한 도구를 클릭하면 새로운 웹 페이지에서 분석 된 데이터를보고합니다. "유전자 기능 분류", "기능 표 주석 '및'기능성 주석 클러스터링 '에서 생성 된 리포트 표로서는 세트 (화살표)로 표시된다.>이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
- 기능 주석 도구 1 : 기능 주석 클러스터링
- 요약 페이지로 이동 '기능 주석 클러스터링'모듈을 클릭하십시오. 기본 주석의 범주를 유지하고 자신의 농축 점수에 의해 순위 유사한 주석 용어의 클러스터를 생성하기 '기능 주석 클러스터링'를 클릭하십시오.
- 그와 'RT'(관련 용어) 범주에 관련된 다른 유사한 용어를 나열에 대한 자세한 내용을 읽어 각 용어의 하이퍼 링크 된 이름을 클릭하십시오.
- 용어 및 클러스터 내의 모든 조건과 관련된 모든 유전자를 나열하는 빨간색 'G'와 관련된 유전자를 나열 보라색 막대를 클릭합니다.
- 클러스터에있는 모든 유전자와 용어의 2 차원보기를 볼 수있는 녹색 아이콘을 클릭합니다.
참고 : 마지막 세 열은 각각에 대한 분석 및 통계 결과를 나열기간. 이에 대한 결과와 다른 모든 분석은 '파일 다운로드'링크를 클릭하여이 .txt 형식으로 다운로드 할 수 있습니다.
- 기능 주석 도구 2 : 기능 주석 차트
- 요약 페이지로 돌아가서 유전자 목록과 관련된 상당히 과대 대표 생물학적 용어 (예를 들어 전사 인자의 활동이나 키나제 활성을) 식별하기 '기능 주석 차트'를 클릭하십시오.
- 더 자세한 정보와 'RT'(관련 용어) 기타 관련 용어를 나열를 얻기 위해 용어의 이름을 클릭합니다.
- 각각의 카테고리에 해당하는 모든 관련 유전자를 나열 보라색 막대를 클릭합니다.
참고 : 마지막 두 열은 각 범주에 대한 통계 - 테스트 '결과를 나열합니다.
- 기능 주석 도구 3 : 기능 주석 표
- 요약 페이지로 돌아가서 능의 '클릭NAL 주석 표는 '통계적 계산없이 목록 유전자와 관련된 모든 주석의리스트를 참조한다.
참고 :이 도구는 목록의 유전자에 의해 유전자 분석에 유용 할 수 있습니다 또는 특정 매우 흥미로운 유전자 볼 수 있습니다.
- 요약 페이지로 돌아가서 능의 '클릭NAL 주석 표는 '통계적 계산없이 목록 유전자와 관련된 모든 주석의리스트를 참조한다.
- 유전자 기능 분류 도구
- '분석 마법사'로 돌아가서 자신의 '농축 점수',리스트의 유전자 그룹의 전체 농축의 측정에 따라 순위가 유전자의 기능과 관련된 그룹으로 입력 유전자 목록을 분리하는 '유전자 기능 분류'모듈을 클릭하십시오.
- 용어의 이름을 클릭하면 유전자 그룹의 기능 관련 유전자를 나타 내기 위해 더 자세한 정보와 'RG'를 얻을 수
- 관련 생물학 모든 유전자와 용어의 2 차원보기를 볼 수있는 녹색 아이콘을 나열 빨간색 'T'(용어 보고서)를 클릭합니다.
- 유전자 이름배치 뷰어
- '분석 마법사'로 돌아와 해당 유전자 이름에 'Wormbase 유전자 ID를'번역 '유전자 이름 일괄 뷰어'를 클릭하십시오. (WBGene00022855 tcer = 1).
- 많은 유전자 관련 정보를 얻기 위해 유전자 이름을 클릭하십시오.
- 유전자를 공개하는 각각의 유전자 옆에있는 'RG'를 클릭합니다 (관련 유전자) 링크는 관심의 유전자 기능적으로 관련이있는 것으로 예측했다.
NCBI의 순서 읽기 아카이브 위에 4. 업로드 RAW 데이터 (SRA)
- NCBI '링크에 로그인에서 SRA 웹 페이지에 액세스하거나 새 계정을 등록합니다.
- 'Bioproject'를 클릭합니다.
- 왼쪽에있는 '사용 Bioproject'제목 아래 '제출'을 클릭합니다.
- 옵션 '새 제출'를 선택합니다. 제출자의 업데이트 정보. 나머지 일곱 개 탭을 계속실험 데이터의 세부 사항을 작성하여 업로드되는. 완료되면 '제출'을 클릭합니다.
참고 : 다섯 번째 '뇨 시료, 척수액 시료'탭에서 '뇨 시료, 척수액 시료'빈의 슬롯을 둡니다. - '내 제출'링크를 클릭하여 결과 페이지를 새로 고칩니다. 제출 된 데이터는 할당 제출 번호, 간략한 설명 및 업로드 상태로 표시됩니다.
- '새 제출을 시작하는'상자에,이 페이지 상단의 '뇨 시료, 척수액 시료'를 클릭하고 '새 제출'을 만들 수 있습니다. 각 샘플에 대해 별도의 제출을 제출합니다.
- 4.4 'Bioproject'의 경우와 마찬가지로, 제출자의 세부 사항을 업데이트하고 각 탭의 세부 사항을 작성 탭의 나머지를 계속합니다. 한 번 검토를 완료하고 '제출'을 클릭합니다.
- 로 이동하여 HTTP : //www.ncbi.nlm.nih.go를V / SRA는 최종 '시퀀스 읽기 아카이브 (SRA)'제출을 만들 수 있습니다.
- '시작하기'에서 'SRA에 로그인'을 클릭합니다.
- 다음 페이지에서 'NCBI PDA'링크를 클릭합니다. 에 '업데이트 환경 설정'링크가 열립니다. 양식을 작성하고 '환경 설정 저장'을 클릭합니다.
- 결과 페이지에서 '새로 만들기 제출'링크를 클릭합니다. '별칭'에 적당한 이름을 입력하고 '저장'을 클릭합니다. 제출 ID 및 기타 세부 사항에 테이블이 생성됩니다.
- '새로운 실험'을 클릭하고 각 ', 뇨 시료, 척수액 시료'에 대해 최소한 하나의 고유 한 시퀀싱 라이브러리를 등록합니다.
- 지정 및 이전에 만든 'BioProject'와 '뇨 시료, 척수액 시료'제출 ID 년대를 연결합니다. A '새로운 실험'이 생성됩니다.
- 페이지 하단의 '새 실행'을 클릭후 SRA 실험이 이루어과 연결해야하는 데이터 파일을 식별하고있다.
- 각 데이터 파일의 MD5 합계를 계산합니다. 매킨토시 터미널에서이 작업을 수행하려면, 응용 프로그램 / 유틸리티 / 터미널로 이동합니다. 단말기 (따옴표 제외) 'MD5'의 유형은 공간 하였다. 드래그 앤 파인더에서 단말기에 업로드하고 '입력'을 클릭해야 할 파일을 놓습니다.
- 터미널은 영숫자 MD5 합계를 반환합니다. 파일 업로드에 대한 제출 과정의 일부로서이를 입력합니다. FTP를 사용하여 파일을 업로드하기 위해 시스템에 의해 제공되는 사용자 이름과 암호를 사용합니다.
Access restricted. Please log in or start a trial to view this content.
결과
C. 엘레 간스에서, 배아 줄기 세포 (GSC를) 수명 연장의 제거는 스트레스 내성을 향상시켜, 체지방 (24), (28)를 상승시킨다. GSC를 손실이 레이저 어블 레이션에 의해 또는 GLP-1과 같은 돌연변이에 의해 초래하거나, 표기의 네트워크 (29)를 인자 활성 수명을 연장시킨다. 하나 개는 이러한 요인, TC...
Access restricted. Please log in or start a trial to view this content.
토론
현대 생물학의 갤럭시 시퀀싱 플랫폼의 중요성
갤럭시 프로젝트 처리하고 빠르고 효율적인 방법으로 높은 처리량 시퀀싱 데이터를 분석하는 생물 정보학 교육을받지 않은 상태에서 생물학을 돕는 수단이되고있다. 일단이 공개적으로 사용 가능한 플랫폼은, 간단 신뢰성, 쉬운 과정 NGS 데이터를 분석하는 복잡한 생물 정보학 알고리즘을 실행했다, 헤라클레스의 작업을 고려했?...
Access restricted. Please log in or start a trial to view this content.
공개
저자가 공개하는 게 없다.
감사의 말
저자는 갤럭시 다윗을 개발, 따라서 과학적인 지역 사회를위한 NGS 광범위하게 액세스 가능하게 한 실험실, 개인과 단체에 감사의 뜻을 전하고 싶습니다. 우리의 생물 정보학 교육 과정 피츠버그 대학의 동료가 제공하는 도움과 조언이 인정된다. 이 작품은 수상 (AG-NS-0879-12)와 국립 보건원 AG에 (R01AG051659)에서 보조금을 노화에 엘리슨 의료 재단 신규 장학생에 의해 지원되었다.
Access restricted. Please log in or start a trial to view this content.
자료
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
참고문헌
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22(2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085(2011).
- Consortium, C. eS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383(2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, Web Server issue W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639(2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788(2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158(2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Access restricted. Please log in or start a trial to view this content.
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유