
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
的转录分析
摘要
银河和大卫已经成为流行的工具,让没有生物信息学培训调查分析和解释RNA-Seq的数据。我们描述了一种协议,用于秀丽隐杆线虫的研究人员进行RNA测序实验,访问和使用银河处理数据集,并获得从使用DAVID的基因列表有意义的生物信息。
摘要
新一代测序(NGS)技术已经彻底改变了生物学研究的性质。在这些中,RNA测序(RNA-SEQ)已经成为用于基因表达分析和转录的映射的有力工具。然而,处理RNA-Seq的数据集需要复杂计算的专业知识和带来的生物学研究人员固有的挑战。这个瓶颈已经缓解由开放接入银河项目,使用户无需生物信息学技术分析RNA-Seq的数据,和注释,可视化和集成发现(DAVID)的数据库,基因本体论(GO)项的分析工具,帮助从大的数据集获得生物学意义。但是,对于初次使用者和生物信息学爱好者,自学习和熟悉这些平台可以是耗时且艰巨。我们描述一个简单的工作流程,这将有助于线虫研究人员分离出病毒RNA,进行了RNA-Seq的实验并分析利用银河DAVID平台的数据。这个协议提供了使用各种银河模块在每个步骤访问原始NGS数据,质量控制检查,对准和差异基因表达分析,指导与参数的用户生成可筛选的富集的基因列表逐步指令基因的类或使用DAVID生物过程。总体而言,我们预计,本文将提供信息,以秀丽隐杆线虫的研究人员正在进行运行的小样本首次RNA-Seq的实验以及频繁的用户。
引言
人类基因组的测序首先,执行使用弗雷德·桑格的双脱氧核苷酸测序方法,用了10年,耗资估计US $ 3十亿1,2。然而,自成立以来一点点过去十年,新一代测序(NGS)技术已经使人们有可能在两周内测序整个人类基因组和US $ 1,000。新NGS仪器的成本是允许日益增长的测序数据采集的速度令人难以置信的效率,与急剧下降一起,以难以想象的方式彻底改变现代生物学的基因组测序项目正在迅速成为家常便饭。此外,这些发展已镀锌在许多其他领域,例如通过RNA测序(RNA-SEQ),的基因组范围的表观遗传修饰的研究中,DNA-蛋白质相互作用的基因表达分析的进步,并筛选在人类宿主微生物多样性。 NGS-基于RNA的硒●在特定使得有可能鉴定和地图转录全面与准确度和灵敏度,并且已取代微阵列技术作为选择的一种绘制表达图谱的方法。虽然微阵列技术已经被广泛使用时,它通过其与已知基因组信息预先存在的阵列,以及其它的缺点的依赖的限制,诸如交叉杂交,并且能够可靠地计量的表达的变化限制范围。 RNA-SEQ,在另一方面,可用于检测已知和未知的转录而产生低的背景噪声,由于其明确的DNA映射性质。 RNA-Seq的,加上通过模式生物如酵母提供了众多的遗传工具,蝇,虫,鱼和小鼠,曾担任许多重要生物医学近期发现的基础。然而,显著挑战依然存在,使以更广泛的科学界NGS无法访问,包括存储的局限性,加工,最重要的是,男大量的测序数据的eaningful生物信息学分析。
在测序技术和指数数据积累的快速发展创造了计算平台,将允许研究人员访问,分析和理解这些信息的巨大需求。早期的系统主要依赖于计算机编程知识,反之,基因组浏览器,比如NCBI,允许非程序员访问和可视化数据不允许复杂的分析。基于Web的,开放的接入平台,银河( https://galaxyproject.org/ ),填补了这一空白,并证明是一个有价值的管道,使研究人员能够处理NGS数据并执行的频谱简单到复杂生物信息学分析。银河初步建立,并保持,由安东Nekrutenko(宾夕法尼亚州立大学)和詹姆斯·泰勒的实验室(约翰霍普金斯大学)F"> 3。银河提供广泛的计算任务使其成为一个‘一站式’为无数生物信息学的需要,包括所有参与的RNA测序研究的步骤。Itallows用户无论是在其服务器或执行数据处理在当地自己的机器上,数据和工作流可以被复制和共享。在线教程,帮助部分和wiki页面 ( https://wiki.galaxyproject.org/Support )专用于银河项目提供持续的支持。但是,对于初次使用的用户,尤其是那些没有生物信息学培训,该管道可以出现艰巨,自主学习和熟悉的过程可能会非常耗时。此外,生物系统研究,实验和方法的具体使用,冲击在几个步骤的分析决策,而这些可能很难没有指令导航。
整体RN A-SEQ银河工作流包括数据上传和质量检查,随后使用Tuxedo的套件4,5,6,7,8,9,这是用于RNA测序数据分析10的不同阶段所需的各种工具的集体分析, 11,12,13,14。典型的RNA测序实验由实验部分(样品制备,分离mRNA和cDNA文库制备)中,并NGS生物信息学分析数据。这些部分,以及涉及在银河管道中的步骤的概述,示于图1。
3fig1.jpg" />
图1:一个RNA测序工作流程的概述。参与的RNA测序实验,以比较两个蠕虫菌株(A和B,橙色和绿色线和箭头,分别地)的基因表达概况的实验和计算步骤图示。银河使用的不同的模块示于盒用在我们的协议以红色表示相应的步骤。各种操作的输出被写在灰色与以蓝色显示的文件格式。 请点击此处查看该图的放大版本。
{kind=link}
在炫套房的第一个工具是一个名为" 高顶礼帽 "的比对程序。它分解的NGS输入读取成较小的片段,然后将它们映射到参考基因组。这种两步处理确保读取跨越内含子区,其比对可以以其他方式二srupted或错过入账和映射。这增加了覆盖范围并促进新的剪接点的鉴定。 顶帽输出被报告为两个文件,一个文件BED(具有约剪接点,其包括基因组位置信息)和一个文件BAM(具有每个读映射的详细信息)。接着,BAM文件针对参照基因组对齐来估计使用于所谓的" 袖扣" Tuxedo的套件随后工具每个样品内的单个转录物的丰度。通过扫描对准来报告全长转录物的片段或"transfrags"跨越所有在为每一个基因的输入数据可能的剪接变体袖扣功能。在此基础上,它会产生一个对于每个样品被测序(每个基因产生的每一个基因的所有转录的组件)"转录"。然后,这些袖扣组件崩溃或再一起合并在一起ference基因组以产生用于使用下一个工具,"Cuffmerge"下游差分分析的单个注释文件。最后,由每个样品的顶帽的输出进行比较,以最终Cuffmerge输出文件( 图1) 的样品之间的"Cuffdiff"工具措施的差异基因表达。 袖扣使用FPKM / RPKM(片段/读取每百万映射成绩单的每千碱基读取)值报告成绩单丰度。这些值反映原始NGS数据的深度归一化和基因长度(基因具有不同的长度,所以计数必须被归一化了的基因,以比较电平的长度(平均数目从对齐到参考基因组中的样本读出)基因之间)。 FPKM和RPKM基本上是相同的与RPKM被用于单端RNA测序,每一个读出对应于一个单一片段,而,FPKM用于配对末端RNA测序,因为它占的事实,两次读取可以对应于相同的片段。最终,这些分析的结果是所测试的条件和/或株之间差异表达的基因的列表。
一旦成功银河运行完成并产生"基因名单",下一个合乎逻辑的步骤需要更多的生物信息学分析,从数据集推断出有意义的知识。许多软件包应运而生,以满足这方面的需求,其中包括公开可用的基于网络的计算包,如DAVID 15(用于注释,可视化和集成发现数据库)。 DAVID利于通过比较基因上传列表,其集成的生物知识库和揭示与基因列表相关的生物注释分配生物学意义,从高通量研究大基因列表。其次是富集分析, 即测试IDEntify如果任何生物过程或基因类是在以统计学显著方式基因列表(一个或多个)过表达。因为一个广泛的,集成的知识基础和强大的分析算法,使研究人员能够检测富含内生物主题的组合,已成为一个流行的选择基因组衍生的"基因列表" 10,16。其它优点包括其处理的任何测序平台和高度友好的用户界面创建基因列表的能力。
所述线虫是一种遗传模型系统中,公知为它的许多优点,例如尺寸小,透明体,简单的身体计划,易于培养的和巨大的顺从的遗传和分子清扫。蠕虫有一个小的,简单的和注释良好的基因组,其包括高达40%保守的基因与已知的人类同系物17。事实上, 线虫是第一个后生动物,其基因组被完全测序18,并且其中RNA测序被用于映射生物体的转录19,20中的第一物种之一。早期的蠕虫的研究涉及的实验用不同的方法进行高通量RNA捕获,文库制备和测序及生物信息学管道是该技术21,22的发展作出了贡献。近年来,在蠕虫基于RNA-Seq的,实验已经司空见惯。但是,传统的蠕虫生物学家通过RNA-Seq的数据的计算分析所带来的挑战仍然是技术的更大和更好地利用的一个主要障碍。
在本文中,我们描述了使用Galaxy平台分析来自秀丽隐杆线虫中产生高通量的RNA测序数据的协议。对于许多第一次和小SCA勒用户来说,最具有成本效益的和直接的方式进行的RNA测序实验是在实验室中分离RNA,并利用商用(或内部)NGS设施用于制备测序的cDNA文库和NGS本身。因此,我们已首次详细涉及隔离的步骤,C.量化和质量评估线虫 RNA样品用于RNA测序。接下来,我们为使用Galaxy接口,用于NGS数据的分析中,有用于测序后质量控制检查,随后对准,装配,和基因表达的定量差试验开始提供一步一步的指示。此外,我们已包括方向审议银河使用DAVID生物富集研究产生的基因列表。由于工作流中的最后的步骤中,我们上传到公共服务器,如序列读数存档(SRA)在NCBI(RNA-SEQ数据提供指令的http://瓦特ww.ncbi.nlm.nih.gov/sra),使其自由科学界访问。总体而言,我们预计,本文将蠕虫生物学家开展运行小样本首次RNA-Seq的实验以及频繁的用户提供全面的,充分的信息。
Access restricted. Please log in or start a trial to view this content.
研究方案
1. RNA分离
- 预防措施
- 向下擦拭整个工作表面,仪器和吸液管使用市售的RNA酶喷雾以消除任何RNA酶存在。
- 在任何时候都戴上手套,定期与在协议的不同步骤新鲜的改变它们。
- 仅使用过滤嘴,并保持所有样品在冰上尽可能避免RNA降解。
注:为了获得从NGS平台的最佳数据,它开始与高品质RNA的关键。 RNA分离和制备方法取决于样品来源,测序和研究者偏好方法而变化。几种市售试剂盒可用于该目的或RNA也可以使用RNA提取的标准的苯酚 - 氯仿法分离。无论使用哪种方法,上面列出的预防措施应遵循的整个过程,以尽量减少污染和OBTAIN原始RNA样品。
- 收获蠕虫
- 通过次氯酸盐漂白处理23同步蜗杆人口以获得每应变1,000-1,500年龄匹配的秀丽隐杆线虫的成虫。
- 在325×g下洗蠕虫关闭使用M9缓冲溶液和旋转板在台式离心机中30秒。吸出M9的缓冲区蠕虫留下的小球落后。重复此步骤至少三次,以消除细菌交叉污染。
- 到蜗杆沉淀,添加〜500μL裂解缓冲液(如果使用商业试剂盒)或Trizol试剂(苯酚和异硫氰酸胍的单相溶液;如果苯酚:在1.3.3中描述氯仿提取进行)以破坏蠕虫组织,停用RNA酶和稳定核酸。
注:该协议可以在这里通过快速冷冻在液氮中,随后储存在-80℃下将样品被暂停。
- RNA分离
- 声处理蠕虫样品在的20秒的周期的45%的振幅。 'ON' 和40秒。 'OFF'(每株8-12个循环)。保持冰样本在任何时候。
注:确保超声仪探头沉浸在缓冲区,并保持在整个恒定的水平。避免样品的泡沫剂和在两者之间彻底清洁样品探头。超声处理周期可以根据所使用超声波仪的类型而改变。建议在超声处理条件上的测试样品在开始实验之前,首先进行了优化。 - 如果使用市售的试剂盒,继续进行RNA分离按照规定的协议。对于使用苯酚 - 氯仿法分离RNA,执行以下步骤。
- 离心机超声处理的样品在16000×g离心10分钟。在4℃下
- 转移上清液到1.5mL无RNase的微量离心管中,加入氯仿100μL(1/5 个 RNA / DNA分离试剂的体积)。
警告:氯仿是有毒的。为了尽量减少接触和吸入避免,处理这种物质时,工作在化学罩。 - 彻底涡样品30 - 60秒。并让样品坐在室温下搅拌3分钟。
- 离心机中以11750×g离心15分钟。在4℃下。仅传送上面的水层到一个新的不含RNA酶的微量离心管中,小心不要吸出含有DNA的白色接口。重复步骤1.3.4至1.3.6。
- 添加250μL的2-丙醇(水相或1/2 RNA / DNA分离试剂体积的70%)和反转管混合。让管坐在室温下10分钟或在-80℃下离开过夜。
- 离心机样品在11750×g离心10分钟。在4℃下。非常小心地倒出上清液,留下了几μL在管的底部,以便沉淀不被打扰。
- 用500μl的75%乙醇(用无RNase水配制)洗涤沉淀并离心在16000×g离心5分钟。一个吨4℃。
- 在不干扰沉淀除去尽可能多的上清液越好。空气在罩中干燥沉淀几分钟。
- 添加无RNase水的30μL,并帮助溶解通过加热RNA沉淀10分钟。在60℃下。
- 检查使用生物分析仪RNA的质量和数量。
注:生物分析仪产生的R NA 我 ntegrityÑ棕土(RIN)的RNA的质量的度量。的至少8的RIN为RNA测序样品推荐的阈值(越高越好)。 RNA的数量和质量,也可以通过分光光度计检查,但也应遵循RNA完整性的视觉评估。要做到这一点,运行在1.2%琼脂糖凝胶样品足够长的时间,以获得28S和18S的核糖体RNA条带的合适的分离。两个不同的频带(1.75 kb的18S为rRNA和3.5kb的在秀丽隐杆线虫的情况下28S rRNA的)的存在是RNA可接受的质量度量。 - 使用〜100毫微克/μLRNA施p和供应商/ NGS设施用于制备测序文库。
注:RNA样品应在干冰上测序服务供应商发货。大多数提供商进行文库制备之前独立的RNA质量控制测试。
- 声处理蠕虫样品在的20秒的周期的45%的振幅。 'ON' 和40秒。 'OFF'(每株8-12个循环)。保持冰样本在任何时候。
2. RNA测序数据分析
- 原始测序数据下载
- 下载在从利用" 文件传输协议"(FTP)的NGS提供商fastq.gz格式编码的压缩的粗的fastq测序数据。

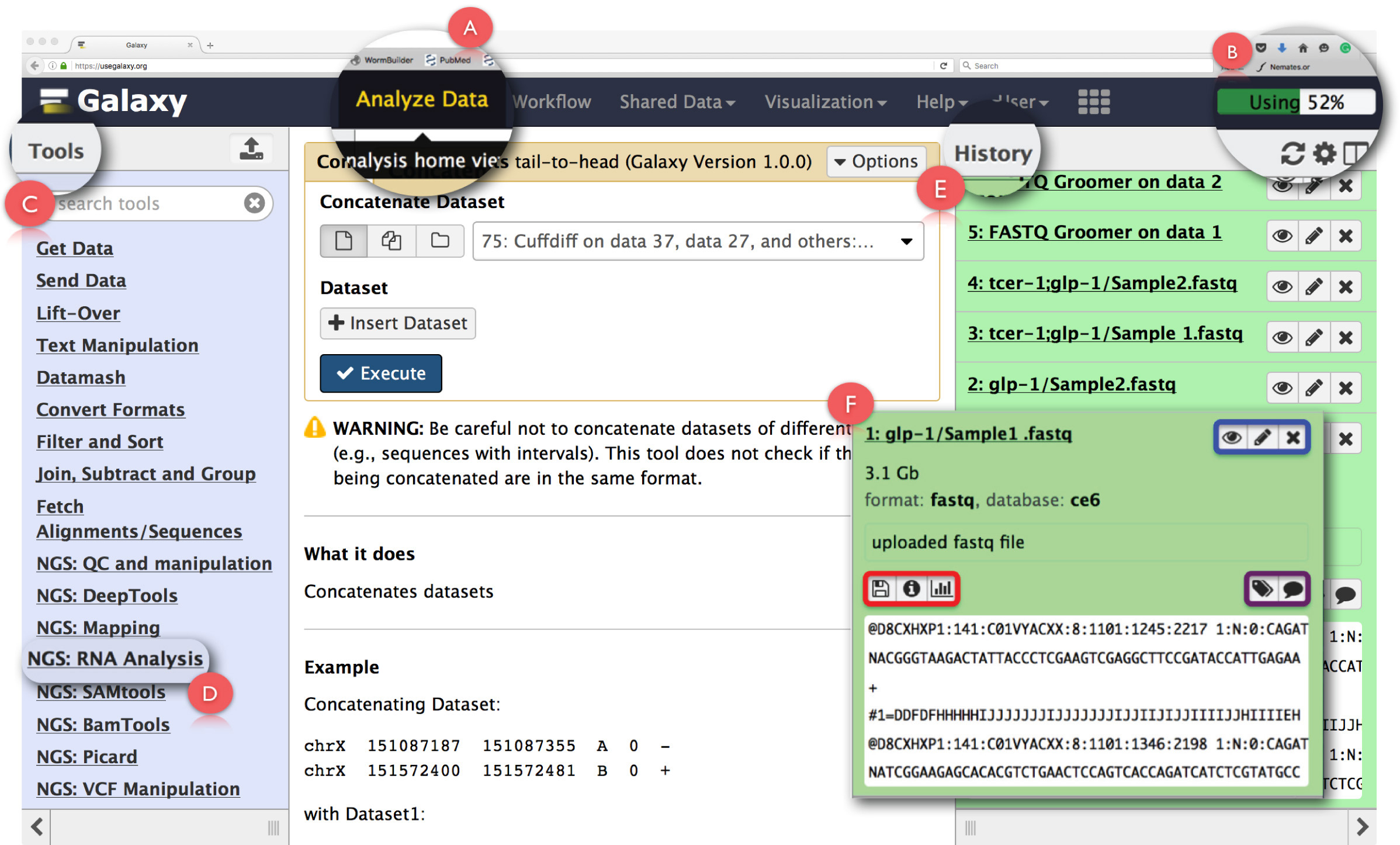
图2:银河用户界面面板和密钥RNA测序函数的布局。页面的主要特点是扩大和突出。 (A)突出用于访问在网页标题" 分析数据 "功能分析首页查看。 (B)是" 进度条 ",指示由该操作使用银河服务器上的空间。 (C)是" 工具条",其中列出所有可以银河界面上运行的工具。 (D)示出了"NGS:RNA分析"用于RNA测序分析工具部分。 (E)描绘了" 历史记录"面板列出了使用银河生成的所有文件。 (F)显示,在历史部分中的任何文件时,点击打开的对话框的例子。在(F),蓝框突出,可用于查看图标,editthe属性或删除数据集,紫色方块强调了可用于"编辑"数据集标记或批注图标,并且,红色框指示图标下载数据, 查看任务的细节进行或重新运行操作。 请点击此处查看该图的放大版本。
{kind=link}
- 入门银河
注:银河可以使用基于Web的平台,提供云服务和免费存储空间有限免费的公共服务器上运行。它也可以被下载到本地用户的机器或机构,但本地处理托管计算集群上运行,可以通过数据存储限制和用户机器的处理能力限制的约束。在下载和安装详情可访问https://wiki.galaxyproject.org/Admin/GetGalaxy 。在这个协议中,我们描述银河管道的基于网络的使用情况。- 在用户的机器上,获得银河下载和存储数据NGS后laxy.org/"目标= "_blank"> https://usegalaxy.org/。
- 在该页面,登录的标题点击"用户"注册一个用户帐户,并通过与用户界面面板结识开始。
注:建议第一次使用的用户提供的主页上的"从这里开始"的教程,以获得与银河的基本设置熟悉( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) 。 - 在头面板点击"数据分析"( 图2A)访问"分析首页查看"这也是对银河的启动画面。
注意:标题还设有其他环节,其方式,在他们将鼠标指针悬停可以看到。头的上右手角部具有监视的任务( 图2B)利用空间的进度条。 - C舔"NGS:RNA分析 "在左边的面板( 图2C)上的"工具菜单"任务能够访问所有RNA-seq的数据分析所需的工具。
注:"工具菜单"编目银河提供的所有操作。该菜单是基于任务,点击任何一个将开辟以完成这项任务所需的所有工具的列表分裂。 - 通过点击齿轮图标右侧( 图2E)的" 历史记录 "面板的顶部创建新的分析历史记录。选择" 新建 ",从弹出菜单中选择。给这个" 历史 "一个合适的名称来识别分析。
注:"历史记录"面板显示上传的分析,以及由银河上运行的任务所产生的所有输出文件中的所有文件。点击此面板中的文件名打开了有关执行的任务的详细信息的对话框和数据集( 图2F)的一个片段。在此框中的图标使用户能够" 视图 "," 编辑属性 "或" 删除 "数据集( 图2F,以蓝色突出显示)。此外,用户还可以" 编辑 "数据集标记或批注( 图2F,突出表现在紫色)," 下载 "的数据,该任务的" 查看详情 "," 重播 "的任务,甚至" 可视化 ",从这个数据集对话框( 图2F,以红色突出显示)。 - 点击"ToolsMenu"下" 获取数据"的" 上传文件"功能上传原始的fastq文件。
注:点击此或任何其他工具打开了操作的简短描述,测试本身,在中间"分析接口"面板。此面板鞋带在一起从左侧面板和"输入文件"从右边" 历史记录 "面板( 图2E)" 工具"。在这里,从" 历史记录 "输入文件选择和确定运行给定的任务的其它参数。从每一个测试所得到的输出数据集在" 历史记录 "救回来。包含在" 分析界面 "面板中的测试是所有可与所有的工具生成的输出文件的详细列表跑着给定的工具的参数说明。 - 任务在"分析界面"打开后,点击" 选择本地文件 "或" 选择FTP文件"(更快的上传),浏览到包含测序文件的文件夹,并选择适当的数据集上传。
- 允许银河" 自动检测 "上传的文件类型(默认设置)。选择" 埃尔C.在基因组中的下拉菜单egans"。
- 点击" 开始 "启动数据上传。一旦文件被上传后,它将被保存在" 历史记录 "面板,并可以从那里访问。
- 如果多个测序数据文件被用于单个样品产生,使用" 串联 "工具将它们结合起来。要做到这一点,打开在"工具菜单"中的" 文本处理"选项。
- 点击"串联"工具,选择需要从下拉框" 分析界面"的中间组合,然后单击" 执行"的文件。
注:使用该任务产生的输出文件的格式的fastq产生。映射程序具有每FASTQ文件,并在达到该上限时的剩余序列生成新的fastq文件16000000组的序列的限制。在';需要在这样的情况下串联"工具将数据集结合起来。 - 通过使用下找到了" 的fastq美容师"工具上传FASTQ格式的文件转换为银河RNA-Seq的分析所需要的fastqsanger格式"NGS:QC和操作 "部分(见补充文件)。
- 选择下适当的fastq数据集" 文件来新郎"选项,并使用默认参数运行该工具。
注:使用该任务产生的输出文件的格式fastqsanger产生。
- fastqsanger数据质量控制测试
- 检查上传fastqsanger质量使用"FastQC"工具位于下写着:在"工具"菜单"NGS QC和操纵 "。
- 选择从' 绍尔下拉菜单中梳理fastqsanger数据文件牛逼从当前库"中读取数据,并使用默认参数运行该工具。
注:要特别注意的读取质量和存在任何适配器序列。适配器通常除去作为由NGS提供商后RNA测序数据处理的一部分,但在一些情况下,可能会留下。对于质量标准的解释去http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ 。 - 请与NGS提供商和适配器是否存在,他们使用从该" 剪辑"工具修剪"NGS:QC和操纵"任务菜单。
注:使用该任务产生的输出文件的原始txt格式,以及在可以在任何Web浏览器中打开HTML生成。
- 与炫Suite数据分析
- 高顶礼帽
- 下载最新版本的℃。线虫参照基因组FASTA和如上文2.2.6所述的GTF(基因转移格式)从上传文件的文件"。
- 打开"NGS:RNA分析"部分,然后点击" 顶礼帽"工具绘制的测序读取到下载的参考基因组。
- 从下拉菜单中的问题的合适的答案"这是单端或末端配对数据?
- 选择适当的fastq文件。
- 选择在接下来的下拉菜单中选择"从历史使用基因组",并选择在步骤2.4.1.1下载参考基因组。
- 选择"默认"为其他参数并单击" 执行"。
注意:在使用该任务产生的输出文件中," 接受点击 "文件用于后续步骤。
- 袖扣和Cuffmerge
- 选择"袖口链接NGS在工具‘:RNA分析’部分来组装转录,估计其对差异表达的丰度和测试。
- 在第一个下拉菜单中,选择映射" 接受命中(BAM格式)"文件从顶礼帽分析获得。
- 在第二个下拉菜单中,设置参考注释在步骤2.4.1.1下载的GTF文件。
- 选择"是"用于" 执行偏差校正"选项,并使用默认设置所有其他参数运行任务。
注意:在使用该任务产生的输出文件中," 接受成绩单"文件用于后续步骤。 - 打开"Cuffmerge"的工具"NGS:RNA分析"合并" 组装成绩单 "生产的所有RNA-Seq的样本。
注:在工具自填充和列出前框中的所有袖扣产生M> GTF文件。 - 选择对于测试的所有菌株/条件,包括相同的应变/条件的生物学重复(参见生物学重复的讨论)的"装配转录"文件。
- 选择"是"的" 使用参考注释",并选择在步骤2.4.1.1下载的GTF文件。
- 在下面的框中,再次选择"是"的" 使用塞克斯·达塔 "选项,并选择在步骤2.4.1.1下载的全基因组FASTA文件。
- 保持其他参数为默认值,单击"执行"。
注:Cuffmerge生成单个GTF输出文件。
- Cuffdiff
- 导航到了"Cuffdiff"工具"NGS:RNA分析"部分。在" 成绩单"菜单,选择Cuffmerge合并后的输出文件。
- 标签条件1和2与两种菌株/条件的名称。
注:Cuffdiff可以多于两个的菌株或条件以及时间过程实验之间进行比较。只需使用" 添加新条件"选项添加的每个新品种/条件,根据需要。 - 对于每个菌株/条件,下" 重复测试"中选择个体从顶帽对应于的菌株/条件的不同生物学重复" 接受命中"的输出文件。按住"CMD"键,如果使用的是Macintosh计算机,和"Ctrl"键,如果使用的是PC,可以选择多个文件。
- 保留所有其他选项为默认参数。点击"执行"运行任务。
注:Cuffdiff以表格的形式作为转录组测序分析的最终读数产生大量的输出文件。这些包括成绩单,基因FPKM跟踪文件(合并转录物分享的基因同一性)的初级转录物序列和编码序列的FPKM值。生成的所有的数据文件可以在任何电子表格应用程序中查看和含有类似的属性,如基因名称,基因座中,倍数变化(在LOG2刻度)以及关于菌株/条件之间进行比较,包括σp值和Q值的统计数据。这些文件中的数据可基于差异的统计学显着性或基因表达的倍数变化(大小和变化的方向,如在上调或下调的基因)进行分类和操纵具体根据用户的要求。如果需要不同的基因标识符之间的转换( 例如 ,Wormbase基因ID 与粘粒号),关于Biomart(可用工具http://www.biomart.org/ )可被利用。
- 高顶礼帽
3.基因本体论(GO)项分析使用DAVID
- 从网站h出入DAVID载荷大小://david.ncifcrf.gov/。在网页的标题,点击"开始分析"。在"步骤1"时,复制和粘贴从所获得的Galaxy,在箱型A.基因名单中"步骤2",选择"Wormbase基因ID"作为标识符用于输入基因。
注:DAVID识别大多数公众可获得的注释类别,因此其他基因标识符(如Entrez基因ID或基因符号)也可以使用。 - 在"步骤3",选择(将要分析的基因)在"列表类型""基因列表",然后点击"提交列表"图标。
注:" 分析向导",将打开列出所有可以上传基因名单上运行的超级链接DAVID工具( 图3)。点击这些链接来访问相关的相应模块按用户的要求。要确定适当的给定任务的工具,点击" 大卫工具使用? "上链接了" ; 分析向导"页面。点击"开始分析"的标题链接的分析过程中,在任何时候返回到" 分析向导"主页。

图3:DAVID 分析向导页面和操作的输出的实例的布局。该" 分析向导 " Web用户界面列出了用于分析基于各种参数富集上传基因列表的工具。在一个新的网页点击这些工具报告分析的数据。的从" 基因功能分类 "," 功能注释图表 "和" 功能注释聚类"所产生的表格报告实例被示出为插图(箭头)。>点击此处查看该图的放大版本。
- 功能注释工具1:功能注释聚类
- 点击" 功能注释聚类 "模块去摘要页面。保留默认的注释类别,点击" 功能注释聚类 "产生通过富集成绩名列同类注释方面的集群。
- 点击每一项的超链接名称,以了解它,"RT"(相关术语)列出相关类别其他类似的条款细节。
- 点击紫色栏上列出与一个术语,红色的"G",列出与集群内的所有方面相关联的所有基因相关的基因。
- 点击绿色图标查看集群中所有的基因和术语的二维视图。
注意:最后三个栏列出每分析和统计结果术语。这个结果和所有其他分析可以在.txt格式通过点击"下载文件"链接下载。
- 功能注释工具2:功能注释图
- 返回到汇总页面,点击" 功能注释图",以确定该基因列表相关显著过表达的生物学术语( 如转录因子活性或激酶活性)。
- 点击项名称获取更详细的信息和"RT"(相关术语)列出其他相关条款。
- 点击紫色栏上列出相应的个别类别的所有相关的基因。
注:最后两个栏列出了统计检验结果为每个类别。
- 功能注释工具3:功能注释表
- 返回到汇总页面,点击" 函数的最终注解表",查看与名单上的基因没有任何统计计算相关联的所有注释的列表。
注:此工具可以用于基因 - 基因列表的分析是有用的还是要看具体的,非常有趣的基因。
- 返回到汇总页面,点击" 函数的最终注解表",查看与名单上的基因没有任何统计计算相关联的所有注释的列表。
- 基因功能分类工具
- 返回到" 分析向导",然后点击" 基因功能分类 "模块来隔离输入基因列表为排名按他们的"富集"分数,在列表中的基因组的整体富集的量度基因的功能有关的团体。
- 点击项名称获取更详细的信息和"RG"揭示基因组功能相关基因
- 点击红色的"T"(项报告)列出相关的生物学和绿色图标看到所有的基因和术语的二维视图。
- 基因名批量浏览器
- 返回到" 分析向导",然后点击" 基因名称批次浏览器"到"Wormbase基因标识"翻译成他们相应的基因名称。 (WBGene00022855 = 于tCER-1)。
- 点击基因名称,以便获得更具体的基因信息。
- 点击"RG"(相关的基因)的链接旁边的每个基因揭示基因预测功能性相关感兴趣的基因。
4.上传原始数据到NCBI序列读数存档(SRA)
- 进入登录时的SRA网页中NCBI"链接或注册一个新账户。
- 点击"Bioproject"。
- 点击" 提交"左侧的" 使用Bioproject"标题下。
- 选择选项" 新投稿"。在提交的更新细节。通过剩余的七个选项卡继续,在实验和数据的细节填充被上传。点击" 提交"完成时。
注:在第五个" 生物标本"选项卡中,保留" 生物样本"空的插槽。 - 通过点击"我提交"链接刷新结果页面。提交的数据将与分配的数提交,简要说明和上传状态上市。
- 点击"生物样本"在此页面的顶部,在"开始新的提交",然后创建一个"新的提交"。提交单独提交每个样品。
- 如在4.4与"Bioproject"的情况下,更新所述提交者的细节,并继续通过凸片在每个标签的细节填充的其余部分。一旦完成审查,并点击" 提交"。
- 导航到HTTP://www.ncbi.nlm.nih.goV / SRA创建最终的"序列读取存档(SRA)"提交。
- 点击"登录SRA"下的"入门"。
- 在接下来的页面点击"NCBI PDA"链接。一个"更新设置"链接将打开。填写表格,然后点击"保存设置"。
- 在结果页面中,点击"创建新提交"链接。在" 别名"输入一个合适的名称,并点击"保存"。与提交ID和其他详细信息的表格将被创建。
- 点击"新实验"和每个"生物样本"至少注册一个独特的测序文库。
- 指定和链接先前创建的"BioProject"和"生物样本"提交的ID。 "新实验"将被创建。
- 点击"新润"在页面的底部之后SRA实验已经取得并确定需要链接到它的数据文件。
- 计算每个数据文件的MD5总和。要做到这一点的是Macintosh终端上,导航到应用程序/实用/终端 。在终端,在"MD5"(没有引号)类型后跟一个空格。拖放需要从取景器上传到终端,然后点击"确认"的文件。
- 终端将返回一个字母MD5总和。输入此作为文件上传提交过程的一部分。使用系统提供的用户名和密码才能使用FTP上传文件。
Access restricted. Please log in or start a trial to view this content.
结果
在秀丽隐杆线虫 ,消去生殖系干细胞(GSC中)延长寿命,提高的应力复原力,和提升体脂肪24,28。 GSC中的损失,或者通过激光烧蚀或通过突变如GLP-1引起,导致寿命延长通过转录的网络因子29活化。一个这样的因素,TCER-1,编码人转录延伸和剪接因子,TCERG1 30的蜗杆同源物...
Access restricted. Please log in or start a trial to view this content.
讨论
在现代生物学银河测序平台的意义
银河项目已成为帮助生物学家没有生物信息学培训,以处理和快速,高效地分析高通量测序数据的工具。曾经被视为一个艰巨的任务,这个公开可用的平台,取得了运行复杂的生物信息学算法来分析NGS数据的简单,可靠,简单的过程。除了托管范围广泛的生物信息学工具,成功的关键银河也是它的用户界面的该鞋带复杂测序分析的各个方面一起...
Access restricted. Please log in or start a trial to view this content.
披露声明
作者什么都没有透露。
致谢
作者想表达自己的感激之情谁开发银河和DAVID,从而取得NGS科学界广泛接受的实验室,团体和个人。在我们的生物信息学的培训,在匹兹堡大学的同事提供的帮助和建议是公认的。这项工作是由一个埃里森医学基金会新学者在老龄化奖(AG-NS-0879-12)和美国国立卫生研究院(R01AG051659),以股份公司的资助。
Access restricted. Please log in or start a trial to view this content.
材料
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
参考文献
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22(2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085(2011).
- Consortium, C. eS. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383(2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, Web Server issue W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639(2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788(2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158(2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Access restricted. Please log in or start a trial to view this content.
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。