Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Transkriptom-Analyse von
In diesem Artikel
Zusammenfassung
Galaxy und DAVID haben als beliebte Werkzeuge entstanden, die Ermittler ohne Bioinformatik Ausbildung ermöglichen RNA-Seq Daten zu analysieren und zu interpretieren. Wir beschreiben ein Protokoll für C. elegans Forscher RNA-Seq Versuche, den Zugang und verarbeiten die Daten - Set mit Galaxy und erhalten aussagekräftige biologische Informationen aus den Gen - Listen mit DAVID auszuführen.
Zusammenfassung
Next Generation Sequencing (NGS) Technologien haben die Art der biologischen Untersuchung revolutioniert. Von diesen hat RNA Sequencing (RNA-Seq) als ein leistungsfähiges Werkzeug für die Gen-Expressionsanalyse und Kartierung Transkriptom entstanden. Allerdings erfordert ausgefeilte Rechenkompetenz RNA-Seq Datensätze Handhabung und inhärente Herausforderungen für Biologie Forscher darstellt. Dieser Engpass wurde von dem Open-Access-Galaxy-Projekt gemildert, die RNA-Seq Daten zu analysieren, und die Datenbank für die Annotation, Visualisierung und integrierte Entdeckung (DAVID), ein Gen Ontology (GO) Zeitanalyse-Suite Benutzer ohne Bioinformatik Fähigkeiten ermöglicht, das hilft biologische Bedeutung von großen Datenmengen abzuleiten. Doch für Erstanwender und Bioinformatik Amateure, selbstlernend und Einarbeitung in diesen Plattformen können zeitaufwendig und entmutigend sein. Wir beschreiben einen einfachen Workflow, den C. elegans Forscher RNA zu isolieren Wurm helfen, führen ein RNA-Seq - Experimentund analysiert die Daten mit Galaxy und DAVID-Plattformen. Dieses Protokoll stellt schrittweise Anweisungen für die Verwendung der verschiedenen Galaxy Modulen für den Zugriff auf rohe NGS Daten, Qualitätskontrollen, Ausrichtung und Differentialgenexpressionsanalyse, Führen des Benutzers mit Parametern bei jedem Schritt ein Gen-Liste zu generieren, die für die Anreicherung gescreent werden können von Genklassen oder biologische Prozesse unter Verwendung von DAVID. Insgesamt erwarten wir , dass diese Artikel Informationen zur Verfügung stellen werden , um C. elegans Forscher RNA-Seq Experimente zum ersten Mal sowie häufige Benutzer eine kleine Anzahl von Proben Unternehmen ausgeführt wird .
Einleitung
Die erste Sequenzierung des menschlichen Genoms führte Fred Sanger-Didesoxynucleotid-Sequenzierungsverfahren verwendet wird , dauerte 10 Jahre und kostete schätzungsweise US $ 1 3000000000, 2. Doch in etwas mehr als ein Jahrzehnt seit seiner Gründung, Next-Generation Sequencing (NGS) Technologie hat es ermöglicht, das gesamte menschliche Genom innerhalb von zwei Wochen sequenzieren und für US $ 1.000. Neue NGS Instrumente, die ständig steigenden Geschwindigkeiten von Sequenzierungs-Datenerfassung mit unglaublicher Effizienz, zusammen mit starken Kostensenkungen ermöglichen, revolutionieren die moderne Biologie in unvorstellbarer Weise wie Genomsequenzierungsprojekte alltäglich schnell geworden sind. in vielen anderen Bereichen, wie zum Beispiel Gen-Expressionsanalyse durch RNA-Sequenzierung (RNA-Seq), Studium der genomweiten epigenetische Modifikationen, DNA-Protein-Wechselwirkungen, und Screening auf mikrobielle Diversität in menschlichen Wirten Zusätzlich haben diese Entwicklungen Fortschritt galvanisiert. NGS-basierte RNA-Sieq insbesondere hat es möglich gemacht zu identifizieren und Karte Transkriptomen umfassend mit Genauigkeit und Empfindlichkeit und hat Microarray-Technologie als Methode der Wahl für die Expressionsprofile ersetzt. Während der Microarray-Technologie extensiv verwendet wurde, wird es durch seine Abhängigkeit von vorbestehenden Arrays mit bekannten genomischen Informationen und anderen Nachteilen, wie eine Kreuzhybridisierung und eingeschränkten Bereich von Expressionsänderungen begrenzt, die zuverlässig gemessen werden können. RNA-Seq, auf der anderen Seite, kann sowohl bekannte als auch unbekannte Transkripte zu erfassen, verwendet werden, während niedriges Hintergrundrauschen zu seiner eindeutigen DNA-Mapping Natur aufgrund erzeugen. RNA-Seq, zusammen mit den zahlreichen genetischen Werkzeuge von Modellorganismen wie Hefe angeboten, Fliegen, Würmer, Fische und Mäuse, hat für viele wichtige neue biomedizinische Entdeckungen als Grundlage diente. Allerdings erhebliche Herausforderungen bleiben, die NGS unzugänglich für die breitere wissenschaftliche Gemeinschaft, einschließlich Beschränkungen der Speicherung, Verarbeitung und vor allem, m eaningful bioinformatische Analyse großer Mengen von Sequenzierungsdaten.
Die schnellen Fortschritte in der Sequenzierungstechnologien und exponentielle Datenakkumulation haben einen großen Bedarf an Rechenplattformen geschaffen, die Forscher ermöglicht den Zugriff auf, analysieren und diese Informationen zu verstehen. Frühe Systeme waren stark abhängig von Computer-Programmierkenntnisse, während Genom-Browser wie NCBI, die Nicht-Programmierer erlaubt den Zugriff und Visualisierung von Daten nicht anspruchsvolle Analysen ermöglichen. Die webbasierte Open-Access - Plattform, Galaxy ( https://galaxyproject.org/ ), hat diese Lücke gefüllt und sich als eine wertvolle Pipeline sein , die Forscher ermöglicht NGS - Daten zu verarbeiten und ein Spektrum von einfach zu komplex durchführen Bioinformatik analysiert. Galaxy wurde ursprünglich gegründet, und aufrecht erhalten wird, von den Labors von Anton Nekrutenko (Penn State University) und James Taylor (Johns Hopkins University)f "> 3. Das Galaxy bietet eine breite Palette von Rechenaufgaben es sich um eine‚one-stop - shop‘für unzählige Bioinformatik Bedürfnisse zu machen, einschließlich aller Schritte , die bei einer RNA-Seq - Studie. Itallows Benutzer Datenverarbeitung auszuführen entweder auf ihren Servern oder lokal auf ihre eigenen Maschinen. Daten und Workflows können wiedergegeben und gemeinsam genutzt werden. Online - Tutorials, Hilfebereich und eine Wiki-Seite ( https://wiki.galaxyproject.org/Support ) an das Galaxy - Projekt konsequent unterstützt gewidmet ist . Allerdings für Erstanwender, vor allem diejenigen, die keine Bioinformatik Ausbildung kann die Pipeline entmutigend erscheinen und der Prozess der Selbstlern und Einarbeitung kann Darüber hinaus untersucht das biologische System, und die Besonderheiten des Experiments und Methoden verwendet, Schlagzeitaufwendig. sein die analytischen Entscheidungen in mehreren Schritten, und diese können schwierig sein, ohne Anleitung zu navigieren.
Die Gesamt RN A-Seq Galaxy Arbeitsablauf besteht aus Daten - Upload und Qualitätskontrolle durch Analyse folgte die Tuxedo Suite 4, 5, 6, 7, 8, 9, mit dem ein Kollektiv von verschiedenen Werkzeugen für verschiedene Stufen von 10 RNA-Seq Datenanalyse erforderlich ist , 11, 12, 13, 14. Ein typisches RNA-Seq Experiment besteht aus dem experimentellen Teil (Probenvorbereitung, mRNA-Isolierung und cDNA-Bibliothek Vorbereitung), die NGS und die Bioinformatics Datenanalyse. Eine Übersicht über diese Abschnitte, und die in der Pipeline Galaxy beteiligten Schritte sind in Abbildung 1 dargestellt.
3fig1.jpg“/>
Abbildung 1: Übersicht über einen RNA-Seq - Workflow. Darstellung der Versuchs- und Berechnungsschritte in einem RNA-Seq Experiment involvierten die Gen-Expressionsprofile von zwei Schnecken Stämmen zu vergleichen (A und B, orange und grüne Linien und Pfeile bezeichnet). Die verschiedenen Module von Galaxy verwendet werden in Kisten mit dem entsprechenden Schritt in unserem Protokoll in rot angezeigt gezeigt. Die Ausgänge der verschiedenen Operationen sind in grau mit den Dateiformaten in blau angezeigt geschrieben. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
Das erste Werkzeug in der Tuxedo - Suite ist ein Ausrichtungsprogramm ‚Tophat‘ genannt. Es bricht die NGS Eingabe liest in kleinere Fragmente nach unten und dann ordnet sie einen Referenzgenom. Dieses zweistufige Verfahren gewährleistet, dass liest intronischen Regionen überspannen, deren Ausrichtung kann anders sein, disrupted oder verpassten werden berücksichtigt und abgebildet. Dies erhöht die Reichweite und erleichtert die Identifizierung von neuen Spleißstellen. Tophat Ausgang als zwei Dateien, eine BETT - Datei (mit Informationen über Spleißstellen , die genomische Lage sind) berichtet und eine BAM - Datei (mit Mapping Details jeder lesen). Als nächstes wird die BAM - Datei gegen einen Referenzgenom ausgerichtet das nachfolgende Werkzeug in der Tuxedo - Suite die Fülle der einzelnen Transkripte innerhalb jeder Probe zu schätzen ‚Manschettenknöpfe‘ genannt werden. Manschettenknöpfe Funktionen durch die Ausrichtung Abtastung in voller Länge Transkript Fragmente oder ‚transfrags‘ zu berichten , die für jedes Gen , alle möglichen Spleißvarianten in den Eingangsdaten überspannt. Auf dieser Grundlage erzeugt er einen ‚Transkriptom‘ (Montag aller Transkripte erzeugen pro Gen für jedes Gen) für jede Probe sequenziert werden. Diese Manschettenknöpfe Anordnungen werden dann kollabiert oder zusammengeführt werden zusammen mit dem Wiederferenz Genom eine einzige Anmerkungsdatei für nachgeschaltete Differentialanalyse mit dem nächsten Werkzeug ‚Cuffmerge‘ zu erzeugen. Schließlich wird der Ausdruck ‚Cuffdiff‘ Werkzeug Maßnahmen Differential Gene zwischen den Proben durch die TopHat Ausgänge von jedem der Proben auf die endgültige Cuffmerge Ausgabedatei zu vergleichen (Abbildung 1). Manschettenknöpfe verwendet FPKM / RPKM (Fragmente / Reads pro Kilobasen von Transkript pro Million abgebildet liest) Werte Transkript Abundanzen zu melden. Diese Werte spiegeln die Normalisierung der Ausgangs NGS Daten für die Tiefe (durchschnittliche Anzahl der von einer Probe liest, die mit dem Bezugsgenom auszurichten) und Gen-Länge (Gene haben unterschiedliche Längen, so Zählungen haben für Länge eines Gens zu normalisierenden Ebenen zu vergleichen, zwischen Genen). FPKM RPKM und ist im Wesentlichen gleich mit RPKM für Single-End-RNA-Seq verwendet wird, wo jedes Lese zu einem einzelnen Fragmente entspricht, während, wird verwendet für FPKMPaired-End-RNA-Seq, da sie die Tatsache berücksichtigt, daß zwei Lesevorgänge auf das gleiche Fragment entsprechen kann. Letztlich ist das Ergebnis dieser Analyse eine Liste von Genen differentiell zwischen den Bedingungen exprimiert und / oder getesteten Stämme.
Sobald ein erfolgreicher Galaxy Lauf beendet ist und eine ‚Gen-Liste‘ erzeugt wird, erfordert der nächste logische Schritt mehr Bioinformatik analysiert aus den Datensatz sinnvoll Wissen abzuleiten. Viele Software - Pakete sind entstanden auf diesen Bedarf gerecht zu werden, einschließlich der öffentlich zugänglichen Web-basierten Rechenpakete wie DAVID (der Datenbank für Annotation, Visualisierung und integrierten Discovery) 15. DAVID erleichtert, indem die hochgeladen Genliste seiner integrierten biologischen Wissensdatenbank und enthüllt die biologischen Anmerkungen im Zusammenhang mit der Genliste biologische Bedeutung zu großen Genlisten von Hochdurchsatz-Studien zuweisen. Dies wird durch Anreicherung Analyse gefolgt, dh Tests identify wenn jedes biologische Prozess oder Gen-Klasse in der Gen-Liste (n) in einer statistisch signifikanten Weise überrepräsentiert ist. Es ist eine beliebte Wahl, weil aus einer Kombination aus einem breiten, integrierten Wissensbasis und leistungsstarke analytische Algorithmen , die den Forschern ermöglichen , biologische Themen innerhalb der Genomik-derived ‚Genlisten‘ 10, 16 angereichert zu erkennen. Weitere Vorteile sind seine Fähigkeit, Genlisten erstellt auf jeder Sequenzierungsplattform und eine sehr benutzerfreundlichen Oberfläche zu verarbeiten.
Der Nematode Caenorhabditis elegans ist ein genetisches Modellsystem, das für seine viele Vorteile, wie geringe Größe, transparente Körper, einfachen Körperplan, einfache Kultur und große amenability zu genetischer und molekularen Präparation bekannt. Würmer haben eine kleine, einfache und gut kommentierten Genom , die mit bekannten humanen Homologe 17 bis 40% konservierte Gene beinhaltet bis. Tatsächlich C. eleganswar das erste Metazoen , dessen Genom sequenziert wurde 18 vollständig, und einer von der ersten Spezies in dem RNA-Seq verwendet wurde , 20 eines Organismus Transkriptom 19, abzubilden. Frühe Wurm Studien Experimentieren mit verschiedenen Methoden für die Hochdurchsatz - RNA - Capture, Bibliothek Vorbereitung und Sequenzierung sowie Bioinformatik - Pipelines beteiligt, die zur Weiterentwicklung der Technologie 21, 22 beigetragen. In den letzten Jahren hat sich RNA-Seq-basierten Experimenten in Würmern alltäglich geworden. Aber für traditionelle Wurm Biologen die durch Computeranalyse von RNA-Seq Daten Herausforderungen wie vor ein großes Hindernis für eine größere und bessere Ausnutzung der Technik.
In diesem Artikel beschreiben wir ein Protokoll der Galaxy - Plattform für den Einsatz von Hochdurchsatz - RNA-Seq Daten von C. elegans erzeugt zu analysieren. Für viele erstmaligen und Klein scale-Nutzer, die kosteneffiziente und einfache Art und Weise einen RNA-Seq Versuch zu unternehmen ist RNA im Labor zu isolieren und eine kommerzielle (oder in-house) NGS-Anlage zur Herstellung von Sequenzierung von cDNA-Bibliotheken und dem NGS selbst zu nutzen. Daher haben wir zuerst die Schritte in Isolierung, Quantifizierung und Qualitätsbewertung von C. elegans - RNA - Proben für die RNA-Seq beteiligt detailliert beschrieben. Weiter stellen wir die Galaxy-Schnittstelle für Analysen der NGS Daten Schritt-für-Schritt-Anweisungen für die Verwendung mit Tests für die post-Sequenzierungsqualitätskontrollprüfungen gefolgt von Ausrichtung, Montage und differentiellen Quantifizierung der Genexpression beginnen. Darüber hinaus haben wir Richtungen enthalten die Genlisten aus Galaxy für die biologische Anreicherung Studien mit DAVID zu prüfen. Als letzter Schritt im Workflow bieten wir Anweisungen RNA-Seq Daten auf öffentlichen Servern wie die Sequenz Archiv lesen (SRA) auf NCBI für das Hochladen ( http: // www.ncbi.nlm.nih.gov/sra), um es an die wissenschaftliche Gemeinschaft frei zugänglich. Insgesamt erwarten wir, dass dieser Artikel umfassende und ausreichende Informationen, um Wurm Biologen liefern RNA-Seq Experimente zum ersten Mal sowie häufige Benutzer Unternehmen eine kleine Anzahl von Proben ausgeführt wird.
Protokoll
1. RNA-Isolierung
- Vorsichtsmaßnahmen
- Wischen der gesamte Arbeitsfläche, Instrumente nach unten und Pipetten mit einem handelsüblichen RNase Spray eventuell vorhandenen RNAsen zu beseitigen.
- Handschuhe tragen zu allen Zeiten, sie regelmäßig mit frischen, um während der verschiedenen Schritte des Protokolls zu ändern.
- Verwenden Sie nur Filterspitzen und hält alle Proben auf Eis so viel wie möglich RNA-Abbau zu vermeiden.
HINWEIS: Um die besten Daten von NGS-Plattformen zu erhalten, ist es wichtig, mit qualitativ hochwertigen RNA zu beginnen. RNA-Isolierung und Herstellungsmethoden variieren je nach Probe Herkunft, Verfahren zur Sequenzierung und Investigator bevorzugt. Mehrere im Handel erhältlichen Kits können für diesen Zweck oder RNA kann auch eine Standard-Phenol-Chloroform-Methode der RNA-Extraktion unter Verwendung isoliert wird verwendet werden. Bei beiden Methoden aufgeführten Vorsichtsmaßnahmen oben sollten während des gesamten Prozesses gefolgt werden, um Verunreinigungen und obt zu minimierenain ursprünglichen RNA-Proben.
- Ernte Worms
- Synchronisieren Sie die Wurmpopulation durch Hypochloritbleich- Behandlung 23 1000-1500 altersangepassten C. elegans erwachsenen Würmer pro Stamm zu erhalten.
- Waschen Sie die Würmer aus Platten unter Verwendung von M9 Pufferlösung und bei 325 xg Spin auf einer Tischzentrifuge für 30 s. Absaugen M9 Puffer aus hinter einem Pellet von Würmern zu verlassen. Wiederholen Sie diesen Schritt mindestens dreimal bakterielle Verschleppung zu beseitigen.
- Um den Wurm Pellet, fügen ~ 500 & mgr; l Lysepuffer (wenn eines kommerziellen Kits verwenden) oder Trizol (eine mono-phasischen Lösung von Phenol und Guanidinisothiocyanat, wenn Phenol: Chloroform-Extraktion in 1.3.3 beschrieben durchgeführt) worm Gewebe zu stören deaktivieren RNasen und Nukleinsäuren stabilisieren.
HINWEIS: Das Protokoll hier pausiert durch Flash werden die Proben in flüssigem Stickstoff durch Lagerung bei -80 ° C, gefolgt einfriert.
- RNA - Isolierung
- Beschallen worm Proben bei 45% Amplitude in Zyklen von 20 s. 'ON' und 40 s. 'OFF' (8-12 Zyklen pro Stamm). Halte Proben auf Eis zu allen Zeiten.
HINWEIS: Stellen Sie sicher, dass die Beschallungsgerät Sonde in dem Puffer eingetaucht und auf einem konstanten Niveau gehalten im gesamten Gebäude. Vermeiden der Probe Aufschäumen und reinigen Sie die Sonde gründlich in-zwischen den Proben. Beschallen Zyklen können in Abhängigkeit von der Art des verwendeten Sonicator variieren. Es wird empfohlen, Beschallung Bedingungen auf einer Testprobe zuerst optimiert werden, bevor ein Experiment zu starten. - Wenn eine im Handel erhältliche Kit, geht mit RNA-Isolierung nach dem vorgeschriebenen Protokoll. Für die RNA-Isolierung eines Phenol-Chloroform-Verfahren, die folgenden Schritte aus.
- Zentrifuge beschallten Proben bei 16.000 × g für 10 min. bei 4 ° C
- Der Überstand in ein 1,5 ml RNase-freien Mikrozentrifugenröhrchen und Zugabe von 100 ul Chloroform (1/5 das Volumen der RNA / DNA - Isolation - Reagenz).
Vorsicht: Chloroform ist giftig. Zur Verringerung der Exposition und Inhalation zu vermeiden, arbeitet in einer chemischen Haube, wenn Umgang mit dieser Substanz. - Vortex, um die Proben gründlich für 30 bis 60 s. und lassen Sie die Proben bei Raumtemperatur für 3 Minuten sitzen.
- Zentrifuge bei 11.750 xg für 15 min. bei 4 ° C. Übertragen Sie nur die obere wässrige Schicht in ein neues RNase-freie Mikrozentrifugenröhrchen dabei nicht die DNA-haltigen weißen Schnittstelle aspirieren. Wiederholen Sie die Schritte 1.3.4 bis 1.3.6.
- Nach Eintragen von 250 & mgr; l (70% der wässrigen Phase oder 1/2 RNA / DNA-Isolation Reagenzes Volumen) von 2-Propanol und invertieren das Rohr zu mischen. Lassen Rohre für 10 Minuten bei Raumtemperatur sitzen oder lassen Sie über Nacht bei -80 ° C.
- Centrifuge Proben bei 11.750 × g für 10 min. bei 4 ° C. Man dekantiert die überstehende Flüssigkeit sehr sorgfältig, hinter ein paar ui am Boden des Röhrchens zu verlassen, so dass das Pellet nicht gestört wird.
- Waschen Sie das Pellet mit 500 ul 75% Ethanol (hergestellt unter Verwendung von RNase-freies Wasser) und Spin-down bei 16000 × g für 5 min. eint 4 & deg; C.
- Entfernen Sie so viel Überstand wie möglich, ohne das Pellet zu stören. Luft trocknet das Pellet in einer Haube für ein paar Minuten.
- In 30 ul RNase-freiem Wasser und helfen, das RNA-Pellet für 10 Minuten durch Erwärmen auflösen. bei 60 ° C.
- Überprüfen RNA-Qualität und Quantität einer Bioanalyzer verwenden.
HINWEIS: Bioanalyzer erzeugt ein R I NA ntegrity N umber (RIN) als Maß für die RNA - Qualität. Ein RIN von mindestens 8 ist der empfohlene Grenzwert für RNA-Seq-Proben (je höher desto besser). RNA-Menge und Qualität können auch spektrophotometrisch geprüft werden, sondern auch durch visuelle Beurteilung der RNA-Integrität befolgt werden sollten. Dazu laufen die Proben auf einem 1,2% Agarose-Gel lang genug, um geeignete Trennung der 28s und 18s ribosomalen RNA-Banden zu erhalten. Das Vorhandensein von zwei distinkten Banden (1,75 kb für 18S - rRNA und 3,5 kb für die 28S rRNA im Fall von C. elegans) ist ein akzeptables Maß der RNA - Qualität. - Verwenden ~ 100 ng / & mgr; l RNA ship an den Anbieter / NGS Anlage zur Vorbereitung von Sequenzierungsbibliotheken.
HINWEIS: Die RNA-Proben sollten auf Trockeneis an den Sequenzierungs Service-Provider versendet werden. Die meisten Anbieter führen einen unabhängigen RNA Qualitätskontrolltest vor der Bibliothek Vorbereitung.
- Beschallen worm Proben bei 45% Amplitude in Zyklen von 20 s. 'ON' und 40 s. 'OFF' (8-12 Zyklen pro Stamm). Halte Proben auf Eis zu allen Zeiten.
2. RNA-Seq Datenanalyse
- Herunterladen von Raw Sequenzierungsdaten
- Laden Sie die komprimierten rohe fastq Sequenzierungsdaten codierten im fastq.gz - Format von dem NGS - Anbieter mit einem „File Transfer Protocol“ (FTP).

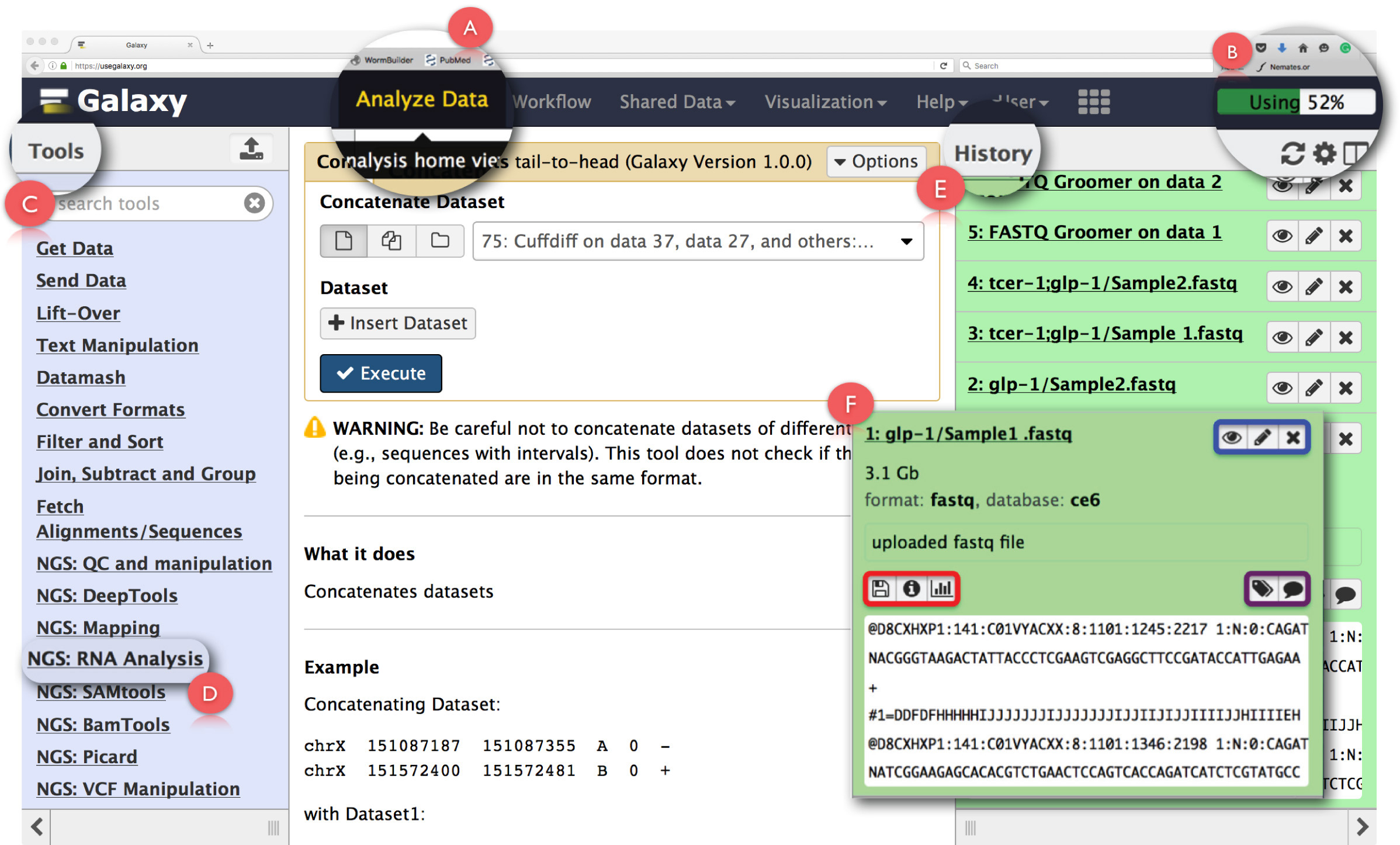
Abbildung 2: Aufbau des Galaxy Benutzeroberfläche Tafel und Key - RNA-Seq - Funktionen. Die wichtigsten Funktionen der Seite werden erweitert und hervorgehoben. (A) betont die ‚Analyse - Daten‘ Funktion auf der Webseite Header für den Zugriff auf Analyse Home Ansicht. (B) ist der Fortschrittsbalken, der den Raum auf der Galaxy - Server durch den Betrieb verwendet anzeigt. (C) ist die ‚Tools Abschnitt‘ , die alle Werkzeuge aufgelistet , die auf dem Galaxy - Schnittstelle ausgeführt werden können. (D) zeigt die 'NGS: RNA Analyse' Werkzeugabschnitt für RNA-Seq - Analyse verwendet. (E) zeigt das Panel ‚Geschichte‘, die alle Dateien auflistet Galaxy erzeugt. (F) zeigt ein Beispiel des Dialogfeld, das sich öffnet , wenn auf eine beliebige Datei in dem Abschnitt Historie klicken. Innerhalb (F), die blaue Box hebt Symbole , die zu betrachten , die verwendet werden können, editthe Attribute oder den Datensatz löschen, die lila Box hebt Symbole , die auf ‚Bearbeiten‘ verwendet werden kann , die Daten - Set - Tags oder Anmerkungen und gibt die rote Box Icons die Daten, Details der Aufgabe zum Download ausgeführt oder den Vorgang erneut auszuführen. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
- Erste Schritte mit Galaxy
HINWEIS: Verwenden einer webbasierten Plattform bietet Cloud-Zugang und kostenloses begrenzte Lager Galaxy kann auf einem öffentlichen Server ausgeführt werden. Es kann auch von Institutionen, sondern der lokalen Verarbeitung gehostet auf dem Computer des Benutzers oder Rechen-Cluster lokal heruntergeladen und ausgeführt werden, kann durch Datenspeichergrenzen und Verarbeitungsleistung Grenzen Benutzermaschinen eingeschränkt werden. Details zum Herunterladen und die Installation kann abgerufen werden unter https://wiki.galaxyproject.org/Admin/GetGalaxy . In diesem Protokoll beschreiben wir die webbasierte Nutzung der Galaxy-Pipeline.- Nach dem Herunterladen und Speichern der NGS-Daten auf den Computer des Benutzers, den Zugang Galaxy auflaxy.org/“target = "_blank"> https://usegalaxy.org/.
- Registrieren Sie ein Benutzerkonto , indem Sie auf ‚Benutzer‘ in der Kopfzeile der Seite, einloggen und beginnen , indem sie mit der Benutzeroberfläche Panel vertraut zu werden .
HINWEIS: Es wird empfohlen , dass Erstanwender die ‚Hier starten‘ Tutorial auf der Homepage nutzen kann mit der grundlegenden Einrichtung von Galaxy vertraut zu machen ( https://github.com/nekrut/galaxy/wiki/Galaxy101-1 ) . - Klicken Sie auf ‚Analysieren von Daten‘ (2A) in der Kopfplatte die ‚Analysis Home Ansicht‘ für den Zugriff auf die auch der Startbildschirm auf Galaxy.
HINWEIS: Der Header befindet sich auch andere Verbindungen, deren Details können durch mit dem Mauszeiger über sie zu sehen. Die obere rechte Ecke des Kopfes hat einen Fortschrittsbalken, den Raum für die Aufgaben (2B) verwendet überwacht. - Clecke auf ‚NGS: RNA - Analyse‘ Aufgabe im ‚Menü Extras‘ auf der linken Seite (2C) , die alle Werkzeuge für die RNA-Seq Datenanalyse erforderlich zuzugreifen.
HINWEIS: Das ‚Menü Extras‘ katalogisiert alle Operationen , die Galaxy - Angebote. Dieses Menü wird aufgeteilt basierend auf Aufgaben, und klicken Sie auf einem eine Liste aller Werkzeuge öffnen benötigt, um diese Aufgabe zu erfüllen. - Erstellen Sie neue Analyse Geschichte , indem Sie auf der rechten Seite am oberen Rand des Fensters ‚Geschichte‘ auf das Zahnrad - Symbol klicken (Abbildung 2E). Wählen Sie ‚Neu erstellen‘ Option aus dem Popup-Menü. Geben Sie diese ‚Geschichte‘ einen geeigneten Namen , um die Analyse zu identifizieren.
HINWEIS: Die Panel ‚History‘ zeigt alle für die Analyse hochgeladenen Dateien sowie alle Ausgabedateien, die von laufenden Tasks auf Galaxy erzeugt werden. Ein Klick auf einen Dateinamen in diesem Fenster öffnet sich ein Dialogfenster mit detaillierten Informationen über die Aufgabe oben ausgeführtund ein Ausschnitt des Datensatzes (Figur 2F). Symbole in dieser Box ermöglichen es dem Benutzer zu ‚Ansicht‘, ‚bearbeiten , um die Attribute‘ oder ‚Löschen‘ der Datensatz (2F, blau hervorgehoben). Darüber hinaus kann der Anwender auch ‚Bearbeiten‘ Daten - Set - Tags oder Annotation (2F, in lila markiert), ‚Download‘ die Daten, ‚Details‘ der Aufgabe, ‚Wiederholung‘ die Aufgabe oder sogar ‚visualisieren‘ den Datensatz aus diesem Dialogbox (2F, rot markiert). - Klicken Sie auf die 'Upload File' Funktion unter 'Get Data' im 'ToolsMenu' raw fastq Dateien zu.
HINWEIS: Wenn Sie auf diesem oder einem anderen Werkzeug öffnet eine kurze Beschreibung des Betriebes nach oben, und der Prüfung selbst, in der Mitte ‚Analysis Interface‘ Panel. Dieses Panel Schnürsenkel zusammen die‚Extras‘ aus dem linken Fenster und dem ‚Input Files‘ auf der rechten ‚Geschichte‘ Panel (Abbildung 2E). Hier Eingabedateien aus ‚Geschichte‘ werden ausgewählt und andere Parameter eine bestimmte Aufgabe auszuführen definiert. Der resultierende Ausgabe - Datensatz aus jedem Test wird in ‚Geschichte‘ gespeichert zurück. Im Lieferumfang der Test im Panel 'Analysis Interface "sind Erklärungen für alle verfügbaren Parameter für den Betrieb eines bestimmten Werkzeugs zusammen mit einer detaillierten Liste aller Ausgabedateien erstellt das Tool. - Nach der Aufgabe in der ‚Analyse - Interface‘ öffnet, klicken Sie auf ‚Wählen Sie Lokale Datei‘ oder ‚Wählen Sie FTP File‘ (schneller Upload), navigieren , um die Sequenzierung von Dateien in den Ordner, und die entsprechende Datenmenge wählen hochgeladen werden.
- Lassen Sie Galaxy zu ‚Auto-detect‘ die hochgeladene Datei - Typ (Standardeinstellung). Wählen Sie 'C elEgans "im Pulldown - Menü für das Genom.
- Klicken Sie auf ‚Start‘ Daten - Upload zu starten. Sobald die Datei hochgeladen wird, wird es im Panel ‚Geschichte‘ gespeichert und können von dort abgerufen werden.
- Wenn mehrere Sequenzierungsdaten - Dateien für eine einzelne Probe erzeugt werden, kombinieren sie das ‚verketten‘ Tool. Um dies zu tun, öffnen Sie die ‚Text Manipulation‘ Option im ‚Menü Extras‘.
- Klicken Sie auf dem ‚verketten‘ Werkzeug, wählen Sie die Dateien , die aus der Drop-Down - Box in der Mitte des ‚Analyse - Schnittstelle‘ und klicken Sie auf ‚Ausführen‘ kombiniert werden müssen.
HINWEIS: Die Ausgabe von Dateien mit dieser Aufgabe erzeugt werden , in dem fastq Format erzeugt. Das Mapping - Programm hat eine Grenze von 16.000.000 Sequenzen pro fastq Datei und wenn diese Grenze eine neue fastq Datei wird für die verbleibenden Sequenzen erzeugt erreicht ist. die '; Concatenate‘Werkzeug ist in solchen Fällen notwendig , um die Datensätze zu kombinieren. - Konvertieren Sie die hochgeladen fastq - Format - Dateien auf das gewünschte fastqsanger Format für Galaxy RNA-Seq - Analyse unter Verwendung des ‚fastq Groomer‘ Werkzeugs unter den gefundenen ‚NGS: QC und Manipulation‘ Abschnitt (siehe ergänzende Datei).
- Wählen Sie den entsprechenden fastq - Datensatz unter ‚Datei zu Groom‘ Option und führen Sie das Tool Standardparameter.
HINWEIS: Ausgabedateien dieser Aufgabe hergestellt unter Verwendung von in der fastqsanger Format erzeugt.
- fastqsanger Data Quality-Control - Tests
- Prüfen Sie die Qualität der hochgeladenen fastqsanger liest mit dem ‚FastQC‘ Tool befindet sich unter ‚NGS: QC und Manipulation‘ im Menü ‚Extras‘.
- Wählen Sie die präparierten fastqsanger Datendatei aus dem Dropdown - Menü für "Short Daten aus der aktuellen Bibliothek lesen und das Werkzeug mit den Standardparametern ausgeführt werden .
HINWEIS: Achten Sie besonders auf die Qualität der liest und Vorhandensein irgendwelcher Adaptersequenzen. Adapter ist in der Regel als Teil der Post-RNA-Seq Datenverarbeitung durch NGS-Anbieter, aber in einigen Fällen entfernt wird, kann zurückgelassen werden. Zur Erläuterung der Qualitätsstandards gehen zu http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ . - Prüfen Sie mit dem NGS - Anbieter und wenn Adapter vorhanden ist, schneiden sie mit dem ‚Clip‘ Werkzeug aus dem ‚NGS: QC und Manipulation‘ Task - Menü.
HINWEIS: Ausgabedateien um diese Aufgabe , hergestellt unter Verwendung werden im unverdünnten txt - Format sowie in HTML erzeugt , die auf einem beliebigen Web - Browser geöffnet werden können.
- Datenanalyse mit Tuxedo Suite
- TopHat
- Laden Sie die neueste Version von C. elegans Referenzgenom fasta und gtf (Gene Transfer Format) Dateien aus Datei hochladen‘ , wie oben in 2.2.6 beschrieben.
- Öffnen Sie die ‚NGS: RNA - Analyse‘ Abschnitt und klicken Sie auf ‚TopHat‘ Werkzeug zur Karte der Sequenzierung liest auf die heruntergeladene Referenzgenom.
- Wählen Sie die entsprechende Antwort aus dem Drop - Down - Menü auf die Frage ‚Ist das Single-End oder Paired-End - Daten?‘
- Wählen Sie die entsprechende Datei fastq.
- Wählen Sie im nächsten Drop - Down - Menü ‚ein Genom aus der Geschichte verwenden‘ und Referenzgenom in Schritt 2.4.1.1 heruntergeladen wählen.
- Wählen Sie ‚Default‘ für die anderen Parameter und klicken Sie auf ‚Ausführen‘.
HINWEIS: Unter den Ausgabedateien diese Aufgabe hergestellt wird, unter der Datei ‚Akzeptierte Hits‘ für die nachfolgenden Schritte verwendet wird.
- Manschettenknöpfe und Cuffmerge
- Wählen Sie den ‚CuffLinks' Werkzeug in den ‚NGS: Abschnitt‘ RNA - Analyse der Transkripte, schätzen ihre Fülle und Test für unterschiedliche Expression zu montieren.
- Im ersten Drop - Down - Menü wählen Sie die abgebildeten ‚Akzeptierte Treffer (BAM - Format)‘ Datei von TopHat - Analyse erhalten.
- Im zweiten Dropdown - Menü eingestellten Referenz Anmerkung an die GTF - Datei in Schritt 2.4.1.1 heruntergeladen.
- Wählen Sie ‚Ja‘ für die ‚Perform Bias - Korrektur‘ Option , und führen Sie die Aufgabe , die Standardeinstellungen für alle anderen Parameter.
HINWEIS: Unter den Ausgabedateien mit dieser Aufgabe betraut wird , die ‚Accepted Transcripts‘ Datei für die nachfolgenden Schritte verwendet wird. - Open 'Cuffmerge' Werkzeug in den 'NGS: RNA - Analyse' , die 'Assembled Transcripts' verschmelzen produziert für alle RNA-Seq - Proben.
Hinweis: das erste Feld in der Werkzeugselbst bevölkert und listet alle gtf Dateien durch Manschettenknöpfe hergestellt. - Wählen Sie den ‚Assembled Transcripts‘ Datei für alle Stämme / Bedingungen getestet wurden, einschließlich biologische Replikate des gleichen Stammes / Zustand (siehe Diskussion für die biologische Replikate).
- Wählen Sie ‚Ja‘ für ‚Use Reference Annotation‘ und wählen Sie die GTF - Datei heruntergeladen in Schritt 2.4.1.1.
- Im folgende Kästchens wieder ‚Ja‘ für die Option ‚Verwenden Sequenzdaten‘ und die gesamte Genom fasta Datei in Schritt 2.4.1.1 heruntergeladen wählen.
- Halten Sie die anderen Parameter als Standard, klicken Sie auf ‚Ausführen‘.
HINWEIS: Cuffmerge erzeugt eine einzelne gtf Ausgabedatei.
- Cuffdiff
- Navigieren Sie zu dem ‚Cuffdiff‘ Werkzeug in dem ‚NGS: RNA - Analyse‘ Abschnitt. Im Menü ‚Transcripts‘, wählen Sie die fusionierte Ausgabedatei von Cuffmerge.
- EtiketteBedingungen 1 und 2 mit den beiden Stämmen / Bedingungsnamen.
HINWEIS: Cuffdiff Vergleiche zwischen mehr als zwei Stämmen oder Bedingungen sowie Zeitverlauf Experimente durchführen können. Nutzen Sie einfach die ‚neue Bedingungen Add‘ Option jede neue Stämme / Bedingung hinzuzufügen, je nach Bedarf. - Für jeden Stamm / Zustand, unter ‚Replikate‘ select individuellen Accepted Hits "Ausgabedateien von TopHat, die auf die verschiedenen biologischen Replikate dieser Stamm / Zustand entsprechen. Halten Sie die Taste ‚cmd‘, wenn ein Macintosh - Computer, und Strg-Taste, wenn ein PC, um mehrere Dateien auszuwählen.
- Lassen Sie alle anderen Optionen als Standardparameter. Klicken Sie auf ‚Ausführen‘ um die Aufgabe auszuführen.
HINWEIS: Cuffdiff zahlreiche Ausgabedateien in tabellarischer Form als die endgültigen Auslesen der RNA-Seq Analyse erzeugt. Dazu gehören Dateien mit FPKM Tracking für Transkripte, Gene (kombiniertFPKM Werte von Transkripten ein Gen Identität teilen), primär und Transkripte kodierenden Sequenzen. Alle Datendateien erzeugt werden, können auf jeder Tabellenkalkulationsanwendung betrachtet werden und enthalten ähnliche Attribute wie Genname, Locus, falzen Änderung (in log2-Skala) sowie statistische Daten über die Vergleiche zwischen den Stämmen / Bedingungen, einschließlich dem p-Wert und Q-Werte. Die Daten in diesen Dateien können auf der Grundlage statistischer Signifikanz der Unterschiede sortiert werden oder eine Änderung in der Genexpression falten (Größe und Richtung der Veränderung, wie in Up- oder regulierte Gene Down-) und gemäß den Anforderungen der Anwender manipuliert. Wenn die Konvertierung zwischen den verschiedenen Gen - Identifikatoren benötigt wird (zB WormBase Gen ID vs. Cosmid - Zahl), verfügbare Werkzeuge auf Biomart ( http://www.biomart.org/ ) verwendet werden.
- TopHat
3. Gene Ontology (GO) Zeitanalyse mit DAVID
- Zugang DAVID von der Website hTGP: //david.ncifcrf.gov/. Klicken Sie auf ‚Start Analysis‘ im Header der Webseite. In ‚Schritt 1‘, kopieren und die Liste der Gene aus Galaxy in Kasten A. In ‚Schritt 2‘ der Option ‚WormBase Gene ID‘ als Kennung für den Eingang Gene erhaltenen Paste.
HINWEIS: DAVID erkennt die meisten öffentlich zugänglichen Anmerkung Kategorien, so dass andere Gen-IDs (wie Entrez Gen ID oder Gen-Symbol) können ebenfalls verwendet werden. - In ‚Schritt 3‘, wählen Sie ‚Gene - Liste‘ (Gene analysiert werden) unter ‚Listentyp‘ und klicken Sie dann auf dem ‚Liste Senden‘ Symbol.
HINWEIS: ‚Analysis Wizard‘, öffnet sich alle verlinkte DAVID Tools aufzulisten , die auf dem hochgeladenen Genliste (Abbildung 3) ausgeführt werden kann. Klicken Sie auf diese Links als relevant entsprechenden Module für den Zugriff pro Anforderung des Benutzers. Zur Identifizierung der Werkzeuge geeignet für eine bestimmte Aufgabe, klicken Sie auf 'Welche DAVID Werkzeuge zu benutzen? ‚Link auf der‘ ; Analyse Wizard‘Seite. Klicken Sie auf den ‚Start Analyse‘ Link in der Kopfzeile der ‚Analysis Wizard‘ Homepage zu jedem Zeitpunkt während der Analyse zurückzukehren.

Abbildung 3: Aufbau des DAVID Analyseassistent Webpage und Beispiele für Betrieb Ausgänge. Die ‚Analyse Wizard‘ Web - User-Interface listet die verwendeten Werkzeuge uploaded Genliste zur Anreicherung auf Grundlage verschiedenen Parameter zu analysieren. Ein Klick auf diesen Tools berichtet über die analysierten Daten in einer neuen Webseite. Beispiele für die tabellarischen Berichte erzeugt von ‚Genen Funktionale Klassifikation‘, ‚Funktionale Annotation - Diagramm‘ und ‚Functional Annotation Clustering‘ wie -einsätze (Pfeile) gezeigt.> Klicken Sie hier um eine größere Version dieser Figur zu sehen.
- Functional Annotation Tool 1: Functional Annotation Clustering
- Klicken Sie auf ‚Functional Annotation Clustering‘ -Modul auf die Übersichtsseite zu gehen. Behalten Sie die Standardanmerkungskategorien und klicken Sie auf ‚Functional Annotation Clustering‘ Cluster ähnlicher Anmerkung Begriffe zu erzeugen durch ihre Anreicherung Punktzahl rangiert.
- Klicken Sie auf die verlinkte Namen jeder Begriff Details zu erfahren und ‚RT‘ (verwandte Begriffe) andere ähnliche Begriffe in die Kategorie im Zusammenhang aufzulisten.
- Klicken Sie auf die lila Balken, die Gene mit einer Laufzeit und dem roten ‚G‘ zugeordnet listet alle Gene zur Liste im Zusammenhang mit allen Ausdrücken innerhalb eines Clusters.
- Klicken Sie auf das grüne Symbol, um eine zweidimensionale Darstellung aller Gene und Begriffe in einem Cluster zu sehen.
HINWEIS: Die letzten drei Spalten zeigen die analytischen und statistische Ergebnisse für jedenBegriff. Die Ergebnisse für diese und alle anderen Analysen können durch Klicken auf die ‚Datei herunterladen‘ Link in einer TXT - Format heruntergeladen werden.
- Functional Annotation Tool 2: Functional Annotation - Diagramm
- Zurück zur Übersichtsseite und klicken Sie auf ‚Functional Annotation Chart‘ zu identifizieren , deutlich überrepräsentiert biologische Begriffe verwenden (zB Transkriptionsfaktor - Aktivität oder Kinase - Aktivität) mit dem Gen - Liste zugeordnet.
- Klicken Sie den Begriff Namen Ausführlichere Informationen und ‚RT‘ (verwandte Begriffe) zur Liste andere verwandte Begriffe zu erhalten.
- Klicken Sie auf die lila Balken um eine Liste aller zugehörigen Gene einzelner Kategorie entspricht.
HINWEIS: Die letzten beiden Spalten der statistischen Tests die Ergebnisse für jede Kategorie auflisten.
- Functional Annotation - Tool 3: Functional Annotation Tabelle
- Zurück zur Übersichtsseite und klicken Sie auf 'Functional Annotation Table "zugeordnet ist, eine Liste aller Anmerkungen zu sehen , mit den Genen auf einer Liste ohne statistische Berechnungen.
HINWEIS: Dieses Tool kann für die Gen-für-Gen-Analyse einer Liste oder suchen an bestimmten, sehr interessante Gene nützlich sein.
- Zurück zur Übersichtsseite und klicken Sie auf 'Functional Annotation Table "zugeordnet ist, eine Liste aller Anmerkungen zu sehen , mit den Genen auf einer Liste ohne statistische Berechnungen.
- Gene Funktionsklassifizierung Werkzeug
- Zurück zu ‚Analysis Wizard‘ und klicken Sie auf ‚Gene Funktionsklassifizierung‘ -Modul die Eingangs Genliste in funktionsbezogenen Gruppen von Genen entmischen rangiert als pro ihre ‚Enrichment Score‘, ein Maß für die allgemeine Bereicherung des Gens Gruppe in der Liste.
- Klicken Sie den Begriff Namen zu erhalten detailliertere Informationen und ‚RG‘ funktionell verwandten Genen des Gens Gruppe zeigen
- Klicken Sie auf den roten ‚T‘ (Begriff Berichte), die Biologie und das grüne Symbol zur Liste eine zweidimensionale Ansicht aller Gene und Begriffe zu sehen.
- Gen-NameBatch-Viewer
- Zurück zu 'Analysis Wizard' und klicken Sie auf 'Gene-name Batch - Viewer' 'WormBase Gene IDs' in ihren entsprechenden Gen - Namen zu übersetzen. (WBGene00022855 = tCER-1).
- Klicken Sie auf Genname mehr Gen-spezifische Informationen zu erhalten.
- Klicken auf den ‚RG‘ ( in Zusammenhang stehenden Genen) Link neben jedem Gen Gene zu offenbaren vorhergesagt zu dem Gen von Interesse funktionell verwandt zu sein.
4. Hochladen von RAW-Daten auf der NCBI Sequence Archiv lesen (SRA)
- Besuchen Sie die SRA - Webseite unter Anmelden bei NCBI‘Link oder ein neues Konto registrieren.
- Klicken Sie auf ‚Bioproject‘.
- Klicken Sie auf ‚Submission‘ unter der ‚Mit Bioproject‘ Überschrift auf der linken Seite.
- Wählen Sie die Option ‚Neue Vorlage‘. Details zum Update des Einreicher. Fahren Sie durch die verbleibenden sieben Registerkarten, In den Einzelheiten des Experiments Abfüll- und Daten hochgeladen werden. Klicken Sie auf ‚Senden‘ , wenn sie abgeschlossen sind .
HINWEIS: In der fünften ‚Bioprobe‘ Reiter, lassen Sie den Steckplatz für ‚Bioprobe‘ leer. - Aktualisieren Sie die resultierende Seite durch einen Klick auf den ‚My Submissions‘ -Link. Die übermittelten Daten werden mit einer zugewiesenen Einreichungsnummer, eine kurze Beschreibung und Upload-Status aufgelistet.
- Klicken Sie auf ‚Bioprobe‘ am oberen Rand der Seite, in der ‚eine neue Vorlage beginnen‘ -Box und schaffen eine ‚neue Vorlage‘. Senden separate Eingaben für jede Probe.
- Wie im Fall mit ‚Bioproject‘ in 4.4, aktualisieren Sie die Einzelheiten der Einreicher und weiter durch den Rest der Registerkarten in den Details der einzelnen Register zu füllen. Nach Überprüfung abgeschlossen und klicken Sie auf ‚Senden‘.
- Navigieren Sie zu http: //www.ncbi.nlm.nih.gov / sra die endgültige 'Sequence Archiv lesen (SRA)' Vorlage zu erstellen.
- Klicken Sie auf 'Login SRA' unter 'Getting Started'.
- Auf der nächsten Seite klicken Sie auf den ‚NCBI PDA‘ Link. Ein ‚Update - Einstellungen‘ Link öffnet sich. Füllen Sie das Formular aus und klicken Sie auf ‚Einstellungen speichern‘.
- Auf der angezeigten Seite klicken Sie auf den Link ‚Create New Submission‘. Geben Sie einen geeigneten Namen unter ‚Alias‘ und klicken Sie auf ‚Speichern‘. Eine Tabelle mit der Vorlage ID und anderen Details wird erstellt.
- Klicken Sie auf ‚New Experiment‘ und registrieren mindestens eine eindeutige Sequenzierung Bibliothek für jeden ‚Bioprobe‘.
- Bezeichnen Sie und verknüpfen Sie die zuvor erstellten ‚Bioproject‘ und ‚Bioprobe‘ Vorlage IDs. A 'New Experiment' wird erstellt.
- Klicken Sie auf ‚New Run‘ am unteren Rand der Seitenach dem SRA Experiment wurde die Datendateien gemacht und identifizieren, die ihn verknüpft werden müssen.
- Berechnen Sie die MD5-Summe von jeder Datendatei. Um dies zu tun auf einem Terminal MacIntosh, navigieren Sie zu Programme / Dienstprogramme / Terminal. In Terminal, Typ in ‚md5‘ (ohne Anführungszeichen), gefolgt von einem Leerzeichen. Drag & Drop die Dateien , die in Terminal von Finder hochgeladen werden müssen, und klicken Sie auf ‚Enter‘.
- Terminal wird eine alphanumerische MD5-Summe zurück. Geben Sie diese als Teil des Einreichungsprozesses für die Datei-Upload. Verwenden Sie den Benutzernamen und das Passwort vom System bereitgestellte Dateien per FTP hochladen.
Ergebnisse
In C. elegans, Eliminierung der Keimbahn - Stammzellen (GSCs) erstreckt Lebensdauer erhöht Stressresistenz, und erhöht Körperfett 24, 28. Verlust von GSCs entweder durch Laserablation oder durch Mutationen , wie GLP-1, verursacht durch die Aktivierung Lebensdauer Erweiterung eines Netzes von Transkriptionsfaktoren 29 hervorgerufen wird . Ein solcher Faktor, TCER-1 kodiert das worm Homo...
Diskussion
Bedeutung der Galaxy Sequencing-Plattform in der modernen Biologie
Die Galaxy-Projekt hat instrumental werden Biologen ohne Bioinformatik Ausbildung helfen Sequenzierungsdaten mit hohem Durchsatz auf eine schnelle und effiziente Art und Weise zu verarbeiten und zu analysieren. Sobald eine Herkules-Aufgabe betrachtet, diese öffentlich zugängliche Plattform komplexe Bioinformatik-Algorithmen gemacht lief NGS-Daten einen einfachen, zuverlässiger und einfacher Prozess zu analysieren. Neben einer...
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Die Autoren möchten ihren Dank an die Laboratorien, Gruppen und Einzelpersonen zum Ausdruck bringen, die Galaxy und DAVID entwickelt hat und damit gemacht NGS für die wissenschaftliche Gemeinschaft allgemein zugänglich. Die Hilfe und Rat von Kollegen an der University of Pittsburgh während unserer Bioinformatik Ausbildung zur Verfügung gestellt werden anerkannt. in Aging Award (AG-NS-0879-12) und einen Zuschuss von der National Institutes of Health (R01AG051659) zu AG Diese Arbeit wurde von einer Ellison Medical Foundation New Scholar unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| RNase spray | Fisher Scientific | 21-402-178 | |
| Trizol | Ambion | 15596026 | |
| Sonicator | Sonics Vibra Cell | VCX130 | |
| Centrifuge | Eppendorf | 5415C | |
| chloroform | Sigma Aldrich | 288306 | |
| 2-propanol | Fisher Scientific | A416P-4 | |
| Ethanol | Decon Labs | 2705HC | |
| RNase-free water | Fisher Scientific | BP561-1 | |
| Bioanalyzer | Agilent | G2940CA | |
| Mac/PC |
Referenzen
- Venter, J. C., et al. The sequence of the human genome. Science. 291 (5507), 1304-1351 (2001).
- Lander, E. S., et al. Initial sequencing and analysis of the human genome. Nature. 409 (6822), 860-921 (2001).
- Afgan, E., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 (W1), W3-W10 (2016).
- Trapnell, C., Pachter, L., Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 25 (9), 1105-1111 (2009).
- Trapnell, C., et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28 (5), 511-515 (2010).
- Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12 (3), R22 (2011).
- Roberts, A., Pimentel, H., Trapnell, C., Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 27 (17), 2325-2329 (2011).
- Trapnell, C., et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7 (3), 562-578 (2012).
- Trapnell, C., et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31 (1), 46-53 (2013).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4 (1), 44-57 (2009).
- Giardine, B., et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15 (10), 1451-1455 (2005).
- Han, Y., Gao, S., Muegge, K., Zhang, W., Zhou, B. Advanced Applications of RNA Sequencing and Challenges. Bioinform Biol Insights. 9 (1), 29-46 (2015).
- Mardis, E. R. Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif). 6, 287-303 (2013).
- Yang, I. S., Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genomics Inform. 13 (4), 119-125 (2015).
- Khatri, P., Draghici, S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 21 (18), 3587-3595 (2005).
- Huang da, W., Sherman, B. T., Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37 (1), 1-13 (2009).
- Shaye, D. D., Greenwald, I. OrthoList: a compendium of C. elegans genes with human orthologs. PLoS One. 6 (5), e20085 (2011).
- Consortium, C. e. S. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 282 (5396), 2012-2018 (1998).
- Agarwal, A., et al. Comparison and calibration of transcriptome data from RNA-Seq and tiling arrays. BMC Genomics. 11, 383 (2010).
- Mortazavi, A., et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20 (12), 1740-1747 (2010).
- Bohnert, R., Ratsch, G. rQuant.web: a tool for RNA-Seq-based transcript quantitation. Nucleic Acids Res. 38, W348-W351 (2010).
- Lamm, A. T., Stadler, M. R., Zhang, H., Gent, J. I., Fire, A. Z. Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome. Genome Res. 21 (2), 265-275 (2011).
- Amrit, F. R., Ratnappan, R., Keith, S. A., Ghazi, A. The C. elegans lifespan assay toolkit. Methods. 68 (3), 465-475 (2014).
- Hsin, H., Kenyon, C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 399 (6734), 362-366 (1999).
- Alper, S., et al. The Caenorhabditis elegans germ line regulates distinct signaling pathways to control lifespan and innate immunity. J Biol Chem. 285 (3), 1822-1828 (2010).
- Steinbaugh, M. J., et al. Lipid-mediated regulation of SKN-1/Nrf in response to germ cell absence. Elife. 4, (2015).
- Lapierre, L. R., Gelino, S., Melendez, A., Hansen, M. Autophagy and lipid metabolism coordinately modulate life span in germline-less. C. elegans. Curr Biol. 21 (18), 1507-1514 (2011).
- Rourke, E. J., Soukas, A. A., Carr, C. E., Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10 (5), 430-435 (2009).
- Ghazi, A. Transcriptional networks that mediate signals from reproductive tissues to influence lifespan. Genesis. 51 (1), 1-15 (2013).
- Ghazi, A., Henis-Korenblit, S., Kenyon, C. A transcription elongation factor that links signals from the reproductive system to lifespan extension in Caenorhabditis elegans. PLoS Genet. 5 (9), e1000639 (2009).
- Amrit, F. R., et al. DAF-16 and TCER-1 Facilitate Adaptation to Germline Loss by Restoring Lipid Homeostasis and Repressing Reproductive Physiology in C. elegans. PLoS Genet. 12 (2), e1005788 (2016).
- Wang, M. C., O'Rourke, E. J., Ruvkun, G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 322 (5903), 957-960 (2008).
- McCormick, M., Chen, K., Ramaswamy, P., Kenyon, C. New genes that extend Caenorhabditis elegans' lifespan in response to reproductive signals. Aging Cell. 11 (2), 192-202 (2012).
- Kartashov, A. V., Barski, A. BioWardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. 16, 158 (2015).
- Goncalves, A., Tikhonov, A., Brazma, A., Kapushesky, M. A pipeline for RNA-seq data processing and quality assessment. Bioinformatics. 27 (6), 867-869 (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten