
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
نهج قائم على قياس الطيف الكتلي لتحديد فوسفاتيز فوسفوبروتين وتفاعلاتها
In This Article
Summary
هنا ، نقدم بروتوكولا لإثراء فوسفاتيز البروتين الفوسفوبروتين الداخلي المنشأ وبروتيناتها المتفاعلة من الخلايا والأنسجة وتحديدها وتحديدها كميا بواسطة البروتيوميات القائمة على قياس الطيف الكتلي.
Abstract
يتم تنظيم معظم العمليات الخلوية عن طريق فسفرة البروتين الديناميكية. أكثر من ثلاثة أرباع البروتينات مفسفرة ، وفوسفاتيز البروتين الفوسفوبروتيني (PPPs) تنسق أكثر من 90٪ من جميع عمليات إزالة الفوسفور الخلوية من السيرين / الثريونين. وقد تورط تحرير فسفرة البروتين في الفيزيولوجيا المرضية لمختلف الأمراض، بما في ذلك السرطان والتنكس العصبي. وعلى الرغم من نشاطها الواسع النطاق، فإن الآليات الجزيئية التي تتحكم في الشراكات بين القطاعين العام والخاص وتلك التي تسيطر عليها الشراكات بين القطاعين العام والخاص لا تتسم بخصائص كافية. هنا ، يتم وصف نهج بروتيني يسمى حبات مثبطات الفوسفاتيز وقياس الطيف الكتلي (PIB-MS) لتحديد وقياس PPPs ، وتعديلاتها بعد الترجمة ، ومفاعلاتها المتفاعلة في أقل من 12 ساعة باستخدام أي خط خلية أو أنسجة. يستخدم PIB-MS مثبطا غير انتقائي لتعادل القوة الشرائية ، microcystin-LR (MCLR) ، مثبتا على حبات السيفاروز لالتقاط وإثراء PPPpps الذاتية المنشأ والبروتينات المرتبطة بها (تسمى PPPome). لا تتطلب هذه الطريقة التعبير الخارجي للإصدارات الموسومة من PPPs أو استخدام أجسام مضادة محددة. يقدم PIB-MS طريقة مبتكرة لدراسة الشراكات بين القطاعين العام والخاص المحفوظة تطوريا وتوسيع فهمنا الحالي لإشارات إزالة الفسفرة.
Introduction
تتحكم فسفرة البروتين في معظم العمليات الخلوية ، بما في ذلك على سبيل المثال لا الحصر الاستجابة لتلف الحمض النووي ، وإشارات عامل النمو ، والمرور عبر الانقسام1،2،3. في خلايا الثدييات ، يتم فسفوريلات غالبية البروتينات في واحد أو أكثر من بقايا سيرين أو ثريونين أو التيروزين في وقت ما ، مع فوسفوسرين وفوسفوثريونين يشكلون حوالي 98 ٪ من جميع مواقع الفسفرة 2,3. في حين تمت دراسة الكينازات على نطاق واسع في الإشارات الخلوية ، فإن دور الشراكات بين القطاعين العام والخاص في تنظيم العمليات الخلوية الديناميكية لا يزال ناشئا.
يتم التحكم في ديناميكيات الفسفرة من خلال التفاعل الديناميكي بين الكينازات والفوسفاتيز. في خلايا الثدييات ، هناك أكثر من 400 كيناز بروتين يحفز فسفرة سيرين / ثريونين. أكثر من 90٪ من هذه المواقع يتم نزع الفوسفوريلات بواسطة فوسفاتيز البروتين الفوسفوبروتيني (PPPs) ، وهي عائلة صغيرة من الإنزيمات التي تتكون من PP1 و PP2A و PP2B و PP4-7 و PPT و PPZ 2,3. PP1 و PP2A مسؤولان عن غالبية إزالة الفوسفوسرين والفوسفوثريونين داخل الخلية2،3،4. أدى الاختلاف الملحوظ في العدد بين الكينازات والفوسفاتيز وعدم خصوصية الوحدات الفرعية الحفازة PPP في المختبر إلى الاعتقاد بأن الكينازات هي المحدد الرئيسي للفسفرة 2,3. ومع ذلك ، فقد أظهرت دراسات متعددة أن الفوسفاتيز يؤسس خصوصية الركيزة من خلال تكوين إنزيمات هولو متعددة الميريك5،6،7،8،9. على سبيل المثال ، PP1 هو غير متجانس يتكون من وحدة فرعية حفازة ، وفي وقت معين ، واحد من أكثر من 150 وحدة فرعية تنظيمية 6,7,8. على العكس من ذلك ، PP2A هو متغاير يتكون من سقالات (A) ، ووحدة تنظيمية (B) ، ووحدة فرعية حفازة (C) 2,3,9. هناك أربع عائلات متميزة من الوحدات الفرعية التنظيمية PP2A (B55 و B56 و PR72 و striatin) ، لكل منها جينات متعددة ، ومتغيرات الربط ، وأنماط التوطين2،3،9. وتسد الطبيعة المتعددة للشراكات بين القطاعين العام والخاص الفجوة في عدد الكينازات والوحدات الفرعية الحفازة لتعادل القوة الشرائية. ومع ذلك ، فإنه يخلق تحديات تحليلية لدراسة إشارات الشراكة بين القطاعين العام والخاص. لتحليل إشارات PPP بشكل شامل ، من الأهمية بمكان التحقيق في مختلف الإنزيمات الهولونية داخل الخلية أو الأنسجة. تم إحراز تقدم كبير في دراسة الكينومي البشري من خلال استخدام حبات مثبطات الكيناز ، التي يطلق عليها اسم حبات مثبطات الإرسال المتعددة أو الخرز ، وهي استراتيجية بروتينية كيميائية حيث يتم تجميد مثبطات الكيناز على الخرز ويستخدم قياس الطيف الكتلي لتحديد الكينازات المخصبة وتفاعلها10،11،12،13.
لقد أنشأنا نهجا مشابها لدراسة بيولوجيا الشراكة بين القطاعين العام والخاص. تتضمن هذه التقنية التقاط التقارب للوحدات الفرعية الحفازة PPP باستخدام الخرز مع مثبط PPP غير انتقائي غير متحرك يسمى microcystin-LR (MCLR) يسمى حبات مثبطات الفوسفاتيز (PIBs) 14,15. على عكس الطرق الأخرى التي تتطلب وضع علامات داخلية أو التعبير عن الوحدات الفرعية لتعادل القوة الشرائية الخارجية التي يمكن أن تغير نشاط البروتين أو توطينه ، يسمح PIB-MS بإثراء الوحدات الفرعية المحفزة لتعادل القوة الشرائية الذاتية المنشأ ، والوحدات الفرعية التنظيمية والسقالات المرتبطة بها ، والبروتينات المتفاعلة (تسمى PPPome) من الخلايا والأنسجة في نقطة زمنية معينة أو في ظل ظروف علاج محددة. يمنع MCLR PP1 و PP2A و PP4-6 و PPT و PPZ بتركيزات نانومولية ، مما يجعل PIBs فعالة للغاية في إثراء PPPome16. يمكن تحجيم هذه الطريقة لاستخدامها على أي مادة أولية من الخلايا إلى العينات السريرية. هنا ، نصف بالتفصيل استخدام PIBs وقياس الطيف الكتلي (PIB-MS) لالتقاط وتحديد وقياس PPPome الداخلي وحالات تعديله بكفاءة.

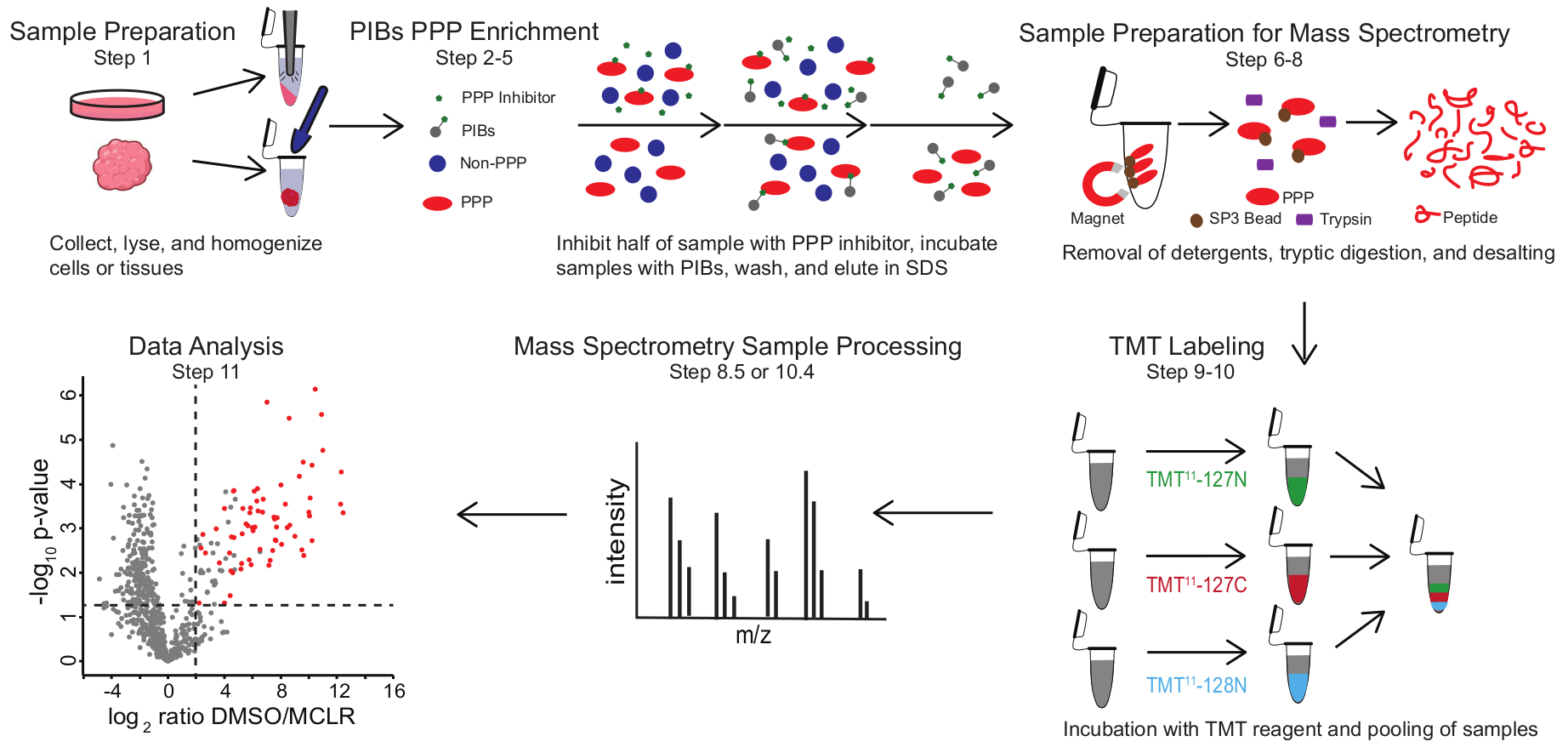
الشكل 1: ملخص مرئي لبروتوكول PIB-MS. في تجربة PIB-MS ، يمكن الحصول على عينات بأشكال مختلفة ، من الخلايا إلى الأورام. يتم جمع العينة وتحليلها وتجانسها قبل إثراء تعادل القوة الشرائية. للإثراء من أجل الشراكات بين القطاعين العام والخاص، يتم احتضان الليزات مع PIBs مع أو بدون مثبط PPP، مثل MCLR. ثم يتم غسل PIBs ، ويتم التخلص من PPPs في ظروف تمسخ. يتم إعداد العينات لتحليل الطيف الكتلي عن طريق إزالة المنظفات من خلال إثراء البروتين SP3 ، والهضم التربتيكي ، وإزالة الملح. يمكن بعد ذلك وضع علامة TMT اختياريا على العينات قبل تحليل مطياف الكتلة. يرجى النقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
يتضمن PIB-MS تحلل وتوضيح الخلايا أو الأنسجة ، وحضانة الليزات باستخدام PIBs ، والاستخلاص ، وتحليل اللوات عبر النشاف الغربي أو النهج القائمة على قياس الطيف الكتلي (الشكل 1). يمكن استخدام إضافة MCLR المجاني كعنصر تحكم لتمييز روابط PIB المحددة عن الجهات الفاعلة غير المحددة. بالنسبة لمعظم التطبيقات ، يمكن استخدام نهج خال من الملصقات لتحديد البروتينات مباشرة في المراوغة. وفي الحالات التي تكون فيها هناك حاجة إلى مزيد من الدقة في التحديد الكمي أو تحديد الأنواع منخفضة الوفرة، يمكن استخدام مزيد من المعالجة باستخدام وسم العلامات الجماعية الترادفية (TMT) لزيادة التغطية وتقليل المدخلات.
Access restricted. Please log in or start a trial to view this content.
Protocol
ملاحظة: يتم توليد PIBs كما هو موضح من قبل Moorhead et al. ، حيث يقترن 1 ملغ من microcystin وحوالي 6 مل من sepharose لتوليد PIBs بقدرة ربط تصل إلى 5 mg / mL17.
1. إعداد العينات
ملاحظة: كمية البدء النموذجية ل PIB-MS هي 1 ملغ من البروتين الكلي لكل حالة. في هذه التجربة ، تم استخدام ما يقرب من 2.5 × 106 خلايا HeLa لاستخراج 1 ملغ من البروتين. يجب إجراء هذا الحساب لكل خط خلية أو نسيج يتم استخدامه في تجربة18. إذا كانت العينة محدودة ولا يمكن الحصول على 1 ملغ ، يمكن تقليل كمية المدخلات مع فقدان طفيف للكشف عن الوحدة الفرعية PPP. بدلا من ذلك ، يمكن استخدام وضع علامات TMT للسماح بخلط جميع الظروف في عينة واحدة ، مما يزيد من حساسية الكشف كما هو موضح في الخطوة 9.
- جمع عينات الأنسجة أو كريات الخلايا. بالنسبة لكريات الخلايا ، قم بجمع الخلايا عن طريق الطرد المركزي عند 277 × g لمدة 2 دقيقة في درجة حرارة الغرفة (RT) ، وإزالة الوسائط ، وغسل الخلايا ب 5 مل من المياه المالحة العازلة بالفوسفات (PBS). يمكن تخزين كريات الخلايا عند -80 درجة مئوية لعدة أشهر.
- تحضير المخزن المؤقت للتحلل (500 mM NaCl ، 50 mM Tris-HCl pH 7.5 ، 0.5٪ Triton X-100 (vol / vol) ، 5 mM beta-glycerophosphoric acid disodium salt pentahydrate ، 1: 500 (vol / vol) protease inhibitor cocktail III) والحفاظ على الثلج. جعل ما يكفي لتحلل وغسل جميع العينات. إذا كنت تبدأ ب 1 ملغ من البروتين لكل حالة ، فقم بعمل حوالي 3 مل من المخزن المؤقت لكل عينة للتحلل والغسيل.
ملاحظة: المخزن المؤقت للتحلل المشار إليه في هذه الخطوة هو محلول منظف خفيف، والذي قد لا يكون كافيا لإذابة الغشاء غير القابل للذوبان أو البروتينات المرتبطة بالهيكل الخلوي. يمكن استكشاف منظفات أخرى لتحسين الذوبان. تم اختبار مجموعة من تركيزات كلوريد الصوديوم وتريتون-X-100 ، ووجد أن التركيزات المذكورة أعلاه هي الأمثل لربط الوحدة الفرعية منخفضة الخلفية وعالية الفوسفاتيز. - أضف مخزن التحلل المبرد إلى العينات. لتحلل 1 ملغ من البروتين ، استخدم 1 مل من المخزن المؤقت. إذا تم تجميد العينة، أضف مخزن التحلل المخزن المؤقت واترك العينة تذوب على الجليد في المخزن المؤقت.
- بالنسبة للخلايا، قم بتجانس العينات عن طريق السونات، مع الحفاظ على الخلايا على الجليد بين البقوليات. سونيك العينات بسعة 15٪ مع ثلاث نبضات 15 ثانية. قد يختلف هذا بناء على السونيكتور المستخدم (انظر جدول المواد).
- بالنسبة للأنسجة، قم بتجانس العينات أولا باستخدام مطحنة أنسجة Dounce لطحن الأنسجة حتى يتم تسييلها قبل الصوتنة كما هو موضح في الخطوة 1.3.1.
- توضيح العينة المتجانسة من الحطام غير القابل للذوبان عن طريق الطرد المركزي عند 21,130 × g لمدة 15 دقيقة عند 4 درجات مئوية. ثم ، دون إزعاج الكريات أو الدهون المشكلة التي تجمعت على جانب الأنابيب ، قم بنقل الليزات إلى أنابيب جديدة. قم بإزالة 100 ميكرولتر من عينة ما قبل التخصيب من المحلول المحلل الموضح إذا رغبت في ذلك وتخزينها عند -20 درجة مئوية. تأكد من الحفاظ على lysates على الجليد.
- حدد إجمالي محتوى البروتين في كل عينة عن طريق إجراء فحص كمي للبروتين ، مثل فحص حمض البيتشينكونينيك (BCA) ، على أليكوت صغير لكل عينة ، وفقا لتعليمات الشركة المصنعة. تأكد من إبقاء الليزات على الجليد أثناء فحص BCA.
- بعد إجراء فحص BCA ، انقل كمية مكافئة من البروتين إلى أنابيب جديدة وقم بتخفيفه باستخدام مخزن التحلل المؤقت لضمان أن كل أنبوب يحتوي على نفس تركيز البروتين (على سبيل المثال ، 1 ملغ / مل). إذا كانت التجربة تتطلب عنصر تحكم في مثبطات تعادل القوة الشرائية، فقم بإعداد اثنين من الأليكوتات لكل عينة تحتوي على نفس محتوى البروتين وانتقل إلى الخطوة 1.7. إذا لم تكن هناك حاجة إلى عناصر تحكم مثبطات PPP، فانتقل إلى الخطوة 2. تأكد من الاحتفاظ بالعينات على الجليد.
ملاحظة: من الأهمية بمكان استخدام تركيزات متساوية من البروتين لكل حالة في تحليل PIB-MS. تستخدم عناصر التحكم في مثبطات PPP للتمييز بين روابط محددة ل PIBs من الخلفية غير المحددة. - بالنسبة للتحكم في مثبطات PPP ، عالج عينة واحدة باستخدام MCLR الحر (1 ميكرومتر) والأخرى بحجم متساو من DMSO كعنصر تحكم. دوامة العينات بلطف واحتضانها على الجليد لمدة 15 دقيقة.
ملاحظة: تمنع معالجة MCLR للليزات ربط الوحدات الفرعية الحفازة PPP ولكن ليس البروتينات التي ترتبط بشكل غير محدد ب PIBs في العينات. كن حذرا عند التعامل مع MCLR لأنه سام. ارجع إلى احتياطات التعامل في جدول المواد.
2. إعداد PIBs
- تحديد كمية PIBs اللازمة للتجربة. بالنسبة ل 1 ملغ من البروتين ، يمكن الحصول على 1-3 ميكروغرام من PPPs والبروتينات المتفاعلة ؛ استخدام ما لا يقل عن 10 ميكرولتر من راتنج PIBs الصلبة لكل عينة لتقليل فقدان الخرزة. قدرة الربط من PIBs هي 3-5 ملغ / مل14,17.
- انقل الكمية المناسبة من PIBs إلى أنبوب 1.5 مل واغسلها 3x مع 0.5 مل من المخزن المؤقت للتحلل عن طريق الدوامة بلطف ثم الطرد المركزي عند 376 × g لمدة 30 ثانية في RT بين الغسيل. تجنب سحب الخرز عند إزالة المخزن المؤقت للتحلل بين الغسيل.
- قم بعمل محلول PIB / المخزن المؤقت بنسبة 50٪ (vol / vol) عن طريق إضافة كمية مناسبة من المخزن المؤقت للتحلل إلى PIBs المغسولة. قم بسحب الماصة بلطف لأعلى ولأسفل وقم بتدوير طرف الماصة في الملاط لإعادة تعليق PIBs.
- انقل 20 ميكرولتر من الملاط إلى أنبوب جديد سعة 1.5 مل يحتوي بالفعل على 0.5 مل من المخزن المؤقت للتحلل. يساعد المخزن المؤقت للتحلل في الأنبوب في طرد الخرز من طرف الماصة. قم بذلك حتى يكون هناك ما يكفي من الأنابيب التي تحتوي على كمية متساوية من PIBs لكل عينة.
- قم بتدوير الأنابيب عند 376 × g لمدة 30 ثانية في RT. تأكد من أن جميع الأنابيب تحتوي على كمية متساوية من راتنج الخرز. تخلص من أي مادة فائقة ، تاركا فقط راتنجا صلبا وبحد أقصى 50 ميكرولتر من المخزن المؤقت للتحلل في كل أنبوب.
3. حضانة PIBs مع lysates
- انقل الليزات من الخطوة 1.6. أو الخطوة 1.7. إلى الأنبوب المسمى بشكل مناسب والذي يحتوي على PIBs من الخطوة 2. قم بتدوير الليزات عند 8 دورات في الدقيقة باستخدام PIBs لمدة 1 ساعة عند 4 درجات مئوية.
4. غسل PIBs
- الطرد المركزي لل PIBs في 376 × g لمدة 30 ثانية عند 4 درجات مئوية لجمع الخرز. قم بإزالة وتجاهل supernatant ، مما يوفر 100 ميكرولتر aliquot لتحليل ما بعد التخصيب ، إذا رغبت في ذلك.
- اغسل PIBs 3x عن طريق إضافة 0.5 مل من المخزن المؤقت للتحلل إلى الخرز ، وعكس الأنابيب (الدوامة غير مستحسن) ، وجمع الخرز عن طريق الطرد المركزي عند 376 × g لمدة 30 ثانية عند 4 درجات مئوية ، وإزالة المخزن المؤقت للتحلل من الخرز المستقر ، مع الحرص على عدم تعطيل حبيبات الخرز.
5. إلغاء الشراكات بين القطاعين العام والخاص من PIBs
- بعد الغسيل النهائي ، قم بإزالة أكبر قدر ممكن من المخزن المؤقت للتحلل دون سحب أي من PIBs. قم بعمل مخزن مؤقت للإزالة يحتوي على 2٪ SDS (vol / vol) وقم بإلغاء PPPs من PIBs عن طريق إضافة حجم كاف من المخزن المؤقت للإزالة ليكون 4x-5x حجم PIBs. على سبيل المثال ، إذا تم استخدام 10 ميكرولتر من PIBs ، فاستخدم 50 ميكرولتر من المخزن المؤقت للاستخراج. احتضن PIBs مع المخزن المؤقت للإزالة عند 65 درجة مئوية لمدة 1 ساعة لإزالة PPPs من PIBs.
- بعد الاستخلاص ، اجمع الإلويات عن طريق الطرد المركزي للأنابيب عند 376 × g لمدة 30 ثانية في RT وسحب اللوات في أنبوب منفصل ، مع الحرص على عدم نقل أي PIBs. استخدم الإيلوات لتحليل اللطخة الغربية أو لتحليلات قياس الطيف الكتلي. يمكن تخزين الشفافيات عند -20 درجة مئوية لمدة تصل إلى عدة أشهر.
- لتجديد PIBs لمزيد من الاستخدام ، احتضن الخرز في 2٪ SDS (vol / vol) ، مع الدوران عند 8 دورة في الدقيقة في RT لمدة 1 ساعة. اغسلها 3x-5x في 25 mM Tris-HCl (درجة الحموضة 7.5) مع الدوران لمدة 30 دقيقة لكل غسلة. بعد كل عمليات الغسيل، قم بتخزين PIBs في مخزن مؤقت للتخزين 25 mM Tris-HCl (الرقم الهيدروجيني 7.5) مع أزيد الصوديوم (0.05٪ وزن/فول).
- تحليل الفتحات عن طريق النشاف الغربي أو قياس الطيف الكتلي. ويرد أدناه وصف لتحليل الطيف الكتلي لمستودعات PIB.
6. إزالة المنظفات
ملاحظة: يمكن استخدام طرق مختلفة لإزالة المنظفات من عينات الإزالة لتحليل التصلب المتعدد. وجدنا أن إعداد العينات المعزز بالطور الصلب (SP3) ، الذي وصفه هيوز وآخرون ، يعمل بشكل جيد19.
- أضف 0.5 ميكرولتر من حبات SP3 إلى 50 ميكرولتر من التوضيح من الخطوة 5.2 أعلاه. استخدم حبات SP3 بنسبة 10: 1 (μg: μg) أو على الأقل 0.5 ميكروغرام / ميكرولتر (محلول المخزون هو 50 ميكروغرام / ميكرولتر). دوامة بلطف الخرز والاستلقاء.
- أضف حجم واحد من الإيثانول بنسبة 100٪ إلى خليط إزالة الخرز (على سبيل المثال ، إذا تم استخدام 50 ميكرولتر من الإيلوثات ، فاستخدم 50 ميكرولتر من الإيثانول بنسبة 100٪). احتضن العينات لمدة 5 دقائق في خلاط حراري مضبوط على الاهتزاز عند 1000 دورة في الدقيقة عند 24 درجة مئوية.
- لجمع كل الخرز ، ضعها في رف أنبوب مغناطيسي. بمجرد جمع الخرز ، تخلص من supernatant واغسل الخرز ب 0.5 مل من الإيثانول 80٪ (vol / vol) 3x. للغسيل ، أعد تعليق الخرز عن طريق الدوامة ، وجمع الخرز عن طريق وضعها في رف الأنبوب المغناطيسي ، وتخلص من supernatant بين الغسيل.
- اغسل الخرز مرة أخرى باستخدام 0.5 مل من الأسيتونيتريل 100٪ (ACN) لإزالة جميع آثار الإيثانول ، وإزالة أكبر قدر ممكن من ACN.
7. هضم البروتينات
- قم بإجراء تخفيف 1:100 من التربسين (التركيز النهائي ل 0.004 ميكروغرام / ميكرولتر) في 166 mM HEPES (الرقم الهيدروجيني 8.5) وأضف 30 ميكرولتر من محلول التربسين هذا إلى كل أنبوب مع الخرز ، مع إعادة التعليق عن طريق الدوامة.
- احتضن خليط حبة SP3-trypsin في الخلاط الحراري عند 1000 دورة في الدقيقة عند 37 درجة مئوية لمدة 5 ساعات أو بين عشية وضحاها عند 30 درجة مئوية. ضع الأنابيب في الرف المغناطيسي لجمع الخرز وإزالة الهضمات إلى أنابيب جديدة.
- للحصول على تحليل خال من الملصقات ، قم بإخماد التفاعل بإضافة 20٪ من حمض ثلاثي فلورو أسيتيك (TFA) (vol / vol) إلى تركيز نهائي قدره 0.2٪ TFA (vol / vol). تحقق من أن الرقم الهيدروجيني لكل عينة يتراوح بين 2-3 باستخدام ورقة الأس الهيدروجيني. إذا لم يكن الأمر كذلك ، أضف أليكوتات إضافية صغيرة بنسبة 20٪ TFA حتى يتم تحقيق ذلك. يجب تحمض العينات بشكل مناسب قبل تحلية المياه. تابع إلى الخطوة 8.
- لوضع علامات TMT على العينات، لا تحمض واستمر في الخطوة 9.
8. تحلية الهضم
- قم بإعداد طرف مرحلة لكل عينة عن طريق تعبئة طرف متوافق مع مذيب MS سعة 200 ميكرولتر مع راتنج C18 كما هو موضح من قبل Rappsilber et al.20. استخدم إبرة حادة النهاية للضغط على قرصين من أقراص المواد C18، مما يضمن بقاء الأقراص في الإبرة. انقل الأقراص إلى الطرف المتوافق مع مذيب MS باستخدام مكبس سلكي رفيع لطرد الأقراص من الإبرة.
ملاحظة: من الأهمية بمكان استخدام نصائح متوافقة مع مذيبات MS من هذه النقطة في البروتوكول لأن نصائح الماصة الأخرى قد تتسرب المواد الكيميائية إلى العينات التي يمكن اكتشافها عبر مطياف الكتلة. - قم بموازنة كل طرف مرحلة مع 30 ميكرولتر من 100٪ MeOH ، ثم مع 30 ميكرولتر من 60٪ MeOH (vol / vol) ، متبوعا ب 30 ميكرولتر من 0.1٪ TFA (vol / vol). ادفع كل حل من خلال طرف المرحلة باستخدام حقنة. قم بتوصيل طرف ماصة بنهاية المحقنة بفيلم شفاف لزيادة الاتصال بين المحقنة وطرف المسرح إذا لزم الأمر. تأكد من عدم السماح أبدا لمادة C18 داخل طرف المسرح بالجفاف.
- أضف هضم الببتيد المحمض من الخطوة 7.3 إلى طرف المرحلة المسمى وادفع من خلال طرف المرحلة باستخدام حقنة ، مع الحرص مرة أخرى على عدم ترك طرف المرحلة يجف تماما.
- اغسل كل عينة 2x ب 30 ميكرولتر من 0.1٪ TFA (vol / vol). قم بالتخلص من الببتيدات من كل طرف مرحلة عن طريق إضافة 30 ميكرولتر من 60٪ MeOH (vol / vol) إلى كل طرف مرحلة وطردها جميعا من الطرف مع ضغط حقنة إلى أنبوب جديد ملصق. هذه هي الخطوة الوحيدة التي يتم فيها تجفيف مادة C18 بالكامل.
- جفف كل عينة عن طريق الطرد المركزي بالتفريغ. يمكن تخزين الببتيدات المجففة عند -20 درجة مئوية لعدة أشهر. العينات جاهزة الآن لتحليل مطياف الكتلة بدون ملصقات. استخدم نصف العينة فقط للتحليل على مطياف الكتلة والنصف الآخر لإعادة الحقن إذا لزم الأمر. ضمان استخدام طريقة مناسبة لمطياف الكتلة للتحليل.
9. وضع العلامات TMT
ملاحظة: يتم استخدام وسم العلامة الترادفية الكتلة لتعدد العينات للتحليل الكمي. قارورة 0.8 ملغ من كاشف TMT كافية لوضع علامات تصل إلى 0.8 ملغ من البروتين21. في تجربة سحب PIB بدءا من 1 ملغ من البروتين ، يتم الحصول على 1-3 ميكروغرام من الوحدات الفرعية للبروتين الفوسفوبروتيني. البروتوكول أدناه هو الأمثل لما يصل إلى 10 ميكروغرام من البروتين.
- إعادة تشكيل قارورة واحدة 0.8 ملغ من كاشف TMT في 80 ميكرولتر من ACN اللامائي.
- قم بتسمية كل عينة من الخطوة 7.4 بتسمية TMT مختلفة لما يصل إلى 18 قناة. تأكد من ملاحظة تسمية TMT التي تتم إضافتها إلى كل عينة. أضف 2 ميكرولتر من كاشف TMT و 2 ميكرولتر من ACN إلى هضم الببتيد ، ودوامة بلطف للخلط ، وأجهزة طرد مركزي عند 376 × g لمدة 30 ثانية في RT لجمع العينة ، واحتضن في RT لمدة 1 ساعة لوضع علامة على العينة.
- لاختبار كفاءة وضع العلامات TMT ، قم بعمل عينة فحص التسمية عن طريق الجمع بين 1 ميكرولتر من كل تفاعل وضع العلامات في أنبوب 0.5 مل يحتوي على 9 ميكرولتر من الماء LC-MS الصف و 1 ميكرولتر من 10٪ هيدروكسيلامين (vol / vol) لإخماد التفاعل. ضع العينات المتبقية غير المروية في ثلاجة -80 درجة مئوية. يمكن تخزين العينات لعدة أيام أثناء تقييم كفاءة وضع العلامات.
- قم بتحمض عينة فحص ملصق TMT عن طريق إضافة 30 ميكرولتر من 0.1٪ TFA (vol / vol). تحقق من أن الرقم الهيدروجيني يتراوح بين 2-3. إذا لم يكن الأمر كذلك ، أضف 20٪ TFA (vol / vol) حتى يتم تحقيق درجة الحموضة هذه. قم بإزالة الملح من عينة فحص الملصق عبر تلميح المرحلة، كما هو موضح في الخطوات من 8.1.إلى 8.5.
- قم بتحليل عينة فحص ملصق TMT على مطياف الكتلة لتقييم كفاءة وضع العلامات. قم بتصفية نتائج البحث إلى معدل اكتشاف خاطئ بنسبة 1٪ (FDR) على مستوى الببتيد وحدد كفاءة وضع العلامات21,22.
- تحتوي الببتيدات التي تحمل علامة TMT بالكامل على كاشف TMT في المحطة N وفي جميع الليسينات. حدد كميا كثافة أيون مراسل TMT وقارن مجموعها الإجمالي عبر جميع القنوات. يتم تصنيف العينة بشكل كاف عندما يتم تصنيف >95٪ من جميع الببتيدات وتكون كثافة المجموع الأيوني لمراسل TMT قابلة للمقارنة
ملاحظة: إلى جانب وضع العلامات غير المكتملة ، يمكن أن تكون الاختلافات في كثافة أيون مراسل TMT المجمعة نتيجة للسحب غير الدقيق لعينة اختبار 1 ميكرولتر. يمكن أن يعكس أيضا ملاحظة بيولوجية حقيقية ، وفي هذه الحالة يجب أن تظهر جميع النسخ المتماثلة نفس السلوك. - قم بإزالة العينات غير المروية من تخزين -80 درجة مئوية وإذابتها. إذا لم يتم تصنيفها بالكامل ، فأضف 1 ميكرولتر من كاشف TMT المناسب إلى العينة كما هو مذكور أعلاه ، واحتضنها لمدة 1 ساعة ، وكرر فحص ملصق TMT. إذا تم تسمية العينات بالكامل، فتابع إلى الخطوة 9.8.
- عند وضع علامة كاملة ، أضف 2 ميكرولتر من 10٪ هيدروكسيلامين (vol / vol) إلى تفاعلات TMT لإخماد وضع العلامات. احتضان العينات في RT لمدة 15 دقيقة. بمجرد إخمادها ، يمكن تخزين العينات عند -80 درجة مئوية لعدة أشهر.
- الجمع بين جميع قنوات TMT المروي وإضافة 2 ميكرولتر من 20٪ TFA (vol / vol) لتحمض التفاعل. تحقق من الرقم الهيدروجيني للتفاعل المشترك ، وتأكد من أنه بين درجة الحموضة 2-3. إذا لم يكن الأمر كذلك ، أضف أليكوتس صغيرة من 20٪ TFA حتى يتم الوصول إلى الرقم الهيدروجيني المطلوب. هذا أمر بالغ الأهمية لتحلية المياه المناسبة.
- قم بإزالة ACN عن طريق الطرد المركزي الفراغي لمدة 30 دقيقة وإزالة الملح من العينة كما هو موضح أدناه.
10. تحلية العينة المدمجة التي تحمل علامة TMT
- استخدم صفيحة تحلية SPE C18 بسعة البروتين المناسبة (2 ملغ من المواد الماصة عادة ما تكون كافية لهذا التطبيق) لإزالة الملح من كاشف TMT المشترك. قم بموازنة البئر مع 200 ميكرولتر من 60٪ MeOH (vol / vol) و 200 μL من 10٪ MeOH / 0.1٪ TFA (vol / vol).
- قم بتحميل الببتيدات المحمضة التي تحمل علامة TMT من الخطوة 9.10. على طبق تحلية المياه. اغسل آبار العينة 2x ب 200 ميكرولتر من 10٪ MeOH/0.1٪ TFA (vol/vol).
- قم بتمييع الببتيدات مع 100 ميكرولتر من 60٪ MeOH (المجلد / المجلد). تجفيف العينات عن طريق الطرد المركزي بالتفريغ. يمكن تخزين العينة المجففة التي تحمل علامة TMT عند -80 درجة مئوية لعدة أشهر.
- لتحليل قياس الطيف الكتلي للعينة المصنفة TMT ، قم بحقن نصف العينة واحفظ النصف الآخر إذا كانت هناك حاجة إلى إعادة الحقن.
ملاحظة: تختلف كميات الحقن في مطياف الكتلة اعتمادا على العمود المحدد وإعداد الأداة ونوع العينة.
11. تحليل البيانات
ملاحظة: تختلف طرق تصفية البيانات وتحليلها وتخرج عن نطاق هذا البروتوكول، ولكن يتم تضمين الملاحظات التالية حول التحليل لتوفير إرشادات خاصة بنوع البيانات الناتجة عن هذا البروتوكول.
- ابحث في بيانات قياس الطيف الكتلي الخام مقابل قاعدة بيانات بروتينات خاصة بالأنواع استنادا إلى أصل خلايا العينة أو الأنسجة المستخدمة. هنا ، تم استخدام المذنب كخوارزمية بحث23.
- قم بتصفية نتائج البحث باستخدام FDR بنسبة 1٪ عن طريق ضبط المعلمات الخاصة بخوارزمية البحث22. للقياس الكمي الخالي من التسميات، استخدم قياسات منطقة الذروة MS1 لتحديد كمية البيانات. بالنسبة للعينات التي تحمل علامة TMT، استخدم كثافة أيون المراسل المشتقة من MSn لتحديد الكمية. للحصول على دلالة إحصائية ، قم بتحليل العينات في ثلاثة أضعاف بيولوجية.
- ومن أجل تحديد الوحدات الفرعية لتعادل القوة الشرائية والجهات الفاعلة البينية الخاصة بها، تأكد من مقارنة العينات البيولوجية الثلاثية للعينات المثبطة للميكروبات المعالجة ب MCLR والعينات المعالجة ب DMSO. لمقارنة PPPome في ظل ظروف مختلفة أو عند العلاج من تعاطي المخدرات ، تأكد من أن ثلاثة أضعاف بيولوجية لكل حالة أو علاج من تعاطي المخدرات قد تم توليدها.
- قم بتصفية البيانات بحيث لا توجد سوى البروتينات التي تحتوي على إجمالي عدد الببتيد >1 في عينتين على الأقل من ثلاث عينات معالجة بالتحكم في DMSO. لا تتنافس منافسة MCLR دائما على جميع الوحدات الفرعية الحفازة الملزمة. أيضا ، قد لا تلتصق بعض الوحدات الفرعية المحفزة PPP على وجه التحديد براتنج السيفاروز. لحساب أي من الاحتمالين أثناء تصفية البروتينات التي ترتبط بشكل غير محدد بالراتنج ، قم بإزالة البروتينات ذات العدد الكلي للببتيد في الحالة المعالجة ب MCLR أعلى من أي وحدة فرعية حفازة PPP.
- استبعاد الملوثات الشائعة، مثل الكيراتين والكولاجين والبروتينات الريبوسومية 40S و60S والبروتينات النووية النووية غير المتجانسة التي ليست وحدات فرعية تعادل القوة الشرائية، من التحليل14.
- استيراد البيانات التي تمت تصفيتها إلى Perseus بالنقر فوق تحميل مصفوفة عامة في قسم التحميل24,25. سجل2 تحويل البيانات عن طريق الانتقال إلى تحويل > الأساسية، وتحديد البيانات، وتحديد وظيفة التحويل، في هذه الحالة log2(x).
- قم بإسناد القيم المفقودة من توزيع عادي بالانتقال إلى الإحالة > استبدال القيم المفقودة من التوزيع العادي، وتحديد البيانات، وتحديد العرض (الافتراضي 0.3) والتحول لأسفل (الافتراضي 1.8) للحساب. قم بإجراء التطبيع الكمي بالانتقال إلى تطبيع > التطبيع الكمي.
- حساب نسب السجل2 وقيم P لاختبار T للطالب للظروف المعنية. أولا، قم بإضافة تعليقات توضيحية إلى البيانات بالانتقال إلى Annot. Rows > Sorts's Categorical Annotation Rows. قم بإجراء اختبار T من خلال الانتقال إلى الاختبارات > الاختبارات المكونة من عينتين ، وتحديد المجموعات المراد مقارنتها ، والاختبار المطلوب إجراؤه ، وطريقة تصحيح اختبار الفرضيات المتعددة كما هو مستخدم في الاقتطاع.
ملاحظة: بالنسبة لتحديد الهوية الجديدة ، يعتبر البروتين بروتينا متفاعلا مع PPP إذا كانت وفرته ذات دلالة إحصائية في حالة MCLR المعالجة مقابل DMSO ، مع تغيير سجل2 أضعاف أكبر من الحد الأدنى لتغيير الطيات لأي وحدة فرعية معروفة مرتبطة تحديدا بتعادل القوة الشرائية.
Access restricted. Please log in or start a trial to view this content.
النتائج

الشكل 2: تحديد روابط PIBs محددة . (أ) يمكن تحليل مجموعة متنوعة من أنواع الأنسجة أو الخلايا عبر PIB-MS. تمت معالجة خلايا HeLa في الثلاثي البيولوجي إما باستخدام DMSO أو MCLR المثبط ل PPP ، وت...
Access restricted. Please log in or start a trial to view this content.
Discussion
PIB-MS هو نهج بروتينات كيميائية يستخدم لتحديد ملامح PPPome كميا من مصادر عينة مختلفة في تحليل واحد. تم القيام بالكثير من العمل باستخدام حبات مثبطات كيناز لدراسة الكينوم وكيف يتغير في السرطان والحالات المرضية الأخرى10،11،12،13. ...
Access restricted. Please log in or start a trial to view this content.
Disclosures
وليس لدى أصحاب البلاغ ما يكشفون عنه ولا تضارب في المصالح.
Acknowledgements
تعترف A.N.K. بالدعم المقدم من NIH R33 CA225458 و R35 GM119455. نشكر مختبري Kettenbach و Gerber على مناقشتهما المفيدة.
Access restricted. Please log in or start a trial to view this content.
Materials
| Name | Company | Catalog Number | Comments |
| Acetonitrile (ACN) | Honeywell | AH015-4 | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Anhydrous Acetonitrile | Sigma-Aldrich | 271004-100ML | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Benchtop centrifuge | Eppendorf | model no. 5424 | |
| Beta-glycerophosphoric acid, disodium salt pentahydrate | Acros Organics | 410991000 | |
| Centrifuge | Eppendorf | model no. 5810 R 15 amp version | |
| Distilled water | |||
| DMSO | Fisher Scientific | BP231-100 | |
| Dounce tissue grinder | Fisherbrand Pellet Pestles | 12-141-363 | |
| Empore solid phase extraction disk, C18 | CDS Analytical | 76333-132 | |
| Eppendorf tubes, 1.5 mL | Eppendorf | 22363204 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Eppendorf tubes, 2 mL | Eppendorf | 22363352 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Extraction plate manifold | Waters | WAT097944 | |
| Falcon tubes, 50 mL | VWR | 21008 | |
| Generic blunt end needle and plunger | |||
| Generic magnetic separation rack | |||
| HEPES | Sigma-Aldrich | H3375 | |
| Hydrogen chloride (HCl) | VWR Chemicals BDH | BDH3028 | CAUTION: HCl is corrosive; wear gloves and work in a chemical fume hood. |
| Hydroxylamine solution 50% (wt/vol) | Sigma-Aldrich | 467804 | |
| Incubator, 65 °C | VWR | model no. 1380FM | |
| Koptec Pure Ethanol, 200 Proof | Decon Labs | V1001 | |
| Methanol for HPLC (MeOH) | Sigma-Aldrich | 34860-4L-R | CAUTION: MeOH is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Microcystin LR (MCLR) | Cayman Chemical | 10007188 | CAUTION: MCLR is toxic; wear gloves when handling and avoid skin contact. |
| PBS, 1× without calcium and magnesium, pH 7.4 ± 0.1 | Corning | 21-040-CV | |
| pH test strips, such as MilliporeSigma MColorpHast pH test strips and indicator papers | Fisher Scientific | M1095310001 | |
| PIBs | For protocol for the generation of PIBs, see Moorhead et al., 2007. | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 | |
| Pipette tips, 10 μL | Eppendorf | 22491504 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 1000 μL | Eppendorf | 22491555 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 200 μL | Eppendorf | 22491539 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| plastic syringe, 10 mL | BD | 309604 | |
| Protease inhibitor cocktail III | Research Products International | P50700-1 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer, Oribtrap Fusion, Orbitrap Fusion Lumos, or Orbitrap Eclipse Tribrid Mass Spectrometer | Thermo Scientific | ||
| Refrigerated benchtop centrifuge | Eppendorf | model no. 5424 R | |
| Rotator (Labquake Shaker Rotisserie) | Thermo Scientific | 13-687-12Q | 8 rpm rotation |
| Sample collection plate, 96- well, 1 mL | Waters | WAT058957 | |
| SDS | Fisher Scientific | BP1311-1 | |
| Sequencing grade modified trypsin | Promega | V511C | |
| Sodium azide | EMD Chemicals | SX0299-1 | CAUTION: Sodium azide is explosive and toxic; wear gloves, work in a chemical fume hood and avoid contact with metals. |
| Sodium chloride (NaCl) | Fisher Chemical | S27110 | |
| Sonicator (Branson digital sonifier) | model no. SFX 250 | ||
| SPE C18 desalting plate | Waters | 186001828BA | |
| SpeedBeads magnetic carboxylate modified particles (SP3 beads) | Cytiva | 6.51521E+13 | |
| Thermomixer | Eppendorf | model no. 5350 | |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent, 3 × 0.8 mg per tag | ThermoFisher | A37725 | |
| Trifluoroacetic acid (TFA) | Honeywell | T6508-25ML | CAUTION: TFA is corrosive and will irritate skin on contact. Wear gloves and eye protection, and work in a chemical fume hood. |
| Tris Base | Research Products International | T60040 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Vacuum centrifuge and vapor trap | Thermo Scientific | model nos. SpeedVac SPD120 and RVT5105 | |
| Vortexer (Vortex-Genie 2) | Scientific Industries | ||
| Water LC-MS | Honeywell | LC365-4 |
References
- Nilsson, J. Protein phosphatases in the regulation of mitosis. Journal of Cell Biology. 218 (2), 395-409 (2019).
- Brautigan, D. L. Protein Ser/Thr phosphatases--the ugly ducklings of cell signalling. The FEBS Journal. 280 (2), 324-345 (2013).
- Brautigan, D. L., Shenolikar, S. Protein serine/threonine phosphatases: keys to unlocking regulators and substrates. Annual Review of Biochemistry. 87, 921-964 (2018).
- Janssens, V., Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochemical Journal. 353, 417-439 (2001).
- Virshup, D. M., Shenolikar, S. From promiscuity to precision: protein phosphatases get a makeover. Molecular Cell. 33 (5), 537-545 (2009).
- Bollen, M., Peti, W., Ragusa, M. J., Beullens, M. The extended PP1 toolkit: designed to create specificity. Trends in Biochemical Sciences. 35 (8), 450-458 (2010).
- Qian, J., Winkler, C., Bollen, M. 4D-networking by mitotic phosphatases. Current Opinion in Cell Biology. 25 (6), 697-703 (2013).
- Heroes, E., et al. The PP1 binding code: a molecular-lego strategy that governs specificity. The FEBS Journal. 280 (2), 584-595 (2013).
- Eichhorn, P. J., Creyghton, M. P., Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et Biophysica Acta. 1795 (1), 1-15 (2009).
- Bantscheff, M., et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature Biotechnology. 25 (9), 1035-1044 (2007).
- Klaeger, S., et al. Chemical proteomics reveals ferrochelatase as a common off-target of kinase inhibitors. ACS Chemical Biology. 11 (5), 1245-1254 (2016).
- Duncan, J. S., et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 149 (2), 307-321 (2012).
- Cooper, M. J., et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE. 8 (6), 66755(2013).
- Lyons, S. P., et al. A quantitative chemical proteomic strategy for profiling phosphoprotein phosphatases from yeast to humans. Molecular and Cellular Proteomics. 17 (12), 2448-2461 (2018).
- Nasa, I., et al. Quantitative kinase and phosphatase profiling reveal that CDK1 phosphorylates PP2Ac to promote mitotic entry. Science Signaling. 13 (648), (2020).
- Swingle, M., Ni, L., Honkanen, R. E. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods in Molecular Biology. 365, 23-38 (2007).
- Moorhead, G. B. G., Haystead, T. A. J., MacKintosh, C. Synthesis and use of the protein phosphatase affinity matrices microcystin-sepharose and microcystin-biotin-sepharose. Methods in Molecular Biology. 365, 39-45 (2007).
- Brauer, B. L., Wiredu, K., Mitchell, S., Moorhead, G. B., Gerber, S. A., Kettenbach, A. N. Affinity-based profiling of endogenous phosphoprotein phosphatases by mass spectrometry. Nature Protocols. 16 (10), 4919-4943 (2021).
- Hughes, C. S., Moggridge, S., Müller, T., Sorensen, P. H., Morin, G. B., Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Zecha, J., et al. TMT labeling for the masses: A robust and cost-efficient, in-solution labeling approach. Molecular and Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Elias, J. E., Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 4 (3), 207-214 (2007).
- Eng, J. K., Jahan, T. A., Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 13 (1), 22-24 (2013).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Yu, S. H., Ferretti, D., Schessner, J. P., Rudolph, J. D., Borner, G. H. H., Cox, J. Expanding the Perseus software for omics data analysis With custom plugins. Current Protocols in Bioinformatics. 71 (1), 1-29 (2020).
Access restricted. Please log in or start a trial to view this content.
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved