
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Масс-спектрометрический подход к идентификации фосфопротеинфосфатаз и их интеракторов
В этой статье
Резюме
Здесь мы представляем протокол обогащения эндогенных фосфопротеинфосфатаз и их взаимодействующих белков из клеток и тканей, а также их идентификацию и количественную оценку с помощью протеомики на основе масс-спектрометрии.
Аннотация
Большинство клеточных процессов регулируются динамическим фосфорилированием белка. Более трех четвертей белков фосфорилируются, а фосфопротеинфосфатазы (ППС) координируют более 90% всего клеточного серо/треонина дефосфорилирования. Дерегуляция фосфорилирования белка была вовлечена в патофизиологию различных заболеваний, включая рак и нейродегенерацию. Несмотря на широкую активность, молекулярные механизмы, контролирующие ПГЧС, и механизмы, контролируемые ПГЧС, плохо характеризуются. Здесь описан протеомный подход, называемый шариками ингибитора фосфатазы и масс-спектрометрией (PIB-MS), для идентификации и количественной оценки ППС, их посттрансляционных модификаций и их интеракторов всего за 12 ч с использованием любой клеточной линии или ткани. PIB-MS использует неселективный ингибитор PPP, микроцистин-LR (MCLR), иммобилизованный на шариках сефарозы для захвата и обогащения эндогенных PPP и связанных с ними белков (называемых PPPome). Этот метод не требует экзогенной экспрессии помеченных версий PPP или использования специфических антител. PIB-MS предлагает инновационный способ изучения эволюционно сохраненных ГЧП и расширения нашего нынешнего понимания сигнализации дефосфорилирования.
Введение
Фосфорилирование белка контролирует большинство клеточных процессов, включая, но не ограничиваясь, реакцией на повреждение ДНК, передачей сигналов фактора роста и прохождением через митоз 1,2,3. В клетках млекопитающих большинство белков фосфорилируются в одном или нескольких остатках серина, треонина или тирозина в какой-то момент времени, причем фосфосерины и фосфотреонины составляют примерно 98% всех участков фосфорилирования 2,3. Хотя киназы широко изучаются в клеточной сигнализации, роль ПГЧС в регуляции динамических клеточных процессов все еще вырисовывается.
Динамика фосфорилирования контролируется динамическим взаимодействием между киназами и фосфатазами. В клетках млекопитающих насчитывается более 400 протеинкиназ, которые катализируют фосфорилирование серина/треонина. Более 90% этих участков дефосфорилируются фосфопротеинфосфатазами (ППС), небольшим семейством ферментов, которое состоит из PP1, PP2A, PP2B, PP4-7, PPT и PPZ 2,3. PP1 и PP2A ответственны за большую часть дефосфосерина и фосфотреонина в клетке 2,3,4. Заметная разница в количестве между киназами и фосфатазами и отсутствие специфичности каталитических субъединиц ППС in vitro привели к убеждению, что киназы являются основным детерминантом фосфорилирования 2,3. Однако многочисленные исследования показали, что фосфатазы устанавливают субстратную специфичность за счет образования многомерных голоферментов 5,6,7,8,9. Например, PP1 представляет собой гетеродимер, который состоит из каталитической субъединицы и, в данный момент времени, одной из более чем 150 регуляторных субъединиц 6,7,8. И наоборот, PP2A представляет собой гетеротример, который образован из строительных лесов (A), регулятора (B) и каталитической (C) субъединицы 2,3,9. Существует четыре различных семейства регуляторных субъединиц PP2A (B55, B56, PR72 и стриатин), каждое из которых имеет несколько генов, вариантов сращивания и паттернов локализации 2,3,9. Многомерный характер ПГЧС заполняет пробел в количестве киназ и каталитических субъединиц ППС. Однако это создает аналитические проблемы для изучения сигнализации ГЧП. Чтобы всесторонне проанализировать передачу сигналов ППС, крайне важно исследовать различные голоферменты в клетке или ткани. Большие успехи были достигнуты в изучении человеческого кинома благодаря использованию шариков ингибитора киназы, называемых бусинами мультиплексных ингибиторов или кинобеодами, химической протеомной стратегии, в которой ингибиторы киназы иммобилизуются на шариках, а масс-спектрометрия используется для идентификации обогащенных киназ и их интеракторов 10,11,12,13.
Мы установили аналогичный подход к изучению биологии ГЧП. Этот метод включает в себя захват аффинности каталитических субъединиц ППС с использованием шариков с иммобилизованным, неселективным ингибитором ППС, называемым микроцистином-LR (MCLR), называемым шариками ингибитора фосфатазы (PIBs)14,15. В отличие от других способов, требующих эндогенной маркировки или экспрессии экзогенных субъединиц ППС, которые могут изменять активность или локализацию белка, PIB-MS позволяет обогащать эндогенные каталитические субъединицы ППС, связанные с ними регуляторные и каркасные субъединицы, а также взаимодействующие белки (называемые PPPome) из клеток и тканей в данный момент времени или при определенных условиях обработки. MCLR ингибирует PP1, PP2A, PP4-6, PPT и PPZ в наномолярных концентрациях, что делает PIB высокоэффективными при обогащении PPPome16. Этот метод может быть масштабирован для использования на любом исходном материале от клеток до клинических образцов. Здесь мы подробно описываем использование PIB и масс-спектрометрии (PIB-MS) для эффективного захвата, идентификации и количественной оценки эндогенного PPPome и его модифицирующих состояний.

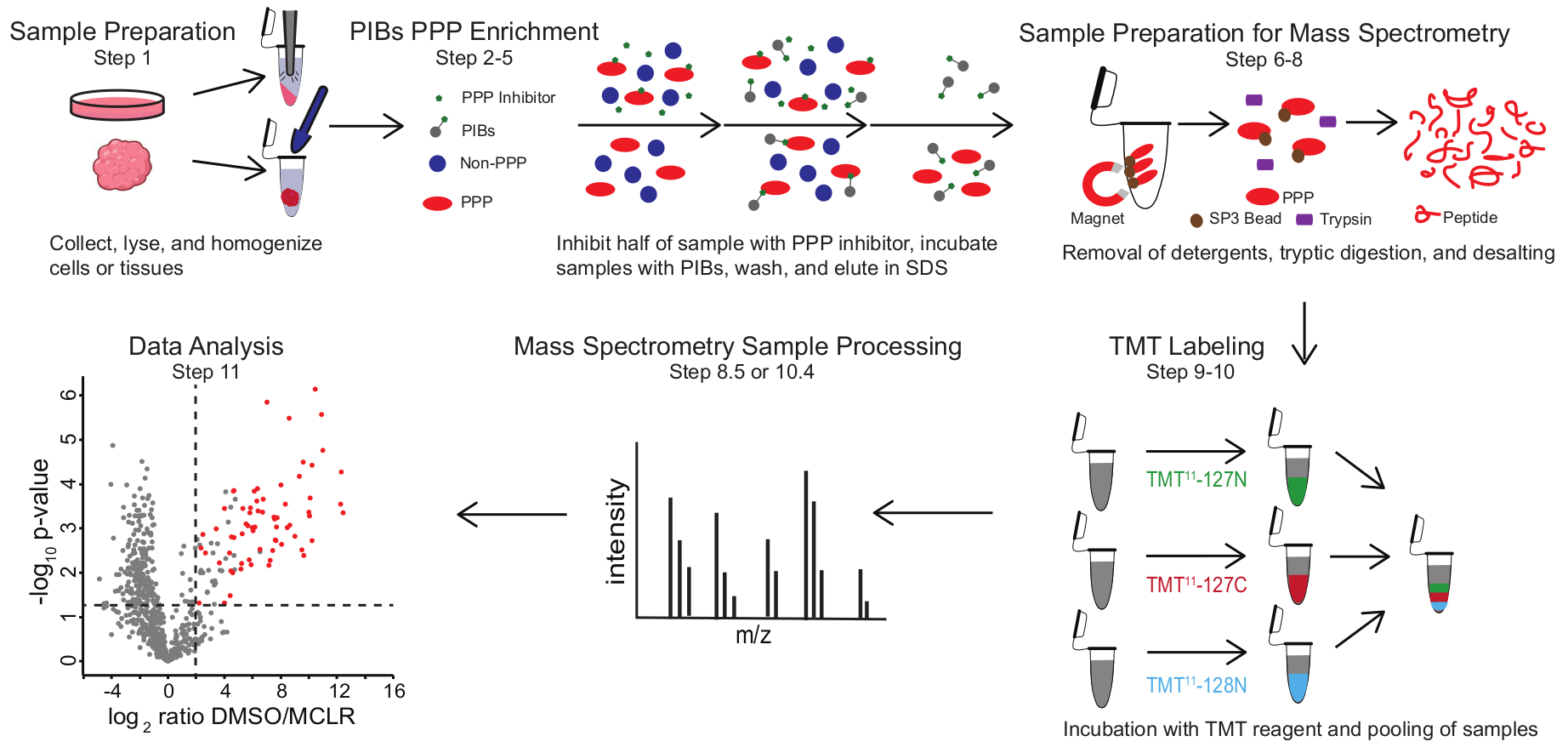
Рисунок 1: Визуальная сводка протокола PIB-MS. В эксперименте PIB-MS образцы могут быть получены в различных формах, от клеток до опухолей. Образец собирается, лизируется и гомогенизируется перед обогащением ППС. Для обогащения для ПГЧС лизат инкубируют с ПИБ с ингибитором ППС или без него, таким как MCLR. Затем ПИБ промывают, а ПГЧС элюируют в условиях денатурации. Образцы подготавливаются для масс-спектрометрического анализа путем удаления моющих средств путем обогащения белка SP3, триптического сбраживания и обессоливания. Затем образцы могут быть опционально помечены TMT перед масс-спектрометрическим анализом. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
PIB-MS включает лизис и осветление клеток или тканей, инкубацию лизата с PIB, элюирование и анализ элюата с помощью подходов, основанных на вестерн-блоттинге или масс-спектрометрии (рисунок 1). Добавление свободного MCLR может быть использовано в качестве элемента управления для различения конкретных связующих PIB от неспецифических интеракторов. Для большинства применений подход без маркировки может быть использован для непосредственной идентификации белков в элюатах. В тех случаях, когда требуется более высокая точность количественной оценки или идентификация видов с низкой численностью, для увеличения охвата и уменьшения вводимых ресурсов может использоваться дальнейшая обработка с помощью маркировки тандемной массы (ТМТ).
Access restricted. Please log in or start a trial to view this content.
протокол
ПРИМЕЧАНИЕ: Генерация PIB осуществляется так, как описано Moorhead et al., где 1 мг микроцистина и около 6 мл сефарозы соединены с образованием PIB с связывающей способностью до 5 мг/мл17.
1. Пробоподготовка
ПРИМЕЧАНИЕ: Типичное начальное количество для PIB-MS составляет 1 мг общего белка на состояние. Для этого эксперимента примерно 2,5 х 106 клеток HeLa использовали для извлечения 1 мг белка. Этот расчет должен быть выполнен для каждой клеточной линии или ткани, используемой в эксперименте18. Если образец ограничен и 1 мг не может быть получен, количество входных данных может быть уменьшено с незначительной потерей обнаружения субъединицы ППС. Альтернативно, маркировка TMT может быть использована для обеспечения смешивания всех условий в одном образце, повышая чувствительность обнаружения, как показано на этапе 9.
- Соберите образцы тканей или гранулы клеток. Для клеточных гранул собирайте клетки центрифугированием при 277 х г в течение 2 мин при комнатной температуре (RT), удаляйте среду и промывайте клетки 5 мл фосфатно-буферного физиологического раствора (PBS). Клеточные гранулы могут храниться при -80 °C в течение нескольких месяцев.
- Готовят лизизный буфер (500 мМ NaCl, 50 мМ Tris-HCl pH 7,5, 0,5% Тритон X-100 (об/об), 5 мМ бета-глицерофосфорной кислоты динатриевой соли пентагидрат, 1:500 (об/об) ингибитор протеазы коктейль III) и держат на льду. Сделайте достаточно, чтобы лизировать и промыть все образцы. Если вы начинаете с 1 мг белка на состояние, сделайте около 3 мл буфера на образец для лизиса и промывки.
ПРИМЕЧАНИЕ: Буфер лизиса, отмеченный на этом этапе, представляет собой мягкий моющий раствор, которого может быть недостаточно для солюбилизации нерастворимой мембраны или цитоскелет-ассоциированных белков. Другие моющие средства могут быть изучены для улучшения солюбилизации. Был протестирован диапазон концентраций NaCl и Triton-X-100, и вышеуказанные концентрации были признаны оптимальными для связывания субъединиц с низким фоном и высоким содержанием фосфатазы. - Добавьте к образцам буфер охлажденного лизиса. Для лизиса 1 мг белка используют 1 мл буфера. Если образец заморожен, добавьте буфер лизиса и дайте образцу оттаять на льду в буфере.
- Для клеток гомогенизируйте образцы путем сонификации, удерживая клетки на льду между импульсами. Обработка образцов ультразвуком с амплитудой 15% тремя импульсами по 15 с. Это может варьироваться в зависимости от используемого ультразвукового аппарата (см. Таблицу материалов).
- Для тканей сначала гомогенизируйте образцы с помощью измельчителя ткани Dounce, чтобы измельчить ткань до сжижения перед обработкой ультразвуком, как описано в Шаге 1.3.1.
- Осветляют гомогенизированный образец нерастворимого мусора центрифугированием при 21 130 х г в течение 15 мин при 4°С. Затем, не нарушая образовавшиеся гранулы или липиды, которые собрались на стороне трубок, переносят лизаты на новые трубки. При желании удалите 100 мкл образца предварительного обогащения осветленного лизата и храните при -20 °C. Обязательно держите лизаты на льду.
- Определите общее содержание белка в каждом образце, выполнив количественный анализ белка, такой как анализ бицинхониновой кислоты (BCA), на небольшой аликвоте каждого образца в соответствии с инструкциями производителя. Убедитесь, что лизаты хранятся на льду во время анализа BCA.
- После выполнения анализа BCA перенесите эквивалентное количество белка в новые пробирки и разбавьте буфером лизиса, чтобы убедиться, что каждая трубка имеет одинаковую концентрацию белка (например, 1 мг / мл). Если эксперимент требует контроля ингибитора ППС, подготовьте две аликвоты каждого образца, которые имеют одинаковое содержание белка, и перейдите к шагу 1.7. Если контроль ингибитора ППС не требуется, перейдите к шагу 2. Убедитесь, что образцы хранятся на льду.
ПРИМЕЧАНИЕ: Крайне важно, чтобы в анализе PIB-MS использовались равные концентрации белка для каждого условия. Контроль ингибиторов ППС используется для различения специфических связующих с PIB от неспецифического фона. - Для контроля ингибитора ППС обработайте один образец свободным MCLR (1 мкМ), а другой - равным объемом DMSO в качестве контроля. Аккуратно вращайте образцы и высиживайте их на льду в течение 15 минут.
ПРИМЕЧАНИЕ: McLR-обработка лизатов блокирует связывание каталитических субъединиц ППС, но не белков, которые связываются неспецифически с ПИБ в образцах. Будьте осторожны при обращении с MCLR, так как он токсичен. Обратитесь к мерам предосторожности при обращении в Таблице материалов.
2. Подготовка PIB
- Определите количество PIB, необходимое для эксперимента. На 1 мг белка можно получить 1-3 мкг ППС и взаимодействующих белков; использовать не менее 10 мкл твердой смолы PIB на образец для минимизации потерь шарика. Связующая способность ПИБ составляет 3-5 мг/мл14,17.
- Переложите соответствующее количество PIB в трубку объемом 1,5 мл и промыть их в 3 раза 0,5 мл лизисного буфера путем осторожного вихрения, а затем центрифугирования при 376 х г в течение 30 с при РТ между промывками. Избегайте пипетки бусин при удалении буфера лизиса между промывками.
- Сделайте 50% PIB/буферный раствор (об/об), добавив соответствующее количество лизисного буфера к промытым PIB. Осторожно пипетируйте вверх и вниз и закрутите наконечник пипетки в навозной жиже, чтобы повторно суспендировать PIB.
- Переложите 20 мкл суспензии в новую трубку объемом 1,5 мл, уже содержащую 0,5 мл лизисного буфера. Буфер лизиса в трубке помогает в изгнании шариков из наконечника пипетки. Делайте это до тех пор, пока не будет достаточно пробирок, содержащих равное количество PIB для каждого образца.
- Вращайте трубки при 376 х г в течение 30 с при RT. Убедитесь, что все трубки содержат равное количество бисерной смолы. Отбросьте любой супернатант, оставив только твердую смолу и максимум 50 мкл лизисного буфера в каждой пробирке.
3. Инкубация ПИБ с лизатами
- Перенесите лизаты из Шага 1.6. или Шаг 1.7. в надлежащим образом маркированную тубу, содержащую ПИБ из этапа 2. Вращайте лизат со скоростью 8 об/мин с PIB в течение 1 ч при 4 °C.
4. Мойка ПИБов
- Центрифугируйте PIB при 376 x g в течение 30 с при 4 °C для сбора шариков. Удалите и выбросьте супернатант, сохранив при желании 100 мкл аликвоту для анализа после обогащения.
- Промывайте PIBs 3x, добавляя 0,5 мл буфера лизиса в бусины, переворачивая трубки (вихрь не рекомендуется), собирая шарики центрифугированием при 376 x g в течение 30 с при 4 °C и удаляя буфер лизиса из осевших шариков, стараясь не нарушить гранулу бисера.
5. Элюирование ГЧП из ПИБ
- После окончательной промывки удалите как можно больше буфера лизиса без пипетки каких-либо плит. Создать буфер элюирования, содержащий 2% SDS (об/об) и элюировать PPP из PIB, добавив достаточный объем буфера элюирования, чтобы он был в 4-5 раз больше объема PIB. Например, если используется 10 мкл ПИБ, используйте 50 мкл буфера элюирования. Инкубировать ПИБ с буфером элюирования при 65°С в течение 1 ч для элюирования ППС из ПИБ.
- После элюирования собирают элюат путем центрифугирования трубок при 376 х г в течение 30 с при РТ и пипетирования элюата в отдельную трубку, стараясь не переносить какие-либо ПИБ. Используйте элюат для анализа вестерн-блот или для масс-спектрометрического анализа. Элюаты могут храниться при -20 °C до нескольких месяцев.
- Для регенерации ПИБ для дальнейшего использования инкубируют шарики в 2% SDS (об/об), вращаясь со скоростью 8 об/мин при RT в течение 1 ч. Мойте их 3x-5x в 25 мМ Tris-HCl (pH 7,5) с вращением в течение 30 мин на стирку. После всех промывок храните PIB в буфере хранения 25 мМ Tris-HCl (pH 7,5) с азидом натрия (0,05% мас./об.).
- Анализ элюатов с помощью вестерн-блоттинга или масс-спектрометрии. Масс-спектрометрический анализ элюатов PIB описан ниже.
6. Удаление моющих средств
ПРИМЕЧАНИЕ: Различные подходы могут быть использованы для удаления моющего средства из образцов элюата для анализа РС. Мы обнаружили, что однокорпусковая, твердофазная усиленная пробоподготовка (SP3), описанная Hughes et al., хорошо работает19.
- Добавьте 0,5 мкл шариков SP3 к 50 мкл элюата на этапе 5.2 выше. Используйте шарики SP3 в соотношении 10:1 (мкг:мкг) или не менее 0,5 мкг/мкл (стандартный раствор составляет 50 мкг/мкл). Аккуратно вихрьте бусины и элюируйте.
- Добавьте один элюционный объем 100% этанола в смесь шарико-элюат (например, если использовалось 50 мкл элюата, используйте 50 мкл 100% этанола). Инкубируйте образцы в течение 5 мин в термомикшере, настроенном на встряхивание при 1000 об/мин при 24 °C.
- Чтобы собрать все бусины, поместите их в стойку с магнитной трубкой. После того, как бусины соберутся, выбросьте супернатант и вымойте бусины 0,5 мл 80% этанола (об/об) 3 раза. Для стирки повторно суспендируйте бусины путем вихря, соберите бусины, поместив их в стойку с магнитной трубкой, и выбросьте супернатант между стирками.
- Вымойте шарики еще раз 0,5 мл 100% ацетонитрила (ACN), чтобы удалить все следы этанола, удалив как можно больше ACN.
7. Переваривание белков
- Сделайте разведение трипсина 1:100 (конечная концентрация 0,004 мкг/мкл) в 166 мМ HEPES (рН 8,5) и добавьте 30 мкл этого раствора трипсина в каждую пробирку с шариками, повторно используя путем вихря.
- Инкубировать смесь ШАРИКОВ SP3-трипсина в термомиксоре при 1000 об/мин при 37 °C в течение 5 ч или на ночь при 30 °C. Поместите трубки в магнитную стойку, чтобы собрать бусины и удалить дайджесты в новые трубки.
- Для анализа без маркировки гасите реакцию, добавляя 20% трифторуксусной кислоты (TFA) (об/об) к конечной концентрации 0,2% ТФА (об/об). Проверьте, что рН каждого образца составляет от 2 до 3 с помощью рН-бумаги. Если нет, добавьте небольшие дополнительные аликвоты в размере 20% TFA, пока это не будет достигнуто. Образцы должны быть соответствующим образом подкислены перед обессоливанием. Перейдите к шагу 8.
- Для маркировки образцов TMT не подкисляйте и переходите к этапу 9.
8. Обессоливание дайджеста
- Подготовьте наконечник ступени для каждого образца, упаковав 200 мкл 200 мкл совместимого с растворителем наконечника со смолой C18, как описано в Rappsilber et al.20. Используйте тупоконечную иглу для прессования двух дисков материала C18, гарантируя, что диски останутся в игле. Переложите диски в наконечник, совместимый с растворителем MS, используя тонкий плунжер, чтобы извлечь диски из иглы.
ПРИМЕЧАНИЕ: С этого момента в протоколе критически важно использовать наконечники, совместимые с растворителями MS, поскольку другие наконечники пипеток могут выщелачивать химические вещества в образцы, которые могут быть обнаружены с помощью масс-спектрометрии. - Уравновешивайте каждый наконечник ступени 30 мкл 100% MeOH, затем 30 мкл 60% MeOH (об/об), а затем 30 мкл 0,1% TFA (об/об). Протолкните каждый раствор через наконечник сцены шприцем. Прикрепите наконечник пипетки к концу шприца прозрачной пленкой, чтобы при необходимости увеличить контакт между шприцем и наконечником сцены. Никогда не позволяйте материалу C18 внутри кончика сцены высохнуть.
- Добавьте подкисленный пептидный переваритель со стадии 7.3 к меченому наконечнику ступени и протолкните наконечник ступени шприцем, снова стараясь не дать кончику ступени полностью высохнуть.
- Промывайте каждый образец в 2 раза с 30 мкл 0,1% TFA (об/об). Элюируйте пептиды из каждого наконечника ступени, добавляя 30 мкл 60% MeOH (об/об) к каждому наконечнику ступени и выталкивая все это из наконечника со шприцем давления в новую, меченую трубку. Это единственный этап, на котором материал C18 полностью высушивается.
- Высушите каждый образец методом вакуумного центрифугирования. Высушенные пептиды могут храниться при -20°C в течение нескольких месяцев. Образцы теперь готовы к масс-спектрометрическому анализу без меток. Используйте только половину образца для анализа на масс-спектрометре, а другую половину для повторной закачки, если это необходимо. Обеспечить использование соответствующего метода масс-спектрометра для анализа.
9. Маркировка ТМТ
ПРИМЕЧАНИЕ: Маркировка тандемных массовых меток используется для мультиплексирования образцов для количественного анализа. Флакона с реагентом ТМТ 0,8 мг достаточно для маркировки до 0,8 мг белка21. В эксперименте по вытягиванию PIB, начиная с 1 мг белка, получают 1-3 мкг субъединиц фосфопротеина. Приведенный ниже протокол оптимален для 10 мкг белка.
- Восстановите один флакон 0,8 мг реагента ТМТ в 80 мкл безводного АКН.
- Пометьте каждый образец из шага 7.4 другой меткой TMT для 18 каналов. Обязательно обратите внимание, какая метка TMT добавляется к каждому образцу. Добавьте 2 мкл реагента TMT и 2 мкл ACN в пептидный реактор, осторожно вихрь для смешивания, центрифугу при 376 х г в течение 30 с на RT для сбора образца и инкубируйте на RT в течение 1 ч, чтобы маркировать образец.
- Чтобы проверить эффективность маркировки TMT, сделайте образец для проверки этикетки, объединив 1 мкл каждой реакции маркировки в пробирке объемом 0,5 мл, содержащей 9 мкл воды класса LC-MS и 1 мкл 10% гидроксиламина (об/об) для гашения реакции. Поместите оставшиеся невыжатые маркированные образцы в морозильную камеру при температуре -80 °C. Образцы могут храниться в течение нескольких дней, пока оценивается эффективность маркировки.
- Подкислить контрольный образец этикетки ТМТ, добавив 30 мкл 0,1% ТФА (об/об). Убедитесь, что рН находится в пределах 2-3. Если нет, добавьте 20% TFA (об/об) до тех пор, пока этот рН не будет достигнут. Опресните контрольный образец этикетки с помощью опрокидывания ступени, как описано в шагах 8.1.-8.5.
- Проанализируйте контрольный образец этикетки TMT на масс-спектрометре для оценки эффективности маркировки. Отфильтруйте результаты поиска до 1% ложного обнаружения (FDR) на пептидном уровне и определите эффективность маркировки21,22.
- Полностью меченые ТМТ пептиды имеют реагент ТМТ на N-конце и на всех лизинах. Количественно оцените интенсивность ионов репортера TMT и сравните их общую сумму по всем каналам. Образец достаточно маркируется, когда мечено >95% всех пептидов, а интенсивность суммы репортерных ионов TMT сопоставима
ПРИМЕЧАНИЕ: Помимо неполной маркировки, различия в суммарной интенсивности ионов TMT могут быть результатом неточного пипетирования испытуемого образца 1 мкл. Это также может отражать истинное биологическое наблюдение, и в этом случае все реплики должны демонстрировать одинаковое поведение. - Извлеките неугашенные образцы из хранилища при температуре -80 °C и разморозьте их. Если они не полностью маркированы, добавьте 1 мкл соответствующего реагента TMT к образцу, как указано выше, инкубируйте в течение 1 ч и повторите проверку этикетки TMT. Если образцы полностью помечены, перейдите к шагу 9.8.
- После полной маркировки добавьте 2 мкл 10% гидроксиламина (об/об) в реакции ТМТ для гашения маркировки. Инкубируйте образцы на RT в течение 15 минут. После закалки образцы могут храниться при -80 °C в течение нескольких месяцев.
- Смешайте все закаленные каналы ТМТ и добавьте 2 мкл 20% ТФА (об/об) для подкисления реакции. Проверьте pH комбинированной реакции, убедившись, что он находится между pH 2-3. Если нет, добавьте небольшие аликвоты 20% TFA до тех пор, пока не будет достигнут желаемый рН. Это имеет решающее значение для правильного обессоливания.
- Удалите ACN методом вакуумного центрифугирования в течение 30 мин и опресните образец, как описано ниже.
10. Обессоливание комбинированного образца, меченного ТМТ
- Используйте обессоливающую пластину SPE C18 с соответствующей белковой емкостью (2 мг сорбента обычно достаточно для этого применения) для обессоливания комбинированного реагента TMT. Уравновешивайте скважину с 200 мкл 60% MeOH (об/об) и 200 мкл 10% MeOH/0,1% TFA (об/об).
- Загрузите подкисленные пептиды, меченые TMT, с шага 9.10. на обессоливающую пластину. Промывайте пробные колодцы 2x с 200 мкл 10% MeOH/0,1% TFA (об/об).
- Элюируют пептиды со 100 мкл 60% MeOH (об/об). Высушите образцы методом вакуумного центрифугирования. Высушенный образец, меченый TMT, может храниться при температуре -80 °C в течение нескольких месяцев.
- Для масс-спектрометрического анализа образца, меченого ТМТ, вводят половину образца и сохраняют другую половину, если требуется повторная инъекция.
ПРИМЕЧАНИЕ: Количество инъекций на масс-спектрометрии будет варьироваться в зависимости от конкретной колонны, настроек прибора и типа образца.
11. Анализ данных
ПРИМЕЧАНИЕ: Методы фильтрации и анализа данных различаются и выходят за рамки настоящего протокола, но следующие примечания по анализу включены в качестве руководства, конкретного для типа данных, полученных в результате этого протокола.
- Поиск необработанных данных масс-спектрометрии по видоспецифической базе данных протеомов на основе происхождения используемых образцов клеток или тканей. Здесь комета использовалась в качестве алгоритма поиска23.
- Отфильтруйте результаты поиска с помощью 1% FDR, настроив параметры, специфичные для алгоритма поиска22. Для количественной оценки без маркировки используйте измерения пиковой области MS1 для количественной оценки данных. Для образцов, меченных TMT, используйте для количественной оценки интенсивность репортерных ионов, полученные из MSn. Для статистической значимости проанализируйте образцы в биологических трипликатах.
- Чтобы de novo идентифицировать субъединицы ППС и их интеракторы, убедитесь, что сравниваются тройные биологические образцы образцов, ингибированных MCLR, и образцов, обработанных ДМСО. Чтобы сравнить PPPome в разных условиях или при медикаментозном лечении, убедитесь, что были получены биологические трипликаты каждого состояния или медикаментозного лечения.
- Отфильтруйте данные таким образом, чтобы присутствовали только белки с общим количеством пептидов >1 по меньшей мере в двух из трех образцов, обработанных контролем ДМСО. Конкуренция MCLR не всегда конкурирует со всеми каталитическими субъединицами. Кроме того, некоторые каталитические субъединицы ППС могут неспецифически прилипать к сефарозной смоле. Чтобы учесть любую возможность при фильтрации белков, которые неспецифически связываются со смолой, удалите белки с общим количеством пептидов в состоянии, обработанном MCLR, выше, чем для любой каталитической субъединицы PPP.
- Исключить из анализа14 распространенные загрязняющие вещества, такие как кератин, коллаген, рибосомные белки 40S и 60S и гетерогенные ядерные рибонуклеопротеины, которые не являются субъединицами ППС.
- Импортируйте отфильтрованные данные в Perseus, щелкнув Загрузка универсальной матрицы в разделе Загрузка24,25. Журнал2 преобразует данные, перейдя в раздел Основные > Преобразование, выбрав данные и указав функцию преобразования, в данном случае log2(x).
- Вмените отсутствующие значения из нормального распределения, перейдя в раздел Условное > Заменить отсутствующие значения из нормального распределения, выбрав данные и указав ширину (по умолчанию 0,3) и сдвиг вниз (по умолчанию 1,8) для вычисления. Выполните квантильную нормализацию, перейдя к нормализации > квантильной нормализации.
- Рассчитайте коэффициенты log2 и P-значения Т-теста Стьюдента для соответствующих условий. Сначала аннотируйте данные, перейдя в Annot. Rows > Categorical Annotation Rows. Выполните Т-тест, перейдя в раздел Тесты > Тесты с двумя образцами и выбрав группы для сравнения, тест для выполнения и метод коррекции нескольких тестов гипотез, используемый для усечения.
ПРИМЕЧАНИЕ: Для идентификации de novo белок считается PPP-взаимодействующим белком, если его обилие статистически значимо в состоянии MCLR-обработанного по сравнению с DMSO, с логарифмическим2-кратным изменением, превышающим минимальное изменение кратности любой конкретно связанной известной субъединицы PPP.
Access restricted. Please log in or start a trial to view this content.
Результаты

Рисунок 2: Идентификация специфических связующих PIB. (A) Различные типы тканей или клеток могут быть проанализированы с помощью PIB-MS. Клетки HeLa в биологическом трипликате л?...
Access restricted. Please log in or start a trial to view this content.
Обсуждение
PIB-MS - это химический протеомический подход, используемый для количественного профилирования PPPome из различных источников образцов в одном анализе. Большая работа была проделана с использованием шариков ингибитора киназы для изучения кинома и того, как он изменяется при раке и других б?...
Access restricted. Please log in or start a trial to view this content.
Раскрытие информации
Авторам нечего раскрывать и нет конфликта интересов.
Благодарности
A.N.K. признает поддержку со стороны NIH R33 CA225458 и R35 GM119455. Мы благодарим лаборатории Кеттенбаха и Гербера за их полезную дискуссию.
Access restricted. Please log in or start a trial to view this content.
Материалы
| Name | Company | Catalog Number | Comments |
| Acetonitrile (ACN) | Honeywell | AH015-4 | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Anhydrous Acetonitrile | Sigma-Aldrich | 271004-100ML | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Benchtop centrifuge | Eppendorf | model no. 5424 | |
| Beta-glycerophosphoric acid, disodium salt pentahydrate | Acros Organics | 410991000 | |
| Centrifuge | Eppendorf | model no. 5810 R 15 amp version | |
| Distilled water | |||
| DMSO | Fisher Scientific | BP231-100 | |
| Dounce tissue grinder | Fisherbrand Pellet Pestles | 12-141-363 | |
| Empore solid phase extraction disk, C18 | CDS Analytical | 76333-132 | |
| Eppendorf tubes, 1.5 mL | Eppendorf | 22363204 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Eppendorf tubes, 2 mL | Eppendorf | 22363352 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Extraction plate manifold | Waters | WAT097944 | |
| Falcon tubes, 50 mL | VWR | 21008 | |
| Generic blunt end needle and plunger | |||
| Generic magnetic separation rack | |||
| HEPES | Sigma-Aldrich | H3375 | |
| Hydrogen chloride (HCl) | VWR Chemicals BDH | BDH3028 | CAUTION: HCl is corrosive; wear gloves and work in a chemical fume hood. |
| Hydroxylamine solution 50% (wt/vol) | Sigma-Aldrich | 467804 | |
| Incubator, 65 °C | VWR | model no. 1380FM | |
| Koptec Pure Ethanol, 200 Proof | Decon Labs | V1001 | |
| Methanol for HPLC (MeOH) | Sigma-Aldrich | 34860-4L-R | CAUTION: MeOH is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Microcystin LR (MCLR) | Cayman Chemical | 10007188 | CAUTION: MCLR is toxic; wear gloves when handling and avoid skin contact. |
| PBS, 1× without calcium and magnesium, pH 7.4 ± 0.1 | Corning | 21-040-CV | |
| pH test strips, such as MilliporeSigma MColorpHast pH test strips and indicator papers | Fisher Scientific | M1095310001 | |
| PIBs | For protocol for the generation of PIBs, see Moorhead et al., 2007. | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 | |
| Pipette tips, 10 μL | Eppendorf | 22491504 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 1000 μL | Eppendorf | 22491555 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 200 μL | Eppendorf | 22491539 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| plastic syringe, 10 mL | BD | 309604 | |
| Protease inhibitor cocktail III | Research Products International | P50700-1 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer, Oribtrap Fusion, Orbitrap Fusion Lumos, or Orbitrap Eclipse Tribrid Mass Spectrometer | Thermo Scientific | ||
| Refrigerated benchtop centrifuge | Eppendorf | model no. 5424 R | |
| Rotator (Labquake Shaker Rotisserie) | Thermo Scientific | 13-687-12Q | 8 rpm rotation |
| Sample collection plate, 96- well, 1 mL | Waters | WAT058957 | |
| SDS | Fisher Scientific | BP1311-1 | |
| Sequencing grade modified trypsin | Promega | V511C | |
| Sodium azide | EMD Chemicals | SX0299-1 | CAUTION: Sodium azide is explosive and toxic; wear gloves, work in a chemical fume hood and avoid contact with metals. |
| Sodium chloride (NaCl) | Fisher Chemical | S27110 | |
| Sonicator (Branson digital sonifier) | model no. SFX 250 | ||
| SPE C18 desalting plate | Waters | 186001828BA | |
| SpeedBeads magnetic carboxylate modified particles (SP3 beads) | Cytiva | 6.51521E+13 | |
| Thermomixer | Eppendorf | model no. 5350 | |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent, 3 × 0.8 mg per tag | ThermoFisher | A37725 | |
| Trifluoroacetic acid (TFA) | Honeywell | T6508-25ML | CAUTION: TFA is corrosive and will irritate skin on contact. Wear gloves and eye protection, and work in a chemical fume hood. |
| Tris Base | Research Products International | T60040 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Vacuum centrifuge and vapor trap | Thermo Scientific | model nos. SpeedVac SPD120 and RVT5105 | |
| Vortexer (Vortex-Genie 2) | Scientific Industries | ||
| Water LC-MS | Honeywell | LC365-4 |
Ссылки
- Nilsson, J. Protein phosphatases in the regulation of mitosis. Journal of Cell Biology. 218 (2), 395-409 (2019).
- Brautigan, D. L. Protein Ser/Thr phosphatases--the ugly ducklings of cell signalling. The FEBS Journal. 280 (2), 324-345 (2013).
- Brautigan, D. L., Shenolikar, S. Protein serine/threonine phosphatases: keys to unlocking regulators and substrates. Annual Review of Biochemistry. 87, 921-964 (2018).
- Janssens, V., Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochemical Journal. 353, 417-439 (2001).
- Virshup, D. M., Shenolikar, S. From promiscuity to precision: protein phosphatases get a makeover. Molecular Cell. 33 (5), 537-545 (2009).
- Bollen, M., Peti, W., Ragusa, M. J., Beullens, M. The extended PP1 toolkit: designed to create specificity. Trends in Biochemical Sciences. 35 (8), 450-458 (2010).
- Qian, J., Winkler, C., Bollen, M. 4D-networking by mitotic phosphatases. Current Opinion in Cell Biology. 25 (6), 697-703 (2013).
- Heroes, E., et al. The PP1 binding code: a molecular-lego strategy that governs specificity. The FEBS Journal. 280 (2), 584-595 (2013).
- Eichhorn, P. J., Creyghton, M. P., Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et Biophysica Acta. 1795 (1), 1-15 (2009).
- Bantscheff, M., et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature Biotechnology. 25 (9), 1035-1044 (2007).
- Klaeger, S., et al. Chemical proteomics reveals ferrochelatase as a common off-target of kinase inhibitors. ACS Chemical Biology. 11 (5), 1245-1254 (2016).
- Duncan, J. S., et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 149 (2), 307-321 (2012).
- Cooper, M. J., et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE. 8 (6), 66755(2013).
- Lyons, S. P., et al. A quantitative chemical proteomic strategy for profiling phosphoprotein phosphatases from yeast to humans. Molecular and Cellular Proteomics. 17 (12), 2448-2461 (2018).
- Nasa, I., et al. Quantitative kinase and phosphatase profiling reveal that CDK1 phosphorylates PP2Ac to promote mitotic entry. Science Signaling. 13 (648), (2020).
- Swingle, M., Ni, L., Honkanen, R. E. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods in Molecular Biology. 365, 23-38 (2007).
- Moorhead, G. B. G., Haystead, T. A. J., MacKintosh, C. Synthesis and use of the protein phosphatase affinity matrices microcystin-sepharose and microcystin-biotin-sepharose. Methods in Molecular Biology. 365, 39-45 (2007).
- Brauer, B. L., Wiredu, K., Mitchell, S., Moorhead, G. B., Gerber, S. A., Kettenbach, A. N. Affinity-based profiling of endogenous phosphoprotein phosphatases by mass spectrometry. Nature Protocols. 16 (10), 4919-4943 (2021).
- Hughes, C. S., Moggridge, S., Müller, T., Sorensen, P. H., Morin, G. B., Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Zecha, J., et al. TMT labeling for the masses: A robust and cost-efficient, in-solution labeling approach. Molecular and Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Elias, J. E., Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 4 (3), 207-214 (2007).
- Eng, J. K., Jahan, T. A., Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 13 (1), 22-24 (2013).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Yu, S. H., Ferretti, D., Schessner, J. P., Rudolph, J. D., Borner, G. H. H., Cox, J. Expanding the Perseus software for omics data analysis With custom plugins. Current Protocols in Bioinformatics. 71 (1), 1-29 (2020).
Access restricted. Please log in or start a trial to view this content.
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены