
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Ein massenspektrometriebasierter Ansatz zur Identifizierung von Phosphoproteinphosphatasen und ihren Interaktoren
In diesem Artikel
Zusammenfassung
Hier stellen wir ein Protokoll zur Anreicherung von endogenen Phosphoproteinphosphatasen und ihren interagierenden Proteinen aus Zellen und Geweben und deren Identifizierung und Quantifizierung durch massenspektrometriebasierte Proteomik vor.
Zusammenfassung
Die meisten zellulären Prozesse werden durch dynamische Proteinphosphorylierung reguliert. Mehr als drei Viertel der Proteine sind phosphoryliert, und Phosphoproteinphosphatasen (PPPs) koordinieren über 90% der gesamten zellulären Serin/Threonin-Dephosphorylierung. Die Deregulierung der Proteinphosphorylierung wurde mit der Pathophysiologie verschiedener Krankheiten, einschließlich Krebs und Neurodegeneration, in Verbindung gebracht. Trotz ihrer weit verbreiteten Aktivität sind die molekularen Mechanismen, die PPPs kontrollieren, und diejenigen, die von PPPs kontrolliert werden, schlecht charakterisiert. Hier wird ein proteomischer Ansatz beschrieben, der als Phosphatase-Inhibitor-Beads und Massenspektrometrie (PIB-MS) bezeichnet wird, um PPPs, ihre posttranslationalen Modifikationen und ihre Interaktoren in nur 12 h unter Verwendung einer Zelllinie oder eines Gewebes zu identifizieren und zu quantifizieren. PIB-MS verwendet einen nicht-selektiven PPP-Inhibitor, Microcystin-LR (MCLR), der auf Sepharosekügelchen immobilisiert wird, um endogene PPPs und ihre assoziierten Proteine (PPPom genannt) einzufangen und anzureichern. Diese Methode erfordert weder die exogene Expression markierter Versionen von PSM noch die Verwendung spezifischer Antikörper. PIB-MS bietet eine innovative Möglichkeit, die evolutionär konservierten PPPs zu untersuchen und unser derzeitiges Verständnis der Dephosphorylierungssignalisierung zu erweitern.
Einleitung
Die Proteinphosphorylierung steuert die meisten zellulären Prozesse, einschließlich, aber nicht beschränkt auf die Reaktion auf DNA-Schäden, die Signalisierung des Wachstumsfaktors und die Passage durch Mitose 1,2,3. In Säugetierzellen wird die Mehrheit der Proteine zu einem bestimmten Zeitpunkt an einem oder mehreren Serin-, Threonin- oder Tyrosinresten phosphoryliert, wobei Phosphosrine und Phosphothreonine etwa 98% aller Phosphorylierungsstellen ausmachen 2,3. Während Kinasen in der zellulären Signalgebung ausführlich untersucht wurden, zeichnet sich die Rolle von PPPs bei der Regulation dynamischer zellulärer Prozesse noch ab.
Die Phosphorylierungsdynamik wird durch das dynamische Zusammenspiel von Kinasen und Phosphatasen gesteuert. In Säugetierzellen gibt es mehr als 400 Proteinkinasen, die die Serin/Threonin-Phosphorylierung katalysieren. Über 90% dieser Stellen werden durch Phosphoproteinphosphatasen (PPPs) dephosphoryliert, eine kleine Familie von Enzymen, die aus PP1, PP2A, PP2B, PP4-7, PPT und PPZ 2,3 besteht. PP1 und PP2A sind für den Großteil der Phosphoserin- und Phosphothreonin-Dephosphorylierung innerhalb einer Zelle verantwortlich 2,3,4. Der bemerkenswerte Unterschied in der Anzahl zwischen Kinasen und Phosphatasen und die mangelnde Spezifität der PPP-katalytischen Untereinheiten in vitro führten zu der Annahme, dass Kinasen die Hauptdeterminante der Phosphorylierungsind 2,3. Mehrere Studien haben jedoch gezeigt, dass Phosphatasen die Substratspezifität durch die Bildung multimerer Holoenzyme 5,6,7,8,9 etablieren. Zum Beispiel ist PP1 ein Heterodimer, das aus einer katalytischen Untereinheit und zu einem bestimmten Zeitpunkt aus einer der mehr als 150 regulatorischen Untereinheiten 6,7,8 besteht. Umgekehrt ist PP2A ein Heterotrimer, der aus einem Gerüst (A), einer regulatorischen (B) und einer katalytischen (C)Untereinheit 2,3,9 besteht. Es gibt vier verschiedene Familien von regulatorischen PP2A-Untereinheiten (B55, B56, PR72 und Striatin), jede mit mehreren Genen, Spleißvarianten und Lokalisationsmustern 2,3,9. Die multimere Natur von PPPs füllt die Lücke in der Anzahl der Kinasen und PPP-katalytischen Untereinheiten. Es schafft jedoch analytische Herausforderungen für die Untersuchung der PPP-Signalgebung. Um die PPP-Signalgebung umfassend analysieren zu können, ist es wichtig, die verschiedenen Holoenzyme in einer Zelle oder einem Gewebe zu untersuchen. Große Fortschritte wurden bei der Untersuchung des menschlichen Kinoms durch die Verwendung von Kinase-Inhibitor-Perlen erzielt, die als Multiplex-Inhibitor-Perlen oder Kinobeads bezeichnet werden, eine chemische Proteomstrategie, bei der Kinase-Inhibitoren auf Perlen immobilisiert werden und Massenspektrometrie verwendet wird, um angereicherte Kinasen und ihre Interaktorenzu identifizieren 10,11,12,13.
Wir haben einen ähnlichen Ansatz für das Studium der PPP-Biologie etabliert. Diese Technik beinhaltet die Affinitätserfassung von PPP-katalytischen Untereinheiten unter Verwendung von Perlen mit einem immobilisierten, nicht-selektiven PPP-Inhibitor namens Microcystin-LR (MCLR), der als Phosphatase-Inhibitor-Perlen (PIBs) bezeichnet wird14,15. Im Gegensatz zu anderen Methoden, die die endogene Markierung oder Expression exogener PPP-Untereinheiten erfordern, die die Proteinaktivität oder -lokalisation verändern könnten, ermöglicht PIB-MS die Anreicherung von endogenen PPP-katalytischen Untereinheiten, ihren assoziierten regulatorischen und gerüstbildenden Untereinheiten und interagierenden Proteinen (als PPPom bezeichnet) aus Zellen und Geweben zu einem bestimmten Zeitpunkt oder unter bestimmten Behandlungsbedingungen. MCLR hemmt PP1, PP2A, PP4-6, PPT und PPZ bei nanomolaren Konzentrationen, wodurch PIBs bei der Anreicherung für dasPPPome 16 hochwirksam sind. Diese Methode kann für den Einsatz auf jedem Ausgangsmaterial von Zellen bis hin zu klinischen Proben skaliert werden. Hier beschreiben wir detailliert den Einsatz von PIBs und Massenspektrometrie (PIB-MS), um das endogene PPPom und seine Modifikationszustände effizient zu erfassen, zu identifizieren und zu quantifizieren.

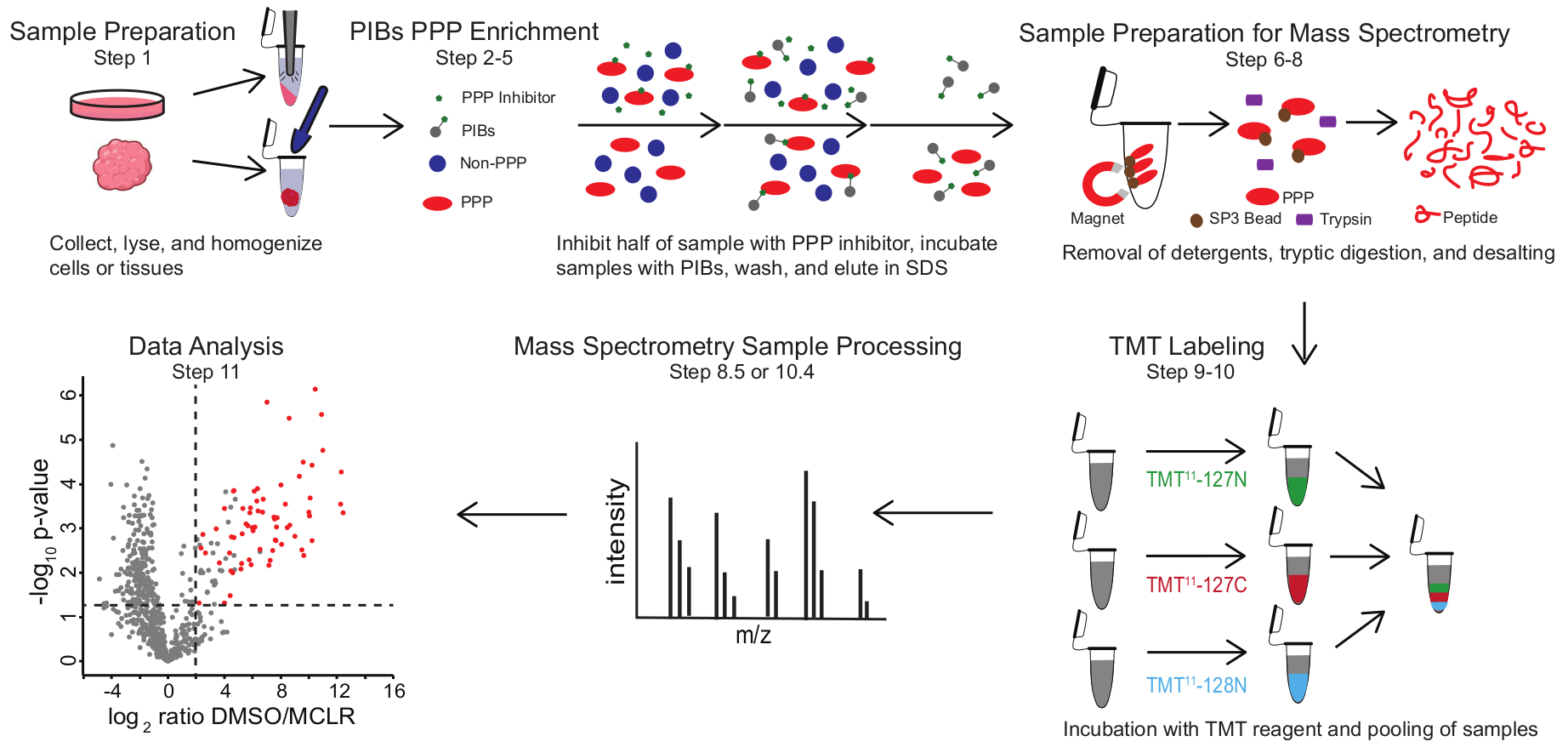
Abbildung 1: Visuelle Zusammenfassung des PIB-MS-Protokolls. In einem PIB-MS-Experiment können Proben in verschiedenen Formen gewonnen werden, von Zellen bis hin zu Tumoren. Die Probe wird vor der PPP-Anreicherung gesammelt, lysiert und homogenisiert. Zur Anreicherung für PPPs wird das Lysat mit PIBs mit oder ohne PPP-Inhibitor, wie MCLR, inkubiert. Die PIBs werden dann gewaschen und PPPs werden unter Denaturierungsbedingungen eluiert. Die Proben werden für die massenspektrometrische Analyse durch Entfernen von Reinigungsmitteln durch SP3-Proteinanreicherung, tryptische Verdauung und Entsalzung vorbereitet. Proben können dann vor der massenspektrometrischen Analyse optional TMT-markiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
PIB-MS umfasst die Lyse und Klärung von Zellen oder Geweben, die Inkubation des Lysats mit PIBs, die Elution und die Analyse des Eluats über westliche Blotting- oder Massenspektrometrie-basierte Ansätze (Abbildung 1). Die Zugabe von freiem MCLR kann als Steuerung verwendet werden, um spezifische PIB-Bindemittel von unspezifischen Interaktoren zu unterscheiden. Für die meisten Anwendungen kann ein markierungsfreier Ansatz verwendet werden, um Proteine in Eluaten direkt zu identifizieren. In Fällen, in denen eine genauere Quantifizierung oder die Identifizierung von Arten mit geringer Häufigkeit erforderlich ist, kann die Weiterverarbeitung mit Tandem-Massen-Tag-Kennzeichnung (TMT) verwendet werden, um die Abdeckung zu erhöhen und den Input zu verringern.
Access restricted. Please log in or start a trial to view this content.
Protokoll
HINWEIS: Die Erzeugung von PIBs erfolgt wie von Moorhead et al. beschrieben, wobei 1 mg Microcystin und etwa 6 ml Sepharis gekoppelt sind, um PIBs mit einer Bindungskapazität von bis zu 5 mg/ml17 zu erzeugen.
1. Probenvorbereitung
HINWEIS: Eine typische Ausgangsmenge für PIB-MS beträgt 1 mg Gesamtprotein pro Zustand. Für dieses Experiment wurden etwa 2,5 x 106 HeLa-Zellen verwendet, um 1 mg Protein zu extrahieren. Diese Berechnung sollte für jede Zelllinie oder jedes Gewebe durchgeführt werden, das in einem Experimentverwendet wird 18. Wenn die Probe begrenzt ist und 1 mg nicht erhalten werden kann, kann die Menge des Inputs mit einem geringfügigen Verlust der PPP-Untereinheitserkennung reduziert werden. Alternativ kann eine TMT-Markierung verwendet werden, um das Mischen aller Bedingungen in einer Probe zu ermöglichen, wodurch die Empfindlichkeit der Detektion erhöht wird, wie in Schritt 9 gezeigt.
- Sammeln Sie Gewebeproben oder Zellpellets. Für Zellpellets sammeln Sie Zellen durch Zentrifugation bei 277 x g für 2 min bei Raumtemperatur (RT), entfernen Sie Medien und waschen Sie die Zellen mit 5 ml phosphatgepufferter Kochsalzlösung (PBS). Zellpellets können mehrere Monate bei -80 °C gelagert werden.
- Lysepuffer (500 mM NaCl, 50 mM Tris-HCl pH 7,5, 0,5% Triton X-100 (vol/vol), 5 mM Beta-Glycerophosphorsäure-Dinatriumsalz-Pentahydrat, 1:500 (vol/vol) Proteaseinhibitor-Cocktail III) vorbereiten und auf Eis legen. Machen Sie genug, um alle Proben zu lysieren und zu waschen. Wenn Sie mit 1 mg Protein pro Bedingung beginnen, machen Sie etwa 3 ml Puffer pro Probe für Lyse und Waschen.
HINWEIS: Der in diesem Schritt notierte Lysepuffer ist eine milde Waschmittellösung, die möglicherweise nicht ausreicht, um unlösliche Membran- oder Zytoskelett-assoziierte Proteine zu lösen. Andere Detergenzien könnten erforscht werden, um die Solubilisierung zu verbessern. Es wurde eine Reihe von NaCl- und Triton-X-100-Konzentrationen getestet, und die oben genannten Konzentrationen erwiesen sich als optimal für eine Bindung von Untereinheiten mit niedrigem Hintergrund und hoher Phosphatase. - Fügen Sie den Proben gekühlten Lysepuffer hinzu. Für die Lyse von 1 mg Protein verwenden Sie 1 ml Puffer. Wenn die Probe gefroren ist, fügen Sie Lysepuffer hinzu und lassen Sie die Probe auf Eis im Puffer auftauen.
- Für Zellen homogenisieren Sie die Proben durch Sonifikation und halten Sie die Zellen zwischen den Impulsen auf Eis. Beschallen Sie die Proben bei 15% Amplitude mit drei 15 s Impulsen. Dies kann je nach verwendetem Ultraschallgerät variieren (siehe Materialtabelle).
- Bei Geweben werden die Proben zuerst mit einem Dounce-Gewebeschleifer homogenisiert, um das Gewebe zu mahlen, bis es verflüssigt ist, bevor es beschallt wird, wie in Schritt 1.3.1 beschrieben.
- Die homogenisierte Probe unlöslicher Ablagerungen wird durch Zentrifugation bei 21.130 x g für 15 min bei 4 °C geklärt. Dann, ohne die gebildeten Pellets oder Lipide zu stören, die sich an der Seite der Röhrchen angesammelt haben, übertragen Sie die Lysate in neue Röhrchen. Entfernen Sie 100 μL der Voranreicherungsprobe des geklärten Lysats, falls gewünscht, und lagern Sie sie bei -20 °C. Achten Sie darauf, die Lysate auf Eis zu halten.
- Bestimmen Sie den Gesamtproteingehalt in jeder Probe, indem Sie einen Proteinquantifizierungsassay, wie z. B. einen Bicchincinsäure-Assay (BCA), an einem kleinen Aliquot jeder Probe gemäß den Anweisungen des Herstellers durchführen. Stellen Sie sicher, dass die Lysate während des BCA-Assays auf Eis gehalten werden.
- Nach der Durchführung des BCA-Assays eine entsprechende Menge Protein in neue Röhrchen überführen und mit Lysepuffer verdünnen, um sicherzustellen, dass jedes Röhrchen die gleiche Proteinkonzentration (z. B. 1 mg/ml) aufweist. Wenn das Experiment eine PPP-Inhibitorkontrolle erfordert, bereiten Sie zwei Aliquots jeder Probe vor, die den gleichen Proteingehalt aufweisen, und fahren Sie mit Schritt 1.7 fort. Wenn keine PPP-Inhibitorkontrollen erforderlich sind, fahren Sie mit Schritt 2 fort. Stellen Sie sicher, dass die Proben auf Eis aufbewahrt werden.
HINWEIS: Es ist wichtig, dass in einer PIB-MS-Analyse gleiche Proteinkonzentrationen pro Bedingung verwendet werden. PPP-Inhibitor-Kontrollen werden verwendet, um spezifische Bindemittel für PIBs vom unspezifischen Hintergrund zu unterscheiden. - Für die PPP-Inhibitor-Kontrolle behandeln Sie eine Probe mit freier MCLR (1 μM) und die andere mit einem gleichen DMSO-Volumen als Kontrolle. Wirbeln Sie die Proben vorsichtig auf und inkubieren Sie sie 15 min lang auf Eis.
HINWEIS: Die MCLR-Behandlung der Lysate blockiert die Bindung von PPP-katalytischen Untereinheiten, nicht jedoch von Proteinen, die unspezifisch an PIBs in den Proben binden. Seien Sie vorsichtig beim Umgang mit MCLR, da es giftig ist. Weitere Informationen finden Sie unter Vorsichtsmaßnahmen für die Handhabung in der Materialtabelle.
2. Erstellung von PIBs
- Bestimmen Sie die Menge an PIBs, die für das Experiment benötigt werden. Für 1 mg Protein können 1-3 μg PPPs und interagierende Proteine erhalten werden; Verwenden Sie mindestens 10 μL festes PIBs-Harz pro Probe, um den Perlenverlust zu minimieren. Die Bindungskapazität von PIBs beträgt 3-5 mg/ml14,17.
- Übertragen Sie die entsprechende Menge an PIBs in ein 1,5-ml-Röhrchen und waschen Sie sie 3x mit 0,5 ml Lysepuffer, indem Sie sie vorsichtig vorwirbeln und dann bei 376 x g für 30 s bei RT zwischen den Wäschen zentrifugieren. Vermeiden Sie es, die Perlen zu pipettieren, wenn Sie den Lysepuffer zwischen den Waschungen entfernen.
- Machen Sie eine 50%ige PIB/Pufferlösung (vol/vol), indem Sie den gewaschenen PIBs eine angemessene Menge Lysepuffer hinzufügen. Pipettieren Sie vorsichtig auf und ab und schwenken Sie die Pipettenspitze in der Aufschlämmung, um die PIBs wieder aufzuhängen.
- Übertragen Sie 20 μL der Aufschlämmung auf ein neues 1,5-ml-Röhrchen, das bereits 0,5 ml Lysepuffer enthält. Der Lysepuffer im Rohr hilft beim Ausstoßen der Perlen aus der Pipettenspitze. Tun Sie dies, bis genügend Röhrchen vorhanden sind, die eine gleiche Menge an PIBs für jede Probe enthalten.
- Drehen Sie die Röhrchen bei 376 x g für 30 s bei RT. Stellen Sie sicher, dass alle Röhrchen eine gleiche Menge an Perlenharz enthalten. Verwerfen Sie jeden Überstand und lassen Sie nur festes Harz und maximal 50 μL Lysepuffer in jedem Röhrchen zurück.
3. Inkubation von PIBs mit Lysaten
- Übertragen Sie die Lysate aus Schritt 1.6. oder Schritt 1.7. zu dem entsprechend beschrifteten Röhrchen, das PIBs aus Schritt 2 enthält. Drehen Sie das Lysat bei 8 U/min mit den PIBs für 1 h bei 4 °C.
4. Waschen von PIBs
- Die PIBs bei 376 x g für 30 s bei 4 °C zentrifugieren, um die Perlen zu sammeln. Entfernen und verwerfen Sie den Überstand, wobei Sie bei Bedarf ein Aliquot von 100 μL für die Analyse nach der Anreicherung speichern.
- Waschen Sie die PIBs 3x, indem Sie 0,5 ml Lysepuffer zu den Perlen geben, die Röhrchen umkehren (Vortexing nicht empfohlen), die Perlen durch Zentrifugation bei 376 x g für 30 s bei 4 ° C sammeln und den Lysepuffer von den abgesetzten Perlen entfernen, wobei darauf zu achten ist, dass das Perlenpellet nicht gestört wird.
5. Gewinnung von ÖPP aus den PIBs
- Entfernen Sie nach der letzten Wäsche so viel wie möglich vom Lysepuffer, ohne eines der PIBs zu pipettieren. Machen Sie einen Elutionspuffer mit 2% SDS (vol/vol) und eluieren Sie die PPPs aus den PIBs, indem Sie genügend Volumen an Elutionspuffer hinzufügen, um das 4x-5-fache des Volumens von PIBs zu erreichen. Wenn beispielsweise 10 μL PIBs verwendet werden, verwenden Sie 50 μL Elutionspuffer. Inkubieren Sie die PIBs mit dem Elutionspuffer bei 65 °C für 1 h, um die PPPs aus den PIBs zu eluieren.
- Nach der Elution sammeln Sie das Eluat, indem Sie die Röhrchen bei 376 x g für 30 s bei RT zentrifugieren und das Eluat in ein separates Rohr pipettieren, wobei darauf zu achten ist, dass keine PIBs übertragen werden. Verwenden Sie das Eluat für die Western-Blot-Analyse oder für Massenspektrometrie-Analysen. Eluate können bis zu mehreren Monaten bei -20 °C gelagert werden.
- Um die PIBs für die weitere Verwendung zu regenerieren, inkubieren Sie die Perlen in 2% SDS (vol/vol) und drehen Sie sich mit 8 U / min bei RT für 1 h. Waschen Sie sie 3x-5x in 25 mM Tris-HCl (pH 7,5) mit Rotation für 30 min pro Waschgang. Nach allen Wäschen PIBs in 25 mM Tris-HCl (pH 7,5) Speicherpuffer mit Natriumazid (0,05% Gew./Vol.) lagern.
- Analysieren Sie die Eluate durch Western Blotting oder Massenspektrometrie. Die massenspektrometrische Analyse von PIB-Eluaten wird im Folgenden beschrieben.
6. Entfernung von Reinigungsmitteln
HINWEIS: Verschiedene Ansätze können verwendet werden, um Reinigungsmittel aus eluatierten Proben für die MS-Analyse zu entfernen. Wir fanden heraus, dass die von Hughes et al. beschriebene Eintopf-, Festphasen-verstärkte Probenvorbereitung (SP3) gut funktioniert19.
- Fügen Sie 0,5 μL SP3-Perlen zu 50 μL Eluat aus Schritt 5.2 oben hinzu. Verwenden Sie SP3-Perlen in einem Verhältnis von 10:1 (μg:μg) oder mindestens 0,5 μg/μL (Stammlösung ist 50 μg/μL). Die Perlen vorsichtig umwirbeln und eluieren.
- Fügen Sie dem Perlen-Eluat-Gemisch ein Elutionsvolumen von 100% Ethanol hinzu (z. B. wenn 50 μL des Eluats verwendet wurden, verwenden Sie 50 μL 100% Ethanol). Inkubieren Sie die Proben für 5 min in einem Thermomixer-Set, um bei 1000 U / min bei 24 ° C zu schütteln.
- Um alle Perlen zu sammeln, legen Sie sie in ein Magnetrohrgestell. Sobald sich die Perlen gesammelt haben, entsorgen Sie den Überstand und waschen Sie die Perlen mit 0,5 ml 80% Ethanol (vol / vol) 3x. Zum Waschen die Perlen durch Vortexen wieder aufhängen, die Perlen sammeln, indem Sie sie in das Magnetrohrgestell legen, und den Überstand zwischen den Waschungen entsorgen.
- Waschen Sie die Perlen noch einmal mit 0,5 ml 100% Acetonitril (ACN), um alle Spuren von Ethanol zu entfernen und so viel ACN wie möglich zu entfernen.
7. Verdauung von Proteinen
- Machen Sie eine 1:100 Verdünnung von Trypsin (Endkonzentration von 0,004 μg/μL) in 166 mM HEPES (pH 8,5) und fügen Sie 30 μL dieser Trypsinlösung zu jedem Röhrchen mit den Perlen hinzu, wobei Sie durch Vortexen resuspendiert werden.
- Inkubieren Sie das SP3-Trypsin-Wulstgemisch im Thermomischer bei 1000 U/min bei 37 °C für 5 h oder über Nacht bei 30 °C. Legen Sie die Röhrchen in das Magnetgestell, um die Perlen zu sammeln und die Digests zu neuen Röhrchen zu entfernen.
- Für eine markierungsfreie Analyse wird die Reaktion durch Zugabe von 20% Trifluoressigsäure (TFA) (vol/vol) zu einer Endkonzentration von 0,2% TFA (vol/vol) abgeschreckt. Überprüfen Sie, ob der pH-Wert jeder Probe zwischen 2-3 mit einem pH-Papier liegt. Wenn nicht, fügen Sie kleine zusätzliche Aliquoten von 20% TFA hinzu, bis dies erreicht ist. Die Proben müssen vor dem Entsalzen entsprechend angesäuert werden. Fahren Sie mit Schritt 8 fort.
- Für die TMT-Kennzeichnung der Proben nicht ansäuern und mit Schritt 9 fortfahren.
8. Entsalzen des Digests
- Bereiten Sie für jede Probe eine Stufenspitze vor, indem Sie eine 200 μL MS-Lösungsmittel-kompatible Spitze mit C18-Harz verpacken, wie von Rappsilber et al.20 beschrieben. Verwenden Sie eine stumpfe Nadel, um zwei C18-Materialscheiben zu pressen, um sicherzustellen, dass die Scheiben in der Nadel verbleiben. Übertragen Sie die Scheiben mit einem dünnen Drahtkolben in die MS-Lösungsmittel-kompatible Spitze, um die Scheiben aus der Nadel zu entfernen.
HINWEIS: Es ist wichtig, MS-Lösungsmittel-kompatible Spitzen von diesem Punkt im Protokoll zu verwenden, da andere Pipettenspitzen Chemikalien in die Proben auslaugen können, die über Massenspektrometrie nachgewiesen werden können. - Gleichen Sie jede Stufenspitze mit 30 μL von 100% MeOH aus, dann mit 30 μL von 60% MeOH (vol/vol), gefolgt von 30 μL von 0,1% TFA (vol/vol). Drücken Sie jede Lösung mit einer Spritze durch die Tischspitze. Befestigen Sie eine Pipettenspitze mit einer transparenten Folie am Ende einer Spritze, um den Kontakt zwischen der Spritze und der Bühnenspitze bei Bedarf zu erhöhen. Achten Sie darauf, das C18-Material niemals in der Bühnenspitze trocknen zu lassen.
- Fügen Sie den angesäuerten Peptidaufschluss aus Schritt 7.3 in die markierte Stufenspitze hinzu und drücken Sie die Stufenspitze mit einer Spritze durch, wobei Sie erneut darauf achten, dass die Stufenspitze nicht vollständig austrocknet.
- Waschen Sie jede Probe 2x mit 30 μL 0,1% TFA (vol/vol). Eluieren Sie die Peptide aus jeder Stufenspitze, indem Sie 30 μL 60% MeOH (vol/vol) zu jeder Stufenspitze hinzufügen und alles mit Spritzendruck aus der Spitze in ein neues, markiertes Röhrchen ausstoßen. Dies ist der einzige Schritt, bei dem das C18-Material vollständig getrocknet wird.
- Trocknen Sie jede Probe durch Vakuumzentrifugation. Getrocknete Peptide können mehrere Monate bei -20°C gelagert werden. Die Proben sind nun bereit für die markierungsfreie Massenspektrometrie-Analyse. Verwenden Sie nur die Hälfte der Probe für die Analyse auf dem Massenspektrometer und die andere Hälfte für die Reinjektion, falls erforderlich. Stellen Sie sicher, dass eine geeignete Massenspektrometermethode für die Analyse verwendet wird.
9. TMT-Kennzeichnung
HINWEIS: Die Tandem-Massen-Tag-Etikettierung wird verwendet, um Proben für die quantitative Analyse zu multiplexen. Eine 0,8 mg Durchstechflasche mit TMT-Reagenz reicht aus, um bis zu 0,8 mg Protein21 zu markieren. In einem PIB-Pulldown-Experiment, beginnend mit 1 mg Protein, werden 1-3 μg Phosphoprotein-Untereinheiten erhalten. Das folgende Protokoll ist optimal für bis zu 10 μg Protein.
- Rekonstituieren Sie eine 0,8 mg Durchstechflasche TMT-Reagenz in 80 μL wasserfreiem ACN.
- Beschriften Sie jedes Sample aus Step 7.4 mit einem anderen TMT-Label für bis zu 18 Kanäle. Notieren Sie sich, welches TMT-Etikett zu jeder Probe hinzugefügt wird. 2 μL des TMT-Reagenzes und 2 μL ACN in den Peptidaufschluss geben, vorsichtig zum Mischen vorwirbeln, bei 376 x g für 30 s bei RT zentrifugieren, um die Probe zu sammeln, und bei RT für 1 h inkubieren, um die Probe zu markieren.
- Um die TMT-Markierungseffizienz zu testen, machen Sie eine Etikettenprüfprobe, indem Sie 1 μL jeder Markierungsreaktion in einem 0,5 ml Röhrchen mit 9 μL LC-MS-Wasser und 1 μL 10% Hydroxylamin (vol/vol) kombinieren, um die Reaktion abzuschrecken. Die restlichen ungelöschten beschrifteten Proben in einen Gefrierschrank von -80 °C geben. Proben können mehrere Tage gelagert werden, während die Etikettiereffizienz bewertet wird.
- Die TMT-Etikettenprüfprobe wird durch Zugabe von 30 μL 0,1 % TFA (vol/vol) angesäuert. Überprüfen Sie, ob der pH-Wert zwischen 2-3 liegt. Wenn nicht, fügen Sie 20% TFA (vol/vol) hinzu, bis dieser pH-Wert erreicht ist. Entsalzen Sie die Etikettenprüfungsprobe durch Stufenkippen, wie in den Schritten 8.1.-8.5 beschrieben.
- Analysieren Sie die TMT-Etikettenprüfprobe auf dem Massenspektrometer, um die Etikettiereffizienz zu bewerten. Filtern Sie die Suchergebnisse auf eine False Discovery Rate (FDR) von 1% auf Peptidebene und bestimmen Sie die Markierungseffizienz21,22.
- Vollständig TMT-markierte Peptide haben TMT-Reagenz am N-Terminus und an allen Lysinen. Quantifizieren Sie die TMT-Reporter-Ionenintensitäten und vergleichen Sie ihre Gesamtsumme über alle Kanäle hinweg. Die Probe ist ausreichend markiert, wenn >95% aller Peptide markiert sind und die TMT-Reporter-Ionensummenintensitäten vergleichbar sind.
HINWEIS: Neben der unvollständigen Beschriftung können Unterschiede in den summierten TMT-Reporterionenintensitäten das Ergebnis eines ungenauen Pipettierens der 1-μL-Testprobe sein. Es könnte auch eine echte biologische Beobachtung widerspiegeln, in diesem Fall sollten alle Replikate das gleiche Verhalten zeigen. - Entfernen Sie die ungelöschten Proben aus der Lagerung von -80 °C und tauen Sie sie auf. Wenn sie nicht vollständig markiert sind, geben Sie 1 μL des entsprechenden TMT-Reagenzes wie oben erwähnt in die Probe, inkubieren Sie für 1 Stunde und wiederholen Sie die TMT-Etikettenprüfung. Wenn die Proben vollständig gekennzeichnet sind, fahren Sie mit Schritt 9.8 fort.
- Wenn vollständig markiert, fügen Sie 2 μL 10% Hydroxylamin (vol/vol) zu den TMT-Reaktionen hinzu, um die Markierung zu löschen. Inkubieren Sie die Proben bei RT für 15 min. Nach dem Abschrecken können die Proben mehrere Monate bei -80 °C gelagert werden.
- Kombinieren Sie alle abgeschreckten TMT-Kanäle und fügen Sie 2 μL 20% TFA (vol/vol) hinzu, um die Reaktion zu säuern. Überprüfen Sie den pH-Wert der kombinierten Reaktion und stellen Sie sicher, dass er zwischen pH 2-3 liegt. Wenn nicht, fügen Sie kleine Aliquots von 20% TFA hinzu, bis der gewünschte pH-Wert erreicht ist. Dies ist entscheidend für die ordnungsgemäße Entsalzung.
- Entfernen Sie ACN durch Vakuumzentrifugation für 30 min und entsalzen Sie die Probe wie unten beschrieben.
10. Entsalzen der TMT-markierten kombinierten Probe
- Verwenden Sie eine SPE C18-Entsalzungsplatte mit der entsprechenden Proteinkapazität (2 mg Sorptionsmittel reichen normalerweise für diese Anwendung aus), um das kombinierte TMT-Reagenz zu entsalzen. Gleichen Sie die Vertiefung mit 200 μL von 60% MeOH (vol/vol) und 200 μL von 10% MeOH/0,1% TFA (vol/vol) aus.
- Laden Sie die angesäuerten TMT-markierten Peptide aus Schritt 9.10. auf die Entsalzungsplatte. Waschen Sie die Probenvertiefungen 2x mit 200 μL 10% MeOH/0,1% TFA (vol/vol).
- Eluieren Sie die Peptide mit 100 μL 60% MeOH (vol/vol). Trocknen Sie die Proben durch Vakuumzentrifugation. Die getrocknete, TMT-gekennzeichnete Probe kann mehrere Monate bei -80 °C gelagert werden.
- Für die massenspektrometriere Analyse der TMT-markierten Probe injizieren Sie die Hälfte der Probe und speichern Sie die andere Hälfte, wenn eine erneute Injektion erforderlich ist.
HINWEIS: Die Injektionsmengen für die Massenspektrometrie variieren je nach Spalte, Geräteaufbau und Probentyp.
11. Datenanalyse
HINWEIS: Die Methoden der Datenfilterung und -analyse variieren und gehen über den Rahmen dieses Protokolls hinaus, aber die folgenden Hinweise zur Analyse sind enthalten, um eine spezifische Anleitung für die Art der Daten zu geben, die sich aus diesem Protokoll ergeben.
- Durchsuchen Sie rohe Massenspektrometriedaten anhand einer speziesspezifischen Proteomdatenbank, basierend auf der Herkunft der verwendeten Probenzellen oder des verwendeten Gewebes. Hier wurde Comet als Suchalgorithmus23 verwendet.
- Filtern Sie die Suchergebnisse mit einem FDR von 1%, indem Sie die suchalgorithmusspezifischen Parameter22 anpassen. Für eine markierungsfreie Quantifizierung verwenden Sie MS1-Peak-Area-Messungen, um die Daten zu quantifizieren. Verwenden Sie für TMT-markierte Proben MSn-abgeleitete Reporterionenintensitäten zur Quantifizierung. Für die statistische Signifikanz analysieren Sie die Proben in biologischen Triplikaten.
- Um PPP-Untereinheiten und ihre Interaktoren zu identifizieren, stellen Sie sicher, dass dreifach biologische Proben von MCLR-inhibierten und DMSO-behandelten Proben verglichen werden. Um das PPPom unter verschiedenen Bedingungen oder bei einer medikamentösen Behandlung zu vergleichen, stellen Sie sicher, dass biologische Triplikate jeder Erkrankung oder medikamentösen Behandlung erzeugt wurden.
- Filtern Sie die Daten so, dass nur Proteine mit einer Gesamtpeptidanzahl von >1 in mindestens zwei von drei DMSO-Kontrollproben vorhanden sind. Die MCLR-Konkurrenz konkurriert nicht immer mit allen katalytischen Untereinheitenbindungen. Außerdem können einige katalytische PPP-Untereinheiten unspezifisch am Sepharoseharz haften. Um eine der beiden Möglichkeiten zu berücksichtigen, während Proteine herausgefiltert werden, die unspezifisch an das Harz binden, entfernen Sie Proteine mit einer Gesamtpeptidzahl im MCLR-behandelten Zustand, der höher ist als die für jede katalytische PPP-Untereinheit.
- Schließen Sie häufige Verunreinigungen wie Keratin, Kollagen, 40S und 60S ribosomale Proteine und heterogene nukleäre Ribonukleoproteine, die keine PPP-Untereinheiten sind, von der Analyseaus 14.
- Importieren Sie gefilterte Daten in Perseus, indem Sie im Abschnitt Laden24,25 auf Generischer Matrix-Upload klicken. Log2 transformieren Sie die Daten, indem Sie zu Basic > Transform wechseln, die Daten auswählen und die Transformationsfunktion angeben, in diesem Fall log2(x).
- Unterstellen Sie fehlende Werte aus einer Normalverteilung, indem Sie zu Imputation > Fehlende Werte aus der Normalverteilung ersetzen, die Daten auswählen und die Breite (Standard 0.3) und die Abwärtsverschiebung (Standard 1.8) für die Berechnung angeben. Führen Sie eine Quantilnormalisierung durch, indem Sie zu Normalisieren > Quantilnormalisierung gehen.
- Berechnen Sie Log-2-Verhältnisse und Student's T-Testp-Werte für die jeweiligen Bedingungen. Kommentieren Sie zunächst die Daten, indem Sie zu Annot wechseln. Zeilen > kategoriale Anmerkungszeilen. Führen Sie den T-Test durch, indem Sie zu Tests > Zweistichprobentests gehen und die zu vergleichenden Gruppen, den durchzuführenden Test und die Methode für die Korrektur mehrerer Hypothesentests auswählen, wie sie für das Abschneiden verwendet wird.
HINWEIS: Für die De-novo-Identifizierung wird ein Protein als PPP-interagierendes Protein betrachtet, wenn seine Häufigkeit in der MCLR-behandelten gegenüber der DMSO-Bedingung statistisch signifikant ist, mit einer logarithmischen2-fachen Änderung, die größer ist als die minimale Faltenänderung einer spezifisch gebundenen bekannten PPP-Untereinheit.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse

Abbildung 2: Identifizierung spezifischer PIBs-Bindemittel. (A) Eine Vielzahl von Gewebetypen oder Zellen können mittels PIB-MS analysiert werden. HeLa-Zellen in biologischer Verdreifachung wurden entweder mit DMSO oder dem PPP-Inhibitor MCLR behandelt, mit PIBs inkubiert und mittels LC-MS/MS analysiert. (
Access restricted. Please log in or start a trial to view this content.
Diskussion
PIB-MS ist ein chemischer Proteomik-Ansatz, der verwendet wird, um das PPPom aus verschiedenen Probenquellen in einer einzigen Analyse quantitativ zu profilieren. Es wurde viel Arbeit mit Kinase-Inhibitor-Perlen geleistet, um das Kinom zu untersuchen und wie es sich bei Krebs und anderen Krankheitszuständenverändert 10,11,12,13. Die Untersuchung des PPPoms hinkt jedoch hinterher. Wir gehen da...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts offenzulegen und keine Interessenkonflikte.
Danksagungen
A.N.K. erkennt die Unterstützung von NIH R33 CA225458 und R35 GM119455 an. Wir danken den Laboren Kettenbach und Gerber für das hilfreiche Gespräch.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| Acetonitrile (ACN) | Honeywell | AH015-4 | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Anhydrous Acetonitrile | Sigma-Aldrich | 271004-100ML | CAUTION: ACN is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Benchtop centrifuge | Eppendorf | model no. 5424 | |
| Beta-glycerophosphoric acid, disodium salt pentahydrate | Acros Organics | 410991000 | |
| Centrifuge | Eppendorf | model no. 5810 R 15 amp version | |
| Distilled water | |||
| DMSO | Fisher Scientific | BP231-100 | |
| Dounce tissue grinder | Fisherbrand Pellet Pestles | 12-141-363 | |
| Empore solid phase extraction disk, C18 | CDS Analytical | 76333-132 | |
| Eppendorf tubes, 1.5 mL | Eppendorf | 22363204 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Eppendorf tubes, 2 mL | Eppendorf | 22363352 | CRITICAL: Other tubes may leach polymer into sample, contaminating the analysis. |
| Extraction plate manifold | Waters | WAT097944 | |
| Falcon tubes, 50 mL | VWR | 21008 | |
| Generic blunt end needle and plunger | |||
| Generic magnetic separation rack | |||
| HEPES | Sigma-Aldrich | H3375 | |
| Hydrogen chloride (HCl) | VWR Chemicals BDH | BDH3028 | CAUTION: HCl is corrosive; wear gloves and work in a chemical fume hood. |
| Hydroxylamine solution 50% (wt/vol) | Sigma-Aldrich | 467804 | |
| Incubator, 65 °C | VWR | model no. 1380FM | |
| Koptec Pure Ethanol, 200 Proof | Decon Labs | V1001 | |
| Methanol for HPLC (MeOH) | Sigma-Aldrich | 34860-4L-R | CAUTION: MeOH is flammable and toxic; wear gloves, and work in a chemical fume hood. |
| Microcystin LR (MCLR) | Cayman Chemical | 10007188 | CAUTION: MCLR is toxic; wear gloves when handling and avoid skin contact. |
| PBS, 1× without calcium and magnesium, pH 7.4 ± 0.1 | Corning | 21-040-CV | |
| pH test strips, such as MilliporeSigma MColorpHast pH test strips and indicator papers | Fisher Scientific | M1095310001 | |
| PIBs | For protocol for the generation of PIBs, see Moorhead et al., 2007. | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23225 | |
| Pipette tips, 10 μL | Eppendorf | 22491504 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 1000 μL | Eppendorf | 22491555 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| Pipette tips, 200 μL | Eppendorf | 22491539 | CRITICAL: Other tips may leach polymer into samples, contaminating the analysis. |
| plastic syringe, 10 mL | BD | 309604 | |
| Protease inhibitor cocktail III | Research Products International | P50700-1 | |
| Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer, Oribtrap Fusion, Orbitrap Fusion Lumos, or Orbitrap Eclipse Tribrid Mass Spectrometer | Thermo Scientific | ||
| Refrigerated benchtop centrifuge | Eppendorf | model no. 5424 R | |
| Rotator (Labquake Shaker Rotisserie) | Thermo Scientific | 13-687-12Q | 8 rpm rotation |
| Sample collection plate, 96- well, 1 mL | Waters | WAT058957 | |
| SDS | Fisher Scientific | BP1311-1 | |
| Sequencing grade modified trypsin | Promega | V511C | |
| Sodium azide | EMD Chemicals | SX0299-1 | CAUTION: Sodium azide is explosive and toxic; wear gloves, work in a chemical fume hood and avoid contact with metals. |
| Sodium chloride (NaCl) | Fisher Chemical | S27110 | |
| Sonicator (Branson digital sonifier) | model no. SFX 250 | ||
| SPE C18 desalting plate | Waters | 186001828BA | |
| SpeedBeads magnetic carboxylate modified particles (SP3 beads) | Cytiva | 6.51521E+13 | |
| Thermomixer | Eppendorf | model no. 5350 | |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent, 3 × 0.8 mg per tag | ThermoFisher | A37725 | |
| Trifluoroacetic acid (TFA) | Honeywell | T6508-25ML | CAUTION: TFA is corrosive and will irritate skin on contact. Wear gloves and eye protection, and work in a chemical fume hood. |
| Tris Base | Research Products International | T60040 | |
| Triton X-100 | Sigma-Aldrich | T9284 | |
| Vacuum centrifuge and vapor trap | Thermo Scientific | model nos. SpeedVac SPD120 and RVT5105 | |
| Vortexer (Vortex-Genie 2) | Scientific Industries | ||
| Water LC-MS | Honeywell | LC365-4 |
Referenzen
- Nilsson, J. Protein phosphatases in the regulation of mitosis. Journal of Cell Biology. 218 (2), 395-409 (2019).
- Brautigan, D. L. Protein Ser/Thr phosphatases--the ugly ducklings of cell signalling. The FEBS Journal. 280 (2), 324-345 (2013).
- Brautigan, D. L., Shenolikar, S. Protein serine/threonine phosphatases: keys to unlocking regulators and substrates. Annual Review of Biochemistry. 87, 921-964 (2018).
- Janssens, V., Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochemical Journal. 353, 417-439 (2001).
- Virshup, D. M., Shenolikar, S. From promiscuity to precision: protein phosphatases get a makeover. Molecular Cell. 33 (5), 537-545 (2009).
- Bollen, M., Peti, W., Ragusa, M. J., Beullens, M. The extended PP1 toolkit: designed to create specificity. Trends in Biochemical Sciences. 35 (8), 450-458 (2010).
- Qian, J., Winkler, C., Bollen, M. 4D-networking by mitotic phosphatases. Current Opinion in Cell Biology. 25 (6), 697-703 (2013).
- Heroes, E., et al. The PP1 binding code: a molecular-lego strategy that governs specificity. The FEBS Journal. 280 (2), 584-595 (2013).
- Eichhorn, P. J., Creyghton, M. P., Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochimica et Biophysica Acta. 1795 (1), 1-15 (2009).
- Bantscheff, M., et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nature Biotechnology. 25 (9), 1035-1044 (2007).
- Klaeger, S., et al. Chemical proteomics reveals ferrochelatase as a common off-target of kinase inhibitors. ACS Chemical Biology. 11 (5), 1245-1254 (2016).
- Duncan, J. S., et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 149 (2), 307-321 (2012).
- Cooper, M. J., et al. Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE. 8 (6), 66755(2013).
- Lyons, S. P., et al. A quantitative chemical proteomic strategy for profiling phosphoprotein phosphatases from yeast to humans. Molecular and Cellular Proteomics. 17 (12), 2448-2461 (2018).
- Nasa, I., et al. Quantitative kinase and phosphatase profiling reveal that CDK1 phosphorylates PP2Ac to promote mitotic entry. Science Signaling. 13 (648), (2020).
- Swingle, M., Ni, L., Honkanen, R. E. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods in Molecular Biology. 365, 23-38 (2007).
- Moorhead, G. B. G., Haystead, T. A. J., MacKintosh, C. Synthesis and use of the protein phosphatase affinity matrices microcystin-sepharose and microcystin-biotin-sepharose. Methods in Molecular Biology. 365, 39-45 (2007).
- Brauer, B. L., Wiredu, K., Mitchell, S., Moorhead, G. B., Gerber, S. A., Kettenbach, A. N. Affinity-based profiling of endogenous phosphoprotein phosphatases by mass spectrometry. Nature Protocols. 16 (10), 4919-4943 (2021).
- Hughes, C. S., Moggridge, S., Müller, T., Sorensen, P. H., Morin, G. B., Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nature Protocols. 14 (1), 68-85 (2019).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature Protocols. 2 (8), 1896-1906 (2007).
- Zecha, J., et al. TMT labeling for the masses: A robust and cost-efficient, in-solution labeling approach. Molecular and Cellular Proteomics. 18 (7), 1468-1478 (2019).
- Elias, J. E., Gygi, S. P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods. 4 (3), 207-214 (2007).
- Eng, J. K., Jahan, T. A., Hoopmann, M. R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 13 (1), 22-24 (2013).
- Tyanova, S., et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods. 13 (9), 731-740 (2016).
- Yu, S. H., Ferretti, D., Schessner, J. P., Rudolph, J. D., Borner, G. H. H., Cox, J. Expanding the Perseus software for omics data analysis With custom plugins. Current Protocols in Bioinformatics. 71 (1), 1-29 (2020).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten