
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
基于 crispr-cas9 的基因组工程, 用于生成 hiv-1 感染与选定的证明病毒整合点的 jurkat 报告模型
摘要
我们提出了一个基因组工程工作流程, 用于生成新的 hiv-1 感染体外模型, 在选定的基因组站点重新构建天际整合。利用 crispr-cas9 介导的、特定地点的基因组操作, 为针对艾滋病毒来源的记者提供了便利。提供了单细胞克隆生成、筛选和正确目标验证的详细协议。
摘要
人类免疫缺陷病毒 (hiv) 将其非随机 dna 整合到复发地点和基因组热点的宿主细胞基因组中。在这里, 我们提出了一个详细的协议, 以生成新的体外模型的艾滋病毒感染与选定的基因组整合位点使用基于 crispr-cas9 的基因组工程技术。通过这种方法, 可以将选择的报告序列集成到有针对性的、选择的基因组位点中, 反映临床相关的整合位点。
该协议描述了 hiv 衍生记者的设计以及目标站点和 grna 序列的选择。构造了具有同源臂的靶向载体, 并将其转染到 jurkat t 细胞中。记者序列是针对选定的基因组位点的, 由在目标站点上的 cas9 介导的双链断裂促成的同源重组。通过流式细胞仪和 pcr 生成单细胞克隆并筛选靶向事件。然后展开选定的克隆体, 并通过 pcr、测序和南方印迹验证正确的靶向。分析了 crispr-cas9 介导的基因组工程的潜在离目标事件。
通过使用该协议, 可以生成在临床相关整合点模拟艾滋病毒感染的新细胞培养系统。尽管单细胞克隆的生成和正确的报告序列集成的验证是非常耗时的, 但由此产生的克隆线是功能上分析天方集成站点选择的强大工具。
引言
感染后将天分 dna 整合到宿主基因组中是人体免疫缺陷病毒 (hiv) 生命周期中的一个关键步骤。整合后, 艾滋病毒持续存在, 在长寿命 cd4+ t 细胞子集 (如记忆 cd4+ t 细胞) 中建立潜伏期。艾滋病毒的整合似乎不是随机的 1,2。通过对急性和慢性感染个体 2, 3,4的 整合位点进行测序, 在几项研究中发现了一些具有递归整合天方 dna 的基因组热点,5,6,7,8。有趣的是, 在其中一些整合位点中, 在很大一部分受感染的细胞中检测到相同的位点, 从而导致了在复发位点的整合可能会对克隆扩展 1产生积极影响的想法。
为了提高我们对经常性集成站点意义的认识, 必须探索天方集成站点的选择。然而, 若干技术方面阻碍了研究艾滋病毒整合地点的选择及其后果。广泛使用的用于艾滋病毒潜伏期的细胞培养模型, 如 jlat 细胞系, 并不反映临床相关的反复整合位点9。对初级患者衍生细胞的研究一方面, 可以通过测序来描述整合场地景观, 但不允许进行功能分析。据我们所知, 没有足够的实验模型可用于功能分析选定的临床相关的集成站点。
在这里, 我们提供了一个详细的工作流程, 以生成新的模型的艾滋病毒感染使用基于 crispr-cas9 为基础的基因组工程技术。本文描述的工作流程可用于生成 t 细胞衍生的病毒感染模型的记者细胞系, 在选定的集成站点携带基因整合的天方记者。因此, 它们是新的工具, 探讨天长整合站点如何影响艾滋病毒生物学, 以及天长如何应对不同的治疗策略 (例如,延迟反转剂的诱导性)。我们的方法利用了基于 crispr-cas9 的基因组工程的优势, 在目标位置上通过同源重组促进了记者序列的整合。根据对艾滋病毒感染者的研究和为 cas9 介导的基因组工程提供合适的 pam 图案的情况, 选择了整合的目标地点。
在我们的示范性结果中, 我们重点研究了 bach2 基因位点, 它编码为 btb 和 cnc 同源转录调节器2。在接受抗逆转录病毒治疗的慢性艾滋病毒感染者中, bach2 是显示艾滋病毒-1 综合序列3、6、7、8、10的丰富情况之一。我们选择了一个最低限度的艾滋病毒衍生的记者组成的 hiv-1-源长终端重复 (ltr), tdTomato 编码序列, 和牛生长激素 (bgh) 多腺苷信号 (pa), 我们已经针对两个特定的地点在 bovine 内含子5。该协议针对 jurkat 细胞进行了优化, jurkat 细胞是人类 cd4+ t 细胞衍生的悬浮细胞系, 但可以使用其他细胞系, 该协议也相应调整。我们提出了一个详细的工作流程, 选择目标地点, 构建目标载体与同源武器, crispr-cas9 介导的目标的记者进入选定的基因组站点, 生成和选择克隆线, 并进行综合验证新生成的、有针对性的记者细胞系。
Access restricted. Please log in or start a trial to view this content.
研究方案
1. 基因组工程的目标策略与定位向量 (tv) 设计
请注意:基因组工程的第一步是选择和生成必要的工具, 以实现 crispr-cas9 介导的目标。在这一步之前, 应选择基因组整合位点, 选择用于定位的细胞类型, 以及设计一个 hiv 衍生的整合记者。该协议描述了针对一个 hiv-ltr _ tdTomato _ bgh-pa 最小记者到 jurkat 目标细胞的目标。基于 crispr-cas9 的克隆线定位、生成、筛选和验证的工作流程流程图如图1所示。所描述的靶向策略使用 s. pyogenes cas9 (spcas9) 在选定的积分位置生成 grna 定向的 dsdna 断裂。然后, 记者通过同源重组被定位到所选择的基因组位点, 提供了一个非线性的目标向量 (tv), 其中包含了由所谓的 5 ' 和 3 ' 同源臂 (ha)11两侧的记者序列。
-
目标位点、grna 和靶向载体设计的选择
- 根据个别科学问题选择有针对性的基因组位点。使用已公布的在不同研究中发现的患者中的艾滋病毒复发性整合位点清单2、3、4、5、6、7 、8 作为指引。在硅胶提取物中, 使用 ucsc 基因组浏览器 (http:///genome.edu.ucsc.edu) 所需的基因组位点 (完整基因序列或至少 5 kb 的基因组序列) 的基因组序列。
- 选择 20 t 的导 rna (grna), 使用 e-crisp 网络工具 (http://www.e-crisp.org) 定位所选的基因组位点。
- 选择 "智人 grch38" 作为生物体。输入 2, 000 bp 的基因组序列覆盖所需的基因组位点, 在步骤1.1.1 中提取。
- 使用中等应用程序设置开始 grna 搜索 (排除任何 pam、任何 5 ' 基、非目标容忍不匹配和内 cpg 岛屿)。将出现一个列表, 其中可能包含 grna 设计, 在特异性和效率方面从最高得分到较低的分数都排在较低的位置。
- 选择一个 grna, 最好显示出一个高的特异性和效率的高分, 并尽可能接近所需的基因组位点, 以成为目标。
请注意:必须在接近所需的基因组位点和设计特定和高效的 grna 之间找到一个妥协。
- 使用 ncbi 爆炸浏览器 (https://blast.ncbi.nlm.nih.gov) 对参考基因组爆破所选择的 grna 序列, 以检查 grna 结合位点的唯一性。
- 选择 "人类" 作为基因组。输入 grna 序列作为查询序列。选择 "高度相似的序列" (超大序列) 作为程序。确保 grna 序列是唯一的。如果没有, 从步骤1.1.2.3 选择不同的 grna, 然后再次爆炸。
- 一旦选择了 grna 序列,就从步骤1.1.1 中提取的 5 ' 和 3 ' ha 中选择 grna 序列的上下游和下游 1, 000 bp。
请注意: grna 应与所选择的基因组整合位点同源, 并位于相邻的质子点 (pam; 例如, spcas9 的 ngg) 附近 (图 2a)。电视中包含的记者序列是 5 ' 和 3 ' 由 ha 侧翼。has 覆盖了 grna 序列11的上游和下游 1000 bp。完整的 grna 序列不应包括在 ha 中。最多 5 nt 的重叠是可以接受的 (图 2a)。
-
grna 和靶向载体的生成
请注意:关于矢量方案, 请参见图 2b。- 要生成 spcas9 和 grna 表达的载体, 请使用 px330-u6-chemeric _ bb-cbhb-hspcas9 作为主干, spcas9 和单引导 rna (sgrna) 可以从该主干中同时表达。要将 grna 序列克隆到主干中, 请使用 bbsi 限制站点12。
- 要生成电视, 请选择高拷贝质粒作为主干 (例如, pmk 或 cdna3.1)。
- 首先, 使用商业程序克隆套件将记者 (在此协议中: ltr _ tdtoamato _ bgh-pa)组装到构造主干中, 使用商业程序集克隆套件, 并引入 5 ' 和 3 ' 侧翼限制站点 (例如, 5 ' paci 和 3 'smai), 用于随后限制消化克隆的 ba。
- 使用具有校对活性的 dna 聚合酶 (pcr 成分见表1和表 2), 1.1.4 从要针对的细胞类型的基因组 dna (gdna) 中选择的 1000 bp 的 ha 片段进行放大 (参见表 1和表 2,以及骑自行车条件)。然后, 在每个 ha 的 5 ' 和 3 ' 端引入记者侧翼限制网站 (pci 在 5 ' ha 两端, smai 在 3 ' ha 两端)。
- 依次克隆 ha 形成构造主干, 通过限制酶克隆14,15, 以包含记者 (在步骤1.2.2.1 中生成)。首先, 在 5 ' ha 中使用 paci 限制站点克隆, 然后在 3 ' ha 中使用 smai 限制站点克隆。
请注意:如果电视主干包含额外的荧光记者, 则可以通过流式细胞仪评估不需要的主干集成 (请参阅步骤3.2.2 和 3.2.3)。如果电视主干不包含荧光记者, 则必须使用 pcr 评估主干集成 (请参阅步骤 3.2.8)。

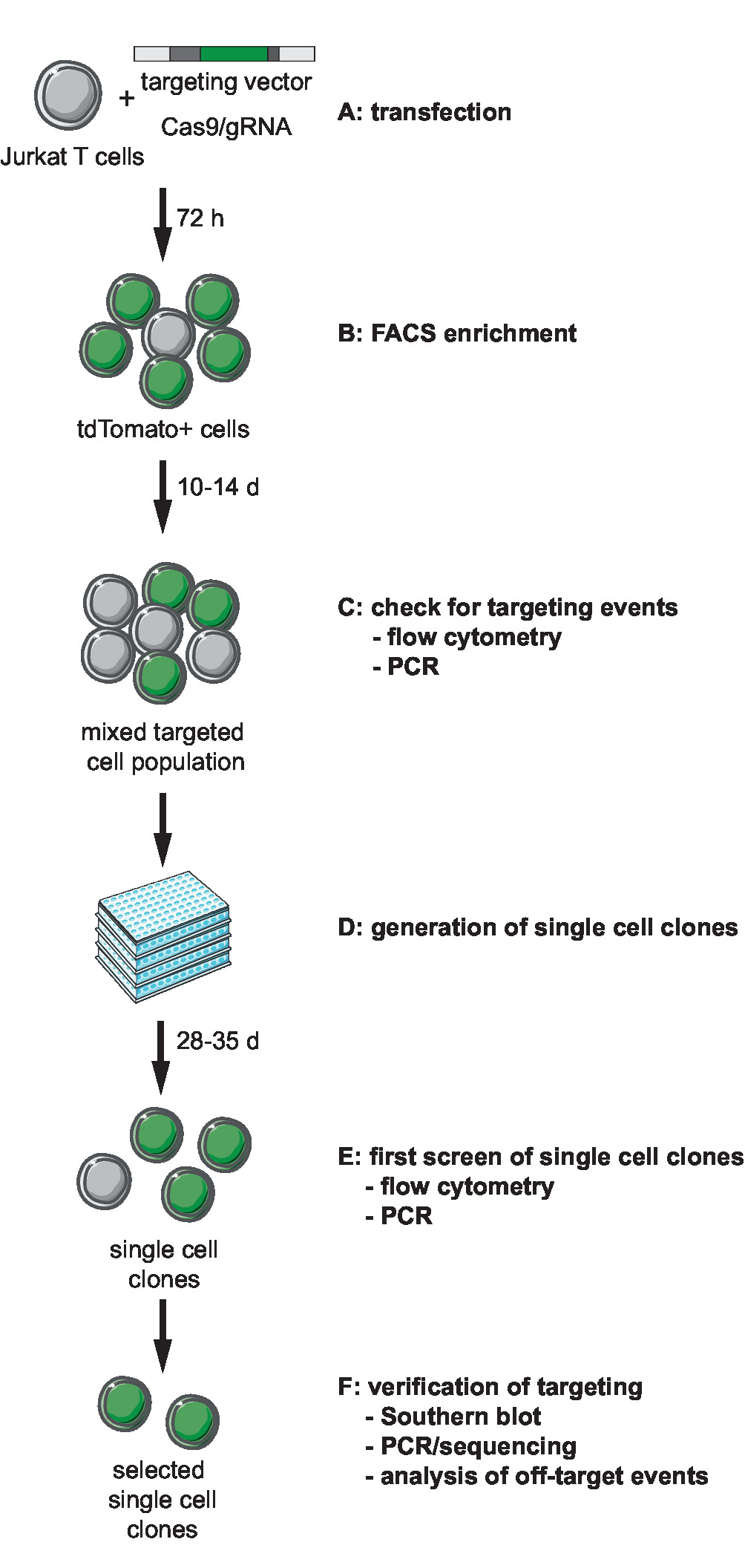
图 1: crispr-cas9 中介定位、生成和选择具有定义的集成站点的克隆记者行的工作流.(a) 生成目标载体, 并利用目标载体和 Cas9/gRNA 表达质粒传递 jurkat t 细胞。(b) 通过流式细胞仪在转染72小时后丰富转染细胞。(c) 让细胞生长 10至14天, 并通过 pcr 和流式细胞术确认靶向事件的发生。(d) 通过限制稀释生成单细胞克隆, 并让克隆体生长3周。(e) 用 pcr 和流式细胞术以96孔的形式筛选克隆, 以确保正确定位。展开选定的克隆。(f) 通过南方印迹、pcr 和测序, 验证选定克隆中的正确靶向, 并分析 cas9 内切酶活性的非靶向事件。请点击这里查看此图的较大版本.
{kind=link}

图 2: 定位策略和矢量设计.(a) grna 和同源武器的选择。20分 grna 与选定的基因组靶点同源, 位于 pam 附近. 同源臂与 grna 的上下游 1, 000 bp 是互补的, 不应包括 grna 序列。(b) 目标向量和 grna cas9 矢量的示意图。目标向量由选择的记者序列组成, 即 5 ' 和 3 ', 由同源臂两侧。grna/cas9 向量是基于 px330-u6-chimeric _ bb-cbhr-hspcas9 主干。(c) 按同源重组确定目标的示意图。目标载体和导引 rna cas9 载体被转染到 jurkat 细胞中。cas9 在基因组靶点进行双链断裂介导 (由 * 表示), 并促进同源重组和将报告者序列整合到基因组靶点。请点击这里查看此图的较大版本.
{kind=link}
2. 基于 crispr-cas9 的 jurkat 细胞目标
-
jurkat 细胞的转染
- 转染前24小时板 1.25 x10 6 jurkat t 细胞在 2.5 ml rpmi 1640 补充 10% (v/v) 胎儿小牛血清 (fcs) 和 4 mL l-谷氨酰胺 [称为 "rpmi. o. 抗生素 (ab)" 每孔6井细胞培养板。对于单个靶向实验, 准备一个完整的6孔板 (即 , 6 口井, 每个井2.5 毫升的细胞悬浮液)。
- 第二天, 用圆形电视和 pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA 联合转染细胞, 使用专门针对 jurkat 细胞的转染试剂。
- 在反应管中加入2μg 的圆形电视和每口2微克的 pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA 到250μl 的商用 rpmi 培养基, 减少血清浓度, 优化为转染 (rpmi, 血清减少 50%), 并在反应管中混合良好。
- 在不接触管壁和旋流的情况下, 慢慢加入12μl 的转染试剂到 dna 介质中。让混合物孵育 15分钟, 滴入一个井的细胞。在37°c 和 5% co 2 下培养细胞.
请注意:转染反应的制备可以扩大。转染后无需更换介质。
-
荧光活化细胞分选法对转染细胞的富集
- 72小时转染后, 将转染的细胞聚集在一起, 对其进行计数, 并为流式细胞仪的富集浓缩做好准备。将细胞收集在一个50毫升的锥形管, 离心机在 300 x x g和室温 (rt) 4分钟, 洗一次细胞在 pbs, 离心机再次, 悬浮颗粒在适当数量的流式细胞仪缓冲液 (pbs 补充 1% fcs + 1 mL edta) 在 1 x 107细胞/ml, 最后转移到一个流式细胞仪管。
- 将细胞与流式细胞管制, 并对那些表达电视荧光记者的细胞进行排序 (例如,本协议中的 tdTomato)。收集 rpmi 1640 中的细胞, 辅以10% 的 fcs、4mm l-谷氨酰胺和 50 u/ml 青霉素和链霉素 (称为 "rpmi w/ab")。
- 在流式细胞调查分类后, 在 rt 以 300 x g 的速度将 rpmi w/ab 的20毫升清洗一次细胞 , 每次. 将细胞颗粒按适当数量的 rpmi w/ab 取出, 并将细胞放入细胞培养板的一个井中, 使其与根据后流式细胞仪的手机号码适当的体积。
请注意:培养对细胞进行小体积分类 (例如, 24 井), 因为在靶向后的第一周观察到相当多的细胞死亡 (高达 80–90%)。 - 在 75 cm²细胞培养瓶中, 将混合靶细胞群扩大到密度为 1 x 10 6 cellsls·ml。这将需要大约10-14天后的外地资产联分类。
-
流式细胞术在混合靶细胞群体中确定靶向事件
- 经过10-14天的扩张 (当细胞在75厘米2细胞培养瓶中达到 1 x10 6 细胞的密度时), 在12井细胞培养中, 在 rpmi w/ab 的 1 毫升中, 在1毫升的 rpmi w/ab 中, 在1毫升的 rpmi w/ab 中, 用 1 x 10 6 细胞的混合靶细胞中装出两口井板。
- 通过添加 50 ngml phorbol 12-myristate 13 醋酸盐 (pma) 和 1μm ionomycin (简称 pma-iono), 在其中一口井中介绍了记者的 hiv ltr (在步骤1.2.2.1 中描述了电视的生成)。使用细胞在第二个和非诱导的控制。培养诱导细胞和非诱导细胞24小时。
- 取非诱导细胞和诱导细胞 (每个) 0.5 ml 的细胞悬浮液, 在 pbs 中清洗一次, 并在200μl 的流式细胞仪缓冲液中悬浮每个细胞悬浮液。
- 流式细胞仪分析 100, 000个细胞。根据前后散射中的大小对活单细胞进行门, 并分析荧光记者基因的表达。
请注意:在这一步 (流式细胞仪分选后 10-14天), 荧光记者通过转染的瞬态表达应该不再是可检测的。此时的荧光表达表明了报告者序列的基因组整合。
-
pcr 检测混合靶向细胞群体基因组 dna 的靶向事件
请注意:要通过 pcr 检测目标事件, 请设计两个特定于 5 ' 积分 (int.) 连接点和 3 ' int. 结点的引物对。对于 5 ' int. 结点 pcr, 正向引物应结合 5 ' ha 的上游和记者 ltr 中的反向引物 (图3a中的引物 p1 和 p2)。3 ' int. 结点 pcr 的引物对应该从记者的 pa 到 3 ' ha 下游的 100–200 bp (图 3a中的引物 p3 和 p4)。引物 p1 和 p4 还将用于扩增混合目标人群中的非目标等位基因。有关示意图, 请参见图 3a。- 从步骤2.2.4 从混合靶向细胞群的2毫升细胞悬浮液中制备 gdna。根据制造商的协议使用 gdna 提取试剂盒。然后制备非靶向细胞的 gdna 作为对照。
- 使用高保真 dna 聚合酶执行 int. 结点 pcr (引物 p半p2 和 p p4 分别为 5 ' 和 3 ' int. 结点) 和非靶向等位基因 pcr (引物 p一点4) ( pcr 成分和循环条件见表 3和表 4).在1.5% 琼脂糖凝胶上分析5μl 的 pcr 产物。
请注意:如果混合目标群体中含有经过基因组工程的细胞, 则应观察到非靶向细胞的 gdna 中无法检测到的特定 pcr 产物 (阴性对照)。对于非靶向等位基因 pcr, 应观察目标细胞和非靶向细胞大小相同的产物 (基因组 p1 和 p4 引物的阳性对照)。如果没有观察到带, 请考虑通过增加循环次数或改变 pcr 缓冲液 (例如通过添加 dmso 或增加 mg2 +的量) 或通过改变聚合酶来改变 pcr 循环条件。
3. 克隆线的生成和正确定位的筛选
请注意:通过流式细胞仪和 pcr 确认混合靶向细胞群中的靶向事件 (第2.2–2.4 节) 后, 生成单细胞克隆 (持续时间: 28至35天) 并进行筛选, 以正确整合报告序列。
- 通过稀释镀生成单细胞克隆
- 提前准备 jurkat 调节培养基: 从健康的、未经处理的 jurkat t 细胞中取出 rpmi w/ab 培养基, 生长到 1 x10 6 cells/mL ml, 离心剂 5分钟, 在 300 x克处, 并使用0.22 微米的注射器过滤器过滤器。
请注意:将有条件的介质保持在 4°c, 以便短期储存, 或在-20°c 下存放超过1周。在稀释电镀之前准备20至30毫升的条件介质。 - 在10-14天的扩张后, 将目标细胞从步骤2.2.4 数一数, 并在 rpmi w/ab 中将其稀释到 1 x10 5 细胞的浓度。取100μl 的 1 x 10 5 cellsl 溶液和9.9 毫升培养基稀释, 达到 1, 000 细胞的浓度。取 1, 000 毫升溶液的1毫升, 用9毫升培养基稀释, 达到100细胞浓度。
- 板96孔板, 每口井1个细胞, 每口井2个细胞。对于每口1细胞, 取100细胞溶液中的1毫升, 在无菌试剂库中轻轻与5毫升的条件培养基和4毫升的新鲜培养基混合。
- 对于每口井2个细胞, 取100细胞溶液中的2毫升, 轻轻与条件培养基的5毫升和新鲜培养基的3毫升轻轻混合。采用多通道管板, 将96孔圆底板与每个井的单孔稀释量为100μl。
注:每个靶向结构5至 10个96孔板足以获得克隆进行筛选。 - 堆叠96孔板, 在每个井中用含有3毫升 pbs 的6孔板覆盖每个堆栈, 并在37°c 的加湿细胞培养孵化器中孵育板, 采用 5% co2, 为期 3周.
请注意:在此期间不要改变细胞培养介质。不要每周打开孵化器超过一两次。在有开水水库的孵化器中观察到了最佳的结果。 - 经过3周的孵化, 用光学显微镜 (4倍放大倍率) 直观地确认生长的菌落的存在, 并用生长的菌落标记, 使它们可以看到在井底的点。
- 准备一个96孔圆底板与100μl 的 rpmi w/ab 每口井。通过移液轻轻重新悬浮有标记的井的细胞。将100μl 的细胞悬浮液转移到新的96孔板的井中, 该板材已经含有100μl 的 rpmi w/ab, 然后通过移液轻轻混合。将该细胞悬浮液的100μl 转移到第二个空的96孔圆底板中复制板。
- 继续与所有有标记的井与生长的殖民地。用200μl 的 rpmi w/ab 介质填充所有的空白井。在37°c 和 5% co 2 下培育板材.
请注意:其中一个板块将用于扩大单细胞克隆体 ("股票板"), 另一个板作为筛选的 "重复板"。
- 提前准备 jurkat 调节培养基: 从健康的、未经处理的 jurkat t 细胞中取出 rpmi w/ab 培养基, 生长到 1 x10 6 cells/mL ml, 离心剂 5分钟, 在 300 x克处, 并使用0.22 微米的注射器过滤器过滤器。
- 流式细胞仪和 pcr 筛选单细胞克隆
请注意:当单细胞克隆体扩展时, 使用重复的板3.1.8 通过 pcr (步骤 3.2.4 3.2.12) 筛选单细胞克隆, 以实现报告序列的存在, 并通过流式细胞术表达荧光记者 (步骤 3.2.2-3.2.3)(图 3c)。- 让复制板孵育 24至48小时, 再复制板。为此, 请在每口井中加入 100μl rpmi w/ab, 通过移液轻轻混合, 并使用多通道移液将100μl 传输到新的96孔圆底板上。使用一个板进行流式细胞仪筛选, 另一个板用于基于 pcr 的筛选。
- 对于流式细胞仪筛选, 用 pma-iono 刺激细胞。准备一个0.1μl 的离子环素 (1 mm 库存), 0.1μl 的 pma (50μgμl 库存) 和 4.8μl rpmi w/ab 每口井, 然后添加5μl 的主混料每口井。
请注意:诱导是必要的, 以成功地识别克隆, 其中 ltr 可能是转录沉默, 因此荧光记者没有表达。 - 让细胞孵育 24小时, 为流式细胞术准备细胞, 如步骤2.3.3 所述。根据前后散射中的大小对任何可行的单细胞进行标记, 并通过流式细胞仪分析荧光记者基因的表达 (例如结果, 见图 3c)。如果电视主干包含第二个荧光记者与启动子 (例如, gfp), 也筛选主干记者表达的任何克隆 (请参阅步骤1.2.2 和以下说明的解释)。
请注意:主干记者表达式表示不需要的主干序列集成。 - 一旦第二个重复板中的克隆充分生长 (通常是96孔板复制后 24至48小时), 准备含有 gdna 的细胞裂解物进行 pcr 筛选。在 rt 以 300 x克的速度离心板 10分钟, 小心地脱下上清液, 而不会干扰细胞颗粒。
请注意:制备裂解物和 pcr 反应的所有步骤都可以用多通道移液器进行。 - 用100微克的 pbs 清洗细胞, 在 rt 用 300 x克的温和移液和离心5分钟。取下 pbs 并加入200μl 的裂解缓冲液 [200 mm ncl, 100 mm Tris-HCl ph 8 8.5, 5 mm edta, 0.1% sds; 然后加入250–1000微米的蛋白酶 k (冻干粉末, 称量新鲜)]。通过移液轻轻混合, 并将悬浮液转移到新的 pcr 板。
- 用石蜡膜密封板, 在55°c 时在热环中孵育1小时。以最大速度离心 10分钟 (3, 000 x g), 将细胞碎片向下旋转, 并将上清液转移到新的 pcr 板上。
请注意:板材中的细胞裂解物可在此阶段保存在 4°c, 直至进一步使用。 - 制备96孔 pcr 板, dh2o为110μl, 并加入10μl 细胞裂解液 (1:12 稀释)。细胞裂解物可能是粘性的, 很难移液。使用至少20μl 管头。
- 在热环器中, 在99°c 孵育 10分钟, 使蛋白酶 k 失活。随后使用灭活和稀释的细胞裂解物进行 pcr 筛选。
- 设计引物用于筛选 pcr (p5 和 p6) 的基础上, 根据选定的记者序列来扩增 500–800 bp 的报告序列。对于阳性控制 pcr, 使用引物 p7 和 p8 扩增 630 bp 的野生类型的、非靶向的基因组位点 (nup188基因) (图 3c和表 5)。设计第三个引物对, 放大 500–600 bp 的电视主干, 作为电视主干序列 (主干 pcr) 的非特异性集成的控制。
- 对于筛选、控制和主干 pcr, 请使用商业 pcr 组合 (pcr 成分和循环条件见表 6和表 7). 使用步骤3.2.8 制备的稀释和灭活裂解液中的2μl 作为模板, 并以96孔格式运行 pcr 扩增过程的38至40个周期。
- 在1.5% 琼脂糖凝胶上分析5μl 的 pcr 产物。
请注意:对于控制 pcr, 应观察到每个样品的特定带 630 bp, 这证实细胞裂解物的质量足以实现 pcr。pcr 筛选中的特定波段 (500–800 bp 取决于引物设计) 表示记者序列的集成。主干 pcr 的特定波段 (500–600 bp, 取决于引物设计) 表示电视主干序列的不需要集成 (例如, 请参见图 3c)。 - 结合流式细胞术 (步骤 3.2.3) 和基于 pcr 的筛选 (步骤 3.2.12) 的结果。在流式细胞术中 pma-iono 诱导后, 选择 pcr 产品大小正确的克隆, 并对荧光记者进行阳性控制 pcr 和表达。排除在主干 pcr 或电视后骨编码荧光蛋白表达中显示任何 pcr 产物的克隆, 表明电视主干序列的不特异性整合。
- 逐步将选定的克隆从96孔的股票板扩大到更大的井形形式 (48/24/126-好), 直到每2至3天添加新鲜培养基, 达到 t75 细胞培养瓶格式。保持细胞密度在 1 x 10 5和 1 x 10 6 cells/mL ml之间。
- 确保在扩张过程中制备克隆细胞库存: 在 rt 上将细胞计数, 离心机在 300 x g 处计数 5分钟, 丢弃上清液, 并在 fcs + 10% dmso 中以 5 x 10 6 细胞/ml轻轻悬挂细胞。在低温小瓶中加入液体, 并使用冷冻容器在 1°c/min 处将细胞冷冻至80°c。对于长期储存, 将它们转移到液体n2。
请注意:建议在 gdna 制剂扩张过程中保留 t75 细胞培养瓶 (即大约 1 x 107 细胞), 以核实南方印迹的靶向性 (见第3.4 节)。
- 利用 pcr/排序在选定克隆体中验证积分位点
请注意: 5 ' 和 3 ' int. 点的选定克隆被 pcr 扩增, 并提交给桑格测序, 以验证正确的目标在 dna 序列水平。- 使用商业 gdna 提取试剂盒制备选定克隆体和 jurkat 野生类型细胞的 gdna。
- 如步骤2.4 所述, 使用引物对绑定 5 ' 端的记者和上游的 5 ' ha 为 5 ' int. int 结 (引物 p1 和 p2) 和 3 ' 端的记者和下游的 3 ' ha 为 3 ' int. int 结 (引物 p3 和 p4)。使用引物 p1 和 p4 放大不带记者集成程序的等位基因表上的目标集成站点 (图 4a)。
- 用 100–200 ng 的 gdna 作为模板准备 pcr 反应, 并使用具有校对活性的聚合酶进行 pcr (pcr 成分和循环条件见表 1和表 2 )。
请注意:如果没有观察到带, 请考虑通过增加循环次数或更改 pcr 缓冲液 (例如, 添加 dmso 或增加 mg2 +量) 或通过改变聚合酶来改变 pcr 循环条件。 - 在1.5% 琼脂糖凝胶上分析5μl 的 pcr 产物。如果观察到正确的带尺寸, 请使用商业试剂盒纯化剩余的 pcr 产品, 并对其进行 sanger 测序。通过将等位基因与预期序列对齐, 验证它们的序列 5 ' int. 交汇点、3 ' int. 交界处和没有记者集成的等位基因的目标站点。
请注意:记者未整合的同源等位基因很可能会在目标站点显示 cas9 介导的变化, 如核苷酸插入或删除 (图 4a)。 - 对于对齐后显示正确 int. 结点序列的克隆, 执行 pcr 放大整个目标记者, 并对其进行 sanger 测序, 以验证积分器的正确序列。
- 用于验证选定克隆中目标的南方印迹分析
请注意:需要对选定的克隆进行南方印迹分析, 以验证正确的定位, 并排除在目标整合站点可能发生的 cas9 介导的重组事件。- 制定适当的 gdna 消化策略, 并在开始实验之前对设计进行探索。
- 选择一种限制酶来限制 gdna, 在目标地点产生2至 10 kb 长度的适当片段。某些限制性酶, 如asp718、 bamhi、 bgli、 bl l ii、eco rv、 hindiii、 nco i、 psti、pvu ii、sca i 、 stui 和斯斯特我已成功地用于消化高分子量 gdna。
- 设计两种不同的南方探针: 一种是报告仪特定探针和基因组探针。报告器特定的探针杂交到一个序列内的报告 (即, tdtomato 特异性探针).基因组探针杂交到基因组区域接近 (但不重叠) 一个 ha。
- 选择基因组探针, 使基因组探针结合所检测到的 gdna 消化片段的长度 (超过 2 kb) 与目标和非靶向等位基因不同 (图 4b)。建议探头长度为400到 1, 000 bp。
- 设计 pcr 引物来扩增两个所需的探针。使用高保真 dna 聚合酶从电视模板中放大报告仪特异性探针 (pcr 成分和循环条件见表 3和表4 )。
- 用具有校对活性的 dna 聚合酶的商业 gdna 提取试剂盒制备的野生型 jurkat gdna 中的基因组探针进行放大 (pcr 成分和循环条件见表 1和表 2 )。在琼脂糖凝胶上纯化 pcr 产品, 并根据制造商的说明使用商业凝胶提取试剂盒提取碎片。
- 从野生类型 jurkat 细胞的 1 x 10 7 细胞和3.2.14 的步骤中提取选定的克隆细胞中的高分子量 gdna。
- 通过离心在 rt 中离心 5分钟, 将细胞颗粒状, 用 pbs 清洗一次, 并将颗粒悬浮在4毫升的裂解缓冲液中 [200 mm nacl, 100 mm tris-hcl ph 值 8, 5 mL edta, 0.1% sds; 然后加入250-1000μml 蛋白酶 k (冻干粉末), 称重的新鲜)。在55°c 下孵化, 在桌面热敏器中以350转/分的速度晃动。
- 加入4毫升异丙醇, 经反转 10 ~ 20次混合。gdna 应该成为可见的白色沉淀。将沉淀的 gdna sprop 放到玻璃移液器的细端上, 在70μl 的 70% etoh 中洗涤, 并在 rt (5至10分钟) 干燥。
- 将沉淀物放入含有500μl 的1xe 缓冲液 (10 mm Tris-HCl ph 值 8.0, 1 mm edta) 的 1.5 ml 反应管中, 在4°c 下溶解, 在350转/时晃动。任何从这个阶段的 gdna 移液应与宽孔尖端, 以避免剪切。
请注意:制备高分子量 gdna 对于南方印迹分析至关重要, 市售 gdna 制备试剂盒不合适。
- 在60μl 反应中, 用6μl 酶 (20 units/μl) 进行60μl 反应, 消化 (两次) 15 微克的克隆细胞和野生类型 jurkat 细胞的 gdna (见步骤 3.4.1.1): 首先, 加入 dna、消化缓冲液和 ddh2o,在37°c 孵育然后在酶特异性消化温度下加入酶和孵育。每个南方探针需要15微克的消化 gdna。
- 使用60μl 限制消化的7μl 对1% 琼脂糖凝胶进行分析凝胶电泳。涂片表明完全消化和良好的 dna 质量为南方印迹分析。
- 通过添加 1:10 3 m 醋酸钠和2卷 100% etoh 来沉淀剩余的限制性消化, 然后在-80°c 孵育 1小时, 在 15600 x克的4°c 下孵育30分钟。
- 丢弃上清液, 用 70% etoh 清洗颗粒。在 15600 x克的4°c 下离心 15分钟, 丢弃上清液, 让颗粒在 rt 短暂干燥, 并溶解在20μl 的 ddh2o中。
- 运行1% 琼脂糖/凝胶, 每条车道加载20升消化的 gdna。在 60 v, 400 毫安时运行凝胶2小时。
请注意:琼脂糖凝胶和运行电压的百分比可根据步进3.4.1.1 计算的南方印迹检测的预期碎片尺寸进行调整。补充协议 (步骤1至 18) 详细介绍了南方印迹分析的以下步骤。这些步骤包括: 清洗印迹凝胶, 涂布尼龙膜, 放射性探针生成, 探针杂交, 和开发的脂肪膜。根据南方策略, 将步骤 18 (补充协议) 中的自动分类开发后获得的条带模式与预期模式进行比较 (例如结果, 请参见图 4b)。
- 制定适当的 gdna 消化策略, 并在开始实验之前对设计进行探索。
- 对目标外事件的分析
请注意:由于 crispr-cas9 介导的基因组工程可以产生非目标效应, pcr 放大了在选定克隆体中硅胶预测的离目标位点中排名最高的10个位点, 并对其进行桑格测序。- 使用 cctop 16 (http://crispr.cos.uni-heidelberg.de) 生成在硅胶预测的非目标序列中排名最高的10个序列的列表。
- 输入 grna 序列, 包括用于定位的 pam 作为查询序列。选择 "ngg" 作为 pam, 选择 "人类基因组" 作为离目标预测的参考。
- 将最大总匹配设置为 "4", 将目标站点长度设置为没有 pam 的 grna 长度。输出文件将为各自的 grna 提供一个基因组外位点的排名列表。
- 在硅胶提取基因组序列 500 bp 上游和下游的10个排名最高的离目标命中使用 ucsc 基因组浏览器 (http://genome.edu.ucsc.edu) 和从 cctop 结果列表中命中的目标的位置。
- 对于要分析的目标地点的每一个, 设计一个 pcr 引物对, 放大长度为600至 700 bp 的片段, 包括预测的目标外位点。
- 使用商业 gdna 提取试剂盒从选定的克隆体和 jurkat 野生类型细胞中提取 gdna。对于每个非目标位点, 使用具有校对活性的 dna 聚合酶 (pcr 成分和循环条件见表 1和表 2 ) 对野生类型和各自的克隆源 gdna 进行 pcr。
- 在1.5% 琼脂糖凝胶上分析5μl 的 pcr 产物。如果观察到正确的带尺寸, 请使用商业 pcr 纯化试剂盒纯化剩余的 pcr 产品, 并对其进行 sanger 测序。比较 jurkat 细胞中的非目标位点序列和目标克隆。
- 使用 cctop 16 (http://crispr.cos.uni-heidelberg.de) 生成在硅胶预测的非目标序列中排名最高的10个序列的列表。
Access restricted. Please log in or start a trial to view this content.
结果
在这个具有代表性的实验中, 我们选择了一个最小的 hiv-1 衍生的记者, 包括一个 ltr, ttomato 编码序列, 和多 a 信号序列到两个位点内含子5的bach2 基因17。根据在对感染艾滋病毒的患者 2,4, 5,6的原代 t 细胞进行不同研究时发现的已公布的复发整合位点的接近度, 选择了靶向定位位?...
Access restricted. Please log in or start a trial to view this content.
讨论
在这里, 我们描述了一个协议, 以生成 hiv-1 衍生 jurkat 记者模型与选择的天方集成站点应用基于 crispr-cas9 的基因组工程。
在规划阶段, 议定书的几个要点需要认真注意。首先, 应仔细选择要针对的位点, 因为某些位点可能比其他位点更容易瞄准 (例如,取决于区域的染色质状态和目标序列本身)。重复序列很难克隆到靶向载体中, 在基因组中通常不是唯一的。用 crispr-cas9 系?...
Access restricted. Please log in or start a trial to view this content.
披露声明
作者没有什么可透露的。
致谢
我们感谢 britta wweeloh 和 bettina abel 的技术援助。我们还感谢 arne düsedau 和 jara hennesen (heinrich pette 研究所流式细胞术技术平台) 的技术支持。
Access restricted. Please log in or start a trial to view this content.
材料
| Name | Company | Catalog Number | Comments |
| pX330-U6-Chimeric_BB-cBh-hSpCas9 | Addgene | 42230 | vector for expression of SpCas9 and gRNA |
| pMK | GeneArt | mammalian expression vector for cloning | |
| cDNA3.1 | Invitrogen | V79020 | mammalian expression vector for cloning |
| BbsI | New England Biolabs | R0539S | restriction enzyme |
| NEBuilder Hifi DNA Assembly Cloning Kit | New England Biolabs | E5520S | Assembly cloning kit used for target vector generation |
| TaqPlus Precision PCR System | Agilent Technologies | 600210 | DNA polymerase with proofreading activity used for amplification of homology arms (step 1.2.2.2), verification of integration site and reporter sequence (step 3.3.3 and 3.3.5), generation of genomic probe for Southern blot (step 3.4.1.5) and analysis of off-target events (step 3.5.4) |
| 96-well tissue culture plate (round-bottom) | TPP | 92097 | tissue culture plates for dilution plating |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530 L | DNA polymerase used for detection of targeting events (step 2.4.2) and generation ofreporter-specific probe for Southern blot (step 3.4.1.4) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D9170 | dimethyl sulfoxide as PCR additive |
| Magnesium Chloride (MgCl2) Solution | New England Biolabs | B9021S | MgCl2 solution as PCR additive |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | dNTP mixture with 10 mM of each nt for PCR reactions |
| 5PRIME HotMasterMix | 5PRIME | 2200400 | ready-to-use PCR mix used for screening PCR (step 3.2.11) |
| QIAamp DNA blood mini kit | Qiagen | 51106 | DNA isolation and purification kit |
| QIAquick PCR Purification Kit | Qiagen | 28106 | PCR Purification Kit |
| RPMI 1640 without glutamine | Lonza | BE12-167F | cell culture medium |
| Fetal Bovine Serum South Africa Charge | PAN Biotech | P123002 | cell culture medium supplement |
| L-glutamine | Biochrom | K 0282 | cell culture medium supplement |
| Penicillin/Streptomycin 10.000 U/mL/ 10.000 µg/mL | Biochrom | A 2212 | cell culture medium supplement |
| Gibco Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | 31985062 | cell culture medium with reduced serum concentration optimized for transfection |
| TransIT-Jurkat | Mirus Bio | MIR2125 | transfection reagent |
| phorbol 12-myristate 13-acetate | Sigma-Aldrich | P8139-1MG | cell culture reagent |
| Ionomycin | Sigma-Aldrich | I0634-1MG | cell culture reagent |
| Syringe-driven filter unit, PES membrane, 0,22 µm | Millex | SLGP033RB | filter unit for sterile filtration |
| Heracell 150i incubator | Thermo Fisher Scientific | 51026280 | tissue culture incubator |
| Amershan Hybond-N+ | GE Healthcare | RPN1520B | positively charged nylon membrane for DNA and RNA blotting |
| Stratalinker 1800 | Stratagene | 400072 | UV crosslinker |
| High Prime | Roche | 11585592001 | kit for labeling of DNA with radioactive dCTP using random oligonucleotides as primers |
| illustra ProbeQuant G-50 Micro Columns | GE Healthcare | 28-9034-08 | chromatography spin-columns for purification of labeled DNA |
参考文献
- Hughes, S. H., Coffin, J. M. What Integration Sites Tell Us about HIV Persistence. Cell Host and Microbe. 19 (5), 588-598 (2016).
- Marini, B., Kertesz-Farkas, A., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 521 (7551), 227-231 (2015).
- Cesana, D., Santoni de Sio, F. R., et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nature Communications. 8 (1), 498(2017).
- Cohn, L. B., Silva, I. T., et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell. 160 (3), 420-432 (2015).
- Han, Y., Lassen, K., et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 78 (12), 6122-6133 (2004).
- Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. The Journal of Infectious Diseases. 195 (5), 716-725 (2007).
- Wagner, T. A., Mclaughlin, S., et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 345 (6196), 570-573 (2014).
- Maldarelli, F., Wu, X., et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 345 (6193), 179-183 (2014).
- Jordan, A., Bisgrove, D., Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 22 (8), 1868-1877 (2003).
- Mack, K. D., Jin, X., et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. Journal of Acquired Immune Deficiency Syndromes. 33 (3), 308-320 (2003).
- Byrne, S. M., Ortiz, L., Mali, P., Aach, J., Church, G. M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Research. 43 (3), 1-12 (2014).
- ZhangLab. CRISPR Genome Engineering Toolbox: Target Sequence Cloning Protocol. Addgene website. , Available from: https://www.addgene.org/static/cms/filer_public/e6/5a/e65a9ef8-c8ac-4f88-98da-3b7d7960394c/zhang-lab-general-cloning-protocol.pdf (2013).
- Moser, F. Addgene. Gibson Assembly Protocol. Addgene website. , Available from: https://www.addgene.org/protocols/gibson-assembly/ (2009).
- Addgene Plasmid Cloning by PCR. Addgene website. , Available from: https://www.addgene.org/protocols/pcr-cloning/ (2014).
- Addgene Plasmid Cloning by Restriction Enzyme Digest (aka Subcloning). Addgene website. , Available from: https://www.addgene.org/protocols/subcloning/ (2013).
- Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., Mateo, J. L. CCTop: An intuitive, flexible and reliable CRISPR-Cas9 target prediction tool. Public Library of Science (PLoS) ONE. 10 (4), (2015).
- Lange, U. C., Bialek, J. K., Walther, T., Hauber, J. Pinpointing recurrent proviral integration sites in new models for latent HIV-1 infection. Virus Research. 249, (2018).
- Bialek, J. K., Dunay, G. A., et al. Targeted HIV-1 Latency Reversal Using CRISPR-Cas9-Derived Transcriptional Activator Systems. PloS ONE. 11 (6), e0158294(2016).
- Lee, C. M., Davis, T. H., Bao, G. Examination of CRISPR-Cas9 design tools and the effect of target site accessibility on Cas9 activity. Experimental Physiology. 103 (4), 456-460 (2018).
- Jensen, K. T., Fløe, L., et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Letters. 591 (13), 1892-1901 (2017).
- Simonetti, F. R., Sobolewski, M. D., et al. Clonally expanded CD4 + T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 113 (7), 1883-1888 (2016).
- Chen, H. C., Martinez, J. P., Zorita, E., Meyerhans, A., Filion, G. J. Position effects influence HIV latency reversal. Nature Structural and Molecular Biology. 24 (1), 47-54 (2017).
Access restricted. Please log in or start a trial to view this content.
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。