
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Basati su CRISPR-Cas9 genoma ingegneria per generare modelli di Jurkat Reporter per infezione da HIV-1 con integrazione Proviral selezionati siti
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Vi presentiamo un flusso di lavoro ingegneria di genoma per la generazione di nuovi modelli in vitro per infezione da HIV-1 che ricapitolano integrazione proviral a selezionati siti genomici. Targeting dei reporter HIV-derivato è facilitato dalla manipolazione del genoma CRISPR-Cas9-mediata, site-specific. Sono disponibili protocolli dettagliati per cella singola clone generazione, lo screening e la corretta verifica targeting.
Abstract
Virus di immunodeficenza umana (HIV) integra il DNA provirale non casualmente nel genoma della cellula ospite alle ricorrenti siti e genomico hotspot. Qui presentiamo un protocollo dettagliato per la generazione di nuovi modelli in vitro per l'infezione da HIV con siti di integrazione genomica selezionate utilizzando la tecnologia di ingegneria basati su CRISPR-Cas9 genoma. Con questo metodo, una sequenza di reporter di scelta può essere integrata in un locus genomico mirato, selezionato, riflettendo siti di integrazione clinicamente rilevanti.
Nel protocollo, la progettazione di un reporter di HIV-derivato e scegliendo di una sequenza di destinazione sito e gRNA sono descritti. Un vettore targeting con braccia di omologia è costruito e transfected nelle cellule di T di Jurkat. La sequenza di reporter si rivolge al sito genomico selezionato tramite la ricombinazione omologa, facilitata da una pausa di doppio filo Cas9-mediata al sito di destinazione. Cloni unicellulari sono generati e proiettati per ottimizzazione degli eventi mediante citometria a flusso e PCR. Cloni selezionati vengono quindi espansi e targeting corretta viene verificato mediante PCR, sequenziamento e macchiare del sud. Vengono analizzati potenziali eventi fuori bersaglio di ingegneria CRISPR-Cas9-mediata del genoma.
Utilizzando questo protocollo, sistemi di coltura cellulare romanzo può essere generato l'infezione da HIV tale modello presso siti di integrazione clinicamente rilevanti. Anche se la generazione di cloni unicellulari e verifica dell'integrazione sequenza corretta reporter è lunghe, le linee clonali risultante sono potenti strumenti per analizzare in modo funzionale scelta del sito di integrazione proviral.
Introduzione
Integrazione del DNA provirale nel genoma ospite al momento dell'infezione è un passo fondamentale nel ciclo di vita del virus di immunodeficenza umana (HIV). A seguito di integrazione, l'HIV persiste attraverso la definizione di latenza in sottoinsiemi di cellule T CD4 + longevi come cellule T CD4 + di memoria. Integrazione di HIV sembra essere non casuale1,2. Un numero di hotspot genomico con DNA provirale ricorrentemente integrato è stato rilevato in parecchi studi attraverso il sequenziamento dei siti di integrazione in individui acutamente e cronicamente infettati2,3,4 ,5,6,7,8. È interessante notare che, in alcuni di questi siti di integrazione, stesso locus è stato rilevato in una grande frazione delle cellule infettate, che conduce all'idea che integrazione alle ricorrenti siti potrebbe influenzare positivamente l'espansione clonale1.
Per avanzare la nostra comprensione del significato dei siti di integrazione ricorrenti, scelta del sito di integrazione proviral deve essere esplorata. Tuttavia, diversi aspetti tecnici ostacolano lo studio integrazione HIV sito scelta e le conseguenze. Ampiamente utilizzati modelli di coltura cellulare per latenza di HIV come linee cellulari JLat non riflettono clinicamente rilevante integrazione ricorrenti siti9. Studi su cellule primarie derivate dal paziente, da un lato, attivare la descrizione di integrazione sito paesaggio dall'ordinamento ma non consentono analisi funzionali. A nostra conoscenza, nessun modello sperimentale adeguato è disponibile per analizzare i siti selezionati clinicamente rilevante integrazione funzionalmente.
Qui presentiamo un flusso di lavoro dettagliato per generare nuovi modelli per l'infezione da HIV usando tecnologia di ingegneria basati su CRISPR-Cas9 genoma. Il flusso di lavoro descritto nel presente documento può essere utilizzato per generare linee cellulari derivati da cellule T reporter che modellano l'infezione da HIV, che trasportano un reporter proviral genomicamente integrato in un sito di integrazione selezionate. Servono così come nuovi strumenti per esplorare come il sito di integrazione proviral può influenzare la biologia di HIV e come provirus risponde a diverse strategie terapeutiche (ad esempio, 270ms dagli agenti inversione di latenza). Il nostro metodo utilizza i vantaggi dell'ingegneria basati su CRISPR-Cas9 genoma, in quale integrazione del reporter sequenza di ricombinazione omologa è facilitato da una pausa di doppio filo indotta da nucleasi Cas9 al sito di destinazione. Siti di destinazione per l'integrazione sono scelti secondo la vicinanza ai siti integrazione ricorrente descritto da studi su individui affetti da HIV e la presenza di motivi di PAM adatti per ingegneria Cas9-mediata del genoma.
Nei nostri risultati di esemplare, ci siamo concentrati sul luogo del gene BACH2, che codifica per il regolatore trascrizionale di BTB e CNC omologia 2. In individui HIV-infettati cronicamente sulla terapia antiretrovirale, BACH2 è uno dei loci risultati arricchimento di HIV-1 integrato sequenze3,6,7,8,10. Abbiamo scelto un reporter HIV-derivato minimo costituito da HIV-1-derivati lungo terminale di ripetizione (LTR), sequenza di codificazione di tdTomato e ormone della crescita bovina (BGH) segnale di poliadenilazione (PA), che siamo presi di mira a due siti specifici in BACH2 introne 5. Il protocollo presentato è ottimizzato per Jurkat cellule, un'umano CD4 + linea cellulare T cellula-derivati sospensione, ma altri cell le linee possono essere utilizzate e il protocollo adattato di conseguenza. Vi presentiamo un flusso di lavoro dettagliato per la selezione del sito di destinazione, costruzione del vettore di destinazione con le braccia di omologia, CRISPR-Cas9-mediata di targeting del reporter nel sito genomico scelto, la generazione e la selezione di linee clonali e complete verifica delle varietà di cellula di reporter appena generato, mirati.
Access restricted. Please log in or start a trial to view this content.
Protocollo
1. strategia di targeting per genoma ingegneria e Targeting disegno vettoriale (tv)
Nota: Il primo passo di ingegneria genoma prevede selezione e generazione degli strumenti necessari per il CRISPR-Cas9-mediated targeting. Selezione di un locus di sito di integrazione genomica, scelta del tipo di cella per il targeting e progettazione di un reporter di HIV-derivati per l'integrazione deve precedere questo passaggio. Questo protocollo descrive il targeting di un reporter di minimo HIV-LTR_tdTomato_BGH-PA in cellule bersaglio Jurkat. Un diagramma di flusso del flusso di lavoro basati su CRISPR-Cas9 targeting, generazione, selezione e verifica delle linee clonali è raffigurato nella Figura 1. La strategia di targeting descritta utilizza il Cas9 di S. pyogenes (SpCas9) per generare interruzioni di dsDNA gRNA-diretto a un sito di integrazione selezionato. Il reporter è quindi mirato nel locus genomico selezionato attraverso ricombinazione omologa fornendo un grezzo targeting vettoriale (tv) che contiene la sequenza di reporter affiancata da cosiddetto 5' e 3' omologia braccia (HA)11.
-
Scelta del luogo mirato, gRNA e targeting per disegno vettoriale
- Scegliere thegenomic locus mirato basato sulla questione scientifica individuale. Uso pubblicati gli elenchi dei siti di integrazione ricorrenti dell'HIV ha trovati nei pazienti in diversi studi2,3,4,5,6,7,8 come un Guida di riferimento. In silico estrarre la sequenza genomic del locus genomico desiderato essere mirata (sequenza del gene completo) o almeno 5 kb della sequenza genomic utilizzando UCSC genome browser (http:// genome.edu.ucsc.edu).
- Scegliere Guida RNAs (gRNAs) di 20 nt per il targeting del locus genomico selezionate con il webtool E-CRISP (http://www.e-crisp.org).
- Selezionare "Homo sapiens GRCh38" come l'organismo. Ingresso 2.000 bp della sequenza genomica che copre il desiderato locus genomico estratto al punto 1.1.1.
- Avviare una ricerca di gRNA tramite impostazioni applicazione medio (qualsiasi PAM, qualsiasi base, 5' off-target tollerano disallineamenti e isole introni/CPG sono esclusi). Verrà visualizzato un elenco con i disegni possibili gRNA, rango dal più alto al più basso punteggi per la specificità e l'efficienza.
- Selezionare una gRNA che preferibilmente Mostra un punteggio alto per la specificità e l'efficienza ed è più vicino possibile al locus genomico desiderato da impostare come destinazione.
Nota: Un compromesso tra la vicinanza al locus genomico desiderato e la progettazione di gRNA specifico ed efficiente deve essere trovato.
- Blast la sequenza selezionate gRNA contro il genoma di riferimento utilizzando il browser NCBI blast (https://blast.ncbi.nlm.nih.gov) per verificare l'unicità del luogo obbligatorio gRNA.
- Selezionare "umana" come il genoma. Inserire la sequenza di gRNA come la sequenza di query. Selezionare "sequenze altamente simili" (megablast) come il programma. Assicurarsi che la sequenza di gRNA sia univoco. Se non, ha scelto una gRNA diverso dal passaggio 1.1.2.3 e blast nuovamente.
- Una volta scelto gRNA sequenza, selezionare in silico 1.000 bp a Monte e a valle della sequenza gRNA da sequenza genomic estratto nel passaggio 1.1.1 come 5' e 3' HA di conseguenza.
Nota: Il gRNAs dovrebbe essere omologa al locus sito selezionate integrazione genomica e adiacente a un motivo adiacente protospacer (PAM; per esempio., NGG per SpCas9) (Figura 2a). La tv contiene la sequenza di reporter che è 5' e 3' affiancato da HAs. Ha copertura 1000 bp a Monte e a valle dei gRNA sequenza11. La sequenza completa gRNA non dovrebbe essere incluso nell'HA. Una sovrapposizione di fino a 5 nt è accettabile (Figura 2a).
-
Generazione di gRNA e vettori di targeting
Nota: Per schemi di vettore, Vedi Figura 2b.- Per generare un vettore per l'espressione di SpCas9 e gRNA, utilizzare il pX330-U6-Chimeric_BB-cBh-hSpCas9 come la spina dorsale da cui può essere contemporaneamente espresso sia SpCas9 e la guida singola RNA (sgRNA). Per clonare la sequenza gRNA nella spina dorsale, utilizzare i siti di restrizione BbsI12.
- Per generare la tv, è possibile scegliere un plasmide di alta-copia come spina dorsale (pMK, o cDNA3.1).
- In primo luogo, montare il reporter (in questo protocollo: LTR_tdTomato_BGH-PA) nel backbone del costrutto di Gibson Assembly clong13 utilizzando un assembly commerciale clonazione kit e introdurre 5' e 3' siti di restrizione di accompagnamento (ad esempio, 5' PacI e 3' SmaI) per la successiva restrizione digestione clonazione dell'HAs.
- Amplificare 1000 bp dei frammenti HA scelto nel passaggio 1.1.4 da DNA genomic (gDNA) del tipo cella di destinazione (in questo protocollo: cellule Jurkat) utilizzando una DNA polimerasi con attività di proofreading (Vedi tabelle 1 e 2 per gli ingredienti PCR e condizioni di ciclismo). Poi, introdurre reporter che fiancheggiano siti di restrizione alle estremità 5' e 3' di ogni HA (PacI su 5' HA su entrambe le estremità, SmaI su 3' HA su entrambe le estremità).
- In sequenza clone ha nella spina dorsale di costrutto già contenente il reporter (generato nel passaggio 1.2.2.1) dall'enzima di restrizione clonazione14,15. In primo luogo, clonare in 5' HA quindi utilizzando siti di restrizione di PacI, clonare in 3' HA utilizzando siti di restrizione SmaI.
Nota: Se spina dorsale tv contiene un ulteriore reporter fluorescente, integrazione backbone indesiderato può essere valutato mediante citometria a flusso (vedere i passaggi 3.2.2 e 3.2.3). Se spina dorsale tv non contiene nessun reporter fluorescente, integrazione di spina dorsale deve essere valutata mediante PCR (Vedi punto 3.2.8).

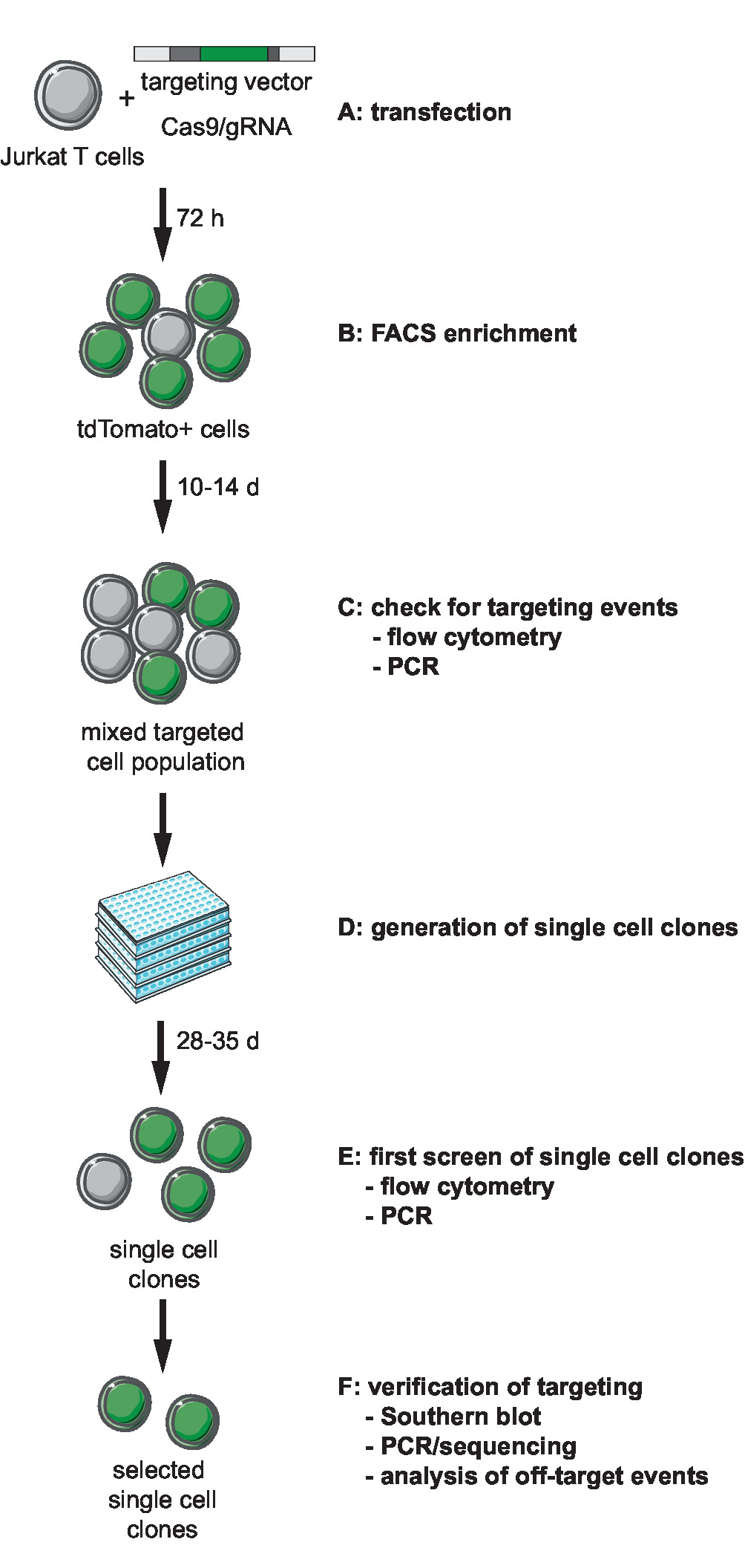
Figura 1: flusso di lavoro per CRISPR-Cas9-mediated targeting, generazione e selezione clonale reporter linee con sito definito integrazione. (A) generare il vettore di destinazione e trasdurre le cellule di T di Jurkat con il vettore di destinazione e il plasmide di espressione Cas9/gRNA. (B) arricchiscono la transfezione di post di 72h transfected le cellule di FACS. (C) lasciare che le cellule crescono per 10-14 giorni e confermano l'avvenimento di ottimizzazione degli eventi mediante PCR e citometria a flusso. (D) generare cloni unicellulari limitando cloni di diluizione e lasciate crescono per 3 settimane. (E) dello schermo i cloni per il targeting corretto mediante PCR e flusso cytometry in formato da 96 pozzetti. Espandere cloni selezionati. (F) verificare corretto targeting in cloni selezionati di Southern blot, PCR e sequenziamento e analisi degli eventi di fuori bersaglio di attività endonucleasica Cas9. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: strategia e vector design di Targeting. (un) gRNA e scelta delle armi di omologia. 20 nt gRNA è omologa al sito target genomici prescelto e adiacente a un'omologia tratteresti bracci sono complementari a 1.000 bp - up e a valle del gRNA e non devono includere la sequenza gRNA. (b) schemi di targeting vettoriale e gRNA/Cas9 vettoriale. Il vettore targeting è costituito dalla sequenza selezionate reporter che è 5' e 3' affiancata le braccia di omologia. Il vettore di gRNA/Cas9 si basa sulla spina dorsale pX330-U6-Chimeric_BB-cBh-hSpCas9. (c) schema di targeting di ricombinazione omologa. Vettore di destinazione e guideRNA/Cas9 vettore sono transfected nelle cellule Jurkat. Cas9 media una pausa doppio filamento al sito di destinazione genomica (indicata da *) e facilita la ricombinazione omologa e integrazione di sequenza di reporter nel locus genomico bersaglio. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
2. CRISPR-Cas9-base di Targeting delle cellule Jurkat
-
Transfezione di cellule Jurkat
- 24 h prima della trasfezione, piastra 1,25 x 106 T di Jurkat cells in 2,5 mL di RPMI 1640 completati con 10% (v/v) siero fetale di vitello (FCS) e 4 mM L-Glutammina [denominato "Antibiotici w.o. RPMI (AB)"] ogni pozzetto di una piastra di coltura delle cellule 6 pozzetti. Per un singolo esperimento targeting, preparare una completa 6 pozzetti (cioè, 6 pozzetti ciascuna con 2,5 mL di sospensione cellulare).
- Il giorno seguente, co-transfect le cellule con circolare tv e pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA utilizzando uno specifico reagente di transfezione per cellule Jurkat.
- Aggiungere 2 µ g di circolare tv e 2 µ g di pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA per pozzetto a 250 µ l di medium RPMI commerciale con concentrazione di siero riduttore ottimizzata per la transfezione (RPMI con riduzione del 50% nel siero) in una provetta di reazione e mescolare bene.
- Aggiungere 12 µ l di reagente di transfezione lentamente al DNA/medium senza toccare la parete del tubo e ricciolo. Lasciare che la miscela Incubare per 15 minuti e aggiungere goccia a goccia in un pozzetto delle cellule. Incubare le cellule a 37 ° C e 5% CO2.
Nota: La preparazione della reazione di transfezione può essere scalata. Nessun cambiamento medio è richiesto dopo la trasfezione.
-
Arricchimento delle cellule transfettate di fluorescenza-attivato delle cellule ordinano (FACS)
- 72 h post-transfezione, piscina le cellule transfettate, contarli e preparare per l'arricchimento di FACS. Raccogliere le cellule in una provetta conica da 50 mL, centrifugare a 300 x g e a temperatura ambiente (TA) per 4 minuti, lavare le cellule una volta in PBS, centrifugare nuovamente, sospendere il pellet in un'appropriata quantità di FACS buffer (PBS completati con 1% FCS + 1 mM EDTA) a 1 x 10 7 cellule/mL e infine trasferire in una provetta di FACS.
- Sottoporre le cellule a FACS e ordinare quelli che esprimono il reporter fluorescente del televisore (ad esempio, tdTomato in questo protocollo). Raccogliere le cellule in RPMI 1640 completati con 10% FCS, 4 mM L-Glutammina e 50 U/mL di penicillina e streptomicina (denominato "RPMI w / AB").
- Dopo FACS ordinamento, lavare le cellule una volta con l'aggiunta di 20 mL di RPMI w / AB alle cellule ordinate e centrifugare a 300 x g per 4 min a RT. Risospendere il pellet cellulare in una quantità appropriata di RPMI w / AB e piastra le cellule in un pozzetto di una piastra di coltura delle cellule con la volume appropriato secondo la cella numero post-FACS.
Nota: Cultura ordinare le celle in un piccolo volume (ad esempio, 24-bene), come sono stati osservati livelli considerevoli di morte delle cellule nella prima settimana post-targeting (fino a 80 – 90%). - Espandere la popolazione di cellule miste mirate fino a una densità di 1 x 106 cellule/mL in un matraccio di cultura cellulare 75 cm ². Questo richiederà circa 10 – 14 giorni post-FACS ordinamento.
-
Conferma di ottimizzazione degli eventi mediante citometria a flusso nella popolazione mista delle cellule mirati
- Dopo 10 – 14 giorni di espansione (quando le cellule hanno raggiunto una densità di 1 x 106 cellule/mL in un matraccio di cultura cellulare2 75 cm), due pozzetti della piastra con 1 x 106 cellule di misto mirati popolazione delle cellule in 1 mL di RPMI w / AB in una coltura cellulare 12-pozzetti piastra.
- Indurre la LTR di HIV del reporter (generazione di tv è descritto al punto 1.2.2.1.) in uno dei pozzi con l'aggiunta di 50 ng/mL phorbol 12-myristate 13-acetato (PMA) e 1 µM ionomicina (denominato PMA-Iono). Utilizzare cellule nel secondo pozzo come il controllo indotto non. Cultura l'indotto e le cellule non-indotta per 24 h.
- Prendere 0,5 mL di sospensione cellulare delle cellule indotto non e indotte (ciascuno), lavarli una volta in PBS e sospendere ciascuno in 200 µ l di tampone di FACS.
- Analizzare 100.000 cellule tramite flusso cytometry. Il singolo-cellule vitali basate sulla dimensione in avanti e lateralmente a dispersione del cancello ed analizzare l'espressione del gene reporter fluorescente.
Nota: In questa fase (10 – 14 giorni post-FACS ordinamento), espressione transitoria del reporter fluorescente di transfezione non deve essere rilevabile. Fluorescente espressione a questo punto del tempo indica integrazione genomica della sequenza di reporter.
-
Rilevamento di ottimizzazione degli eventi mediante PCR su DNA genomico della popolazione cellulare mirata misto
Nota: Per rilevare eventi targeting tramite PCR, disegno due coppie di primer specifici per il bivio di integrazione (int.) 5' e 3' int. junction. Per 5' int. svincolo PCR, il primer in avanti deve essere associato a Monte del 5' HA e il primer reverse in LTR del reporter (primer P1 e P2 in Figura 3a). La coppia di primer per la giunzione di int. 3' PCR deve estendersi dal PA del reporter a 100 – 200 bp a valle del 3' HA (primer P3 e P4 in Figura 3a). Primer P1 e P4 servirà anche per l'amplificazione dell'allele nella popolazione mista mirata non mirati. Per uno schema, vedere Figura 3a.- Preparare gDNA da 2 mL di sospensione cellulare della popolazione cellulare mirata mista dal punto 2.2.4. Utilizzare un kit di estrazione gDNA secondo il protocollo del produttore. Quindi preparare il gDNA delle cellule non mirati come controllo.
- Eseguire la giunzione int. PCRs (primer P1/P2 e P3/P4 per 5' e 3' int. giunzione, rispettivamente) e non mirati allele PCR (primer P1/P4) utilizzando una DNA polimerasi ad alta fedeltà (Vedi tabelle 3 e 4 per PCR ingredienti e condizioni in bicicletta) . Analizzare 5 µ l di prodotti di PCR su un gel di agarosio/TAE 1,5%.
Nota: Se la popolazione mirata mista contiene cellule che hanno subito ingegneria di genoma, un prodotto PCR specifico dovrebbe essere osservato che non è rilevabile nel gDNA delle cellule non mirati (controllo negativo). Per l'allele non mirati PCR, uno dovrebbe osservare un prodotto della stessa dimensione per entrambe le celle mirate e non mirati (controllo positivo per genomica primer P1 e P4). Se si osservano senza bande, considera alterando in bicicletta di PCR, aumentando il numero di cicli o alterando il PCR buffer (ad esempio mediante aggiunta di DMSO o gli importi aumentati di Mg2 +), o modificando la polimerasi.
3. generazione di linee clonali e Screening per il Targeting corretto
Nota: Dopo la conferma degli eventi targeting nella popolazione misto delle cellule mirata mediante citometria a flusso e PCR (sezioni 2.2-2.4), generare cloni unicellulari (durata: 28-35 giorni) e schermo per integrare correttamente la sequenza di reporter.
- Generazione di cloni unicellulari attraverso placcatura di diluizione
- Preparare in anticipo in supporto di Jurkat-condizionati: decollare RPMI w / mezzo di AB da cellule T di Jurkat sane e non trattate, cresciuta fino a 1 x 106 cellule/mL, centrifugare per 5 minuti a 300 x ge filtrare il surnatante utilizzando un'unità filtrante di siringa da 0,22 µm.
Nota: Tenere il medium condizionato a 4 ° C per la conservazione a breve termine o a-20 ° C per più di 1 settimana di deposito. Preparare il 20-30 mL di medium condizionato prima della placcatura di diluizione. - Contare le celle mirate dal punto 2.2.4 dopo 10 – 14 giorni di espansione e diluirli in RPMI w / AB ad una concentrazione di 1 x 105 cellule/mL. Prendere 100 µ l di soluzione di 1 x 105 cellule/mL e diluire con 9,9 mL di medium per ottenere una concentrazione di 1.000 cellule/mL. Prelevare 1 mL della soluzione di cellule/mL 1.000 e diluire con 9 mL di medium per ottenere una concentrazione di 100 cellule/mL.
- Piastra 96 pozzetti piastre contenenti 1 delle cellule per pozzetto e 2 cellule per pozzetto. Per 1 delle cellule per pozzetto, prendere 1 mL di soluzione di 100 cellule/mL e mescolare delicatamente con 5 mL di medium condizionato e 4 mL di terreno fresco in un serbatoio di reagente sterile.
- Per 2 cellule per pozzetto, prendere 2 mL di soluzione di 100 cellule/mL e mescolare delicatamente con 5 mL di medium condizionato e 3 mL di terreno nuovo. Piastra 96 pozzetti turno-fondo piastre con 100 µ l della diluizione delle rispettive cellule per bene usando una pipetta multicanale.
Nota: da 5 a 10 piastre da 96 pozzetti al costrutto di targeting sono sufficienti per ottenere cloni per lo screening. - Impilare le piastre da 96 pozzetti, coprire ogni stack con una piastra a 6 pozzetti contenenti 3 mL di PBS in ciascun pozzetto e incubare le piastre a 37 ° C in un'incubatrice di coltura cellulare umidificata con 5% CO2 per 3 settimane.
Nota: Non cambiare il mezzo di coltura cellulare durante questo tempo. Non aprire l'incubatrice più di una volta o due volte a settimana. I migliori risultati sono osservati in incubatrice con serbatoio dell'acqua aperta. - Dopo 3 settimane di incubazione, confermare visivamente la presenza di colonie coltivate usando microscopia (ingrandimento 4x) e contrassegnare i pozzetti con colonie coltivate in modo da sono visibili come punti sul fondo dei pozzetti.
- Preparare una piastra 96 pozzetti turno-parte inferiore con 100 µ l di RPMI w / AB. Risospendere delicatamente le cellule di un marcato bene pipettando. Trasferire 100 µ l di sospensione cellulare in un pozzetto della piastra 96 pozzetti nuovo già contenente 100 µ l di RPMI w / AB, quindi mescolare delicatamente pipettando. Trasferire 100 µ l di sospensione cellulare in un vuoto secondo piastra 96 pozzetti turno-inferiore per duplicare la piastra.
- Continuare con tutti i pozzetti contrassegnati con colonie coltivate. Riempire tutti i pozzetti del bianco con 200 µ l di RPMI w / medio di AB. Incubare le piastre a 37° C e 5% CO2.
Nota: Uno di questi piatti servirà per espansione dei cloni unicellulari ("piastra di stock") e l'altro come un piatto"duplicato" per lo screening.
- Preparare in anticipo in supporto di Jurkat-condizionati: decollare RPMI w / mezzo di AB da cellule T di Jurkat sane e non trattate, cresciuta fino a 1 x 106 cellule/mL, centrifugare per 5 minuti a 300 x ge filtrare il surnatante utilizzando un'unità filtrante di siringa da 0,22 µm.
- Screening di cloni unicellulari mediante citometria a flusso e PCR
Nota: Mentre i cloni unicellulari sono in espansione, è possibile utilizzare la piastra duplicata dal punto 3.1.8 ai cloni unicellulari schermo per presenza di reporter sequenza mediante PCR (misure 3.2.4–3.2.12) e l'espressione del reporter fluorescente da citometria a flusso (misure 3.2.2–3.2.3) (Figura 3C).- Incubare per 24-48 h e duplicare nuovamente la piastra, lasciate che la piastra di duplicati. Per effettuare questa operazione, aggiungere 100 µ l di RPMI w / AB in ogni pozzetto, mescolare delicatamente pipettando e trasferire 100 µ l di una nuova piastra 96 pozzetti turno-fondo utilizzando una pipetta multicanale. Utilizzare una piastra per lo screening di citometria a flusso e l'altro per screening basato su PCR.
- Per lo screening di citometria a flusso, stimolano le cellule con PMA-Iono. Preparare un mastermix di 0.1 µ l di Ionomocin (1 mM stock), 0.1 µ l di PMA (50 µ g / µ l stock), e 4,8 µ l di RPMI w / AB per il numero di pozzetti, quindi aggiungere 5 µ l di mastermix per pozzetto.
Nota: Ad induzione è necessario identificare correttamente cloni, dove la LTR potrebbe essere trascrizionalmente silenziosa e pertanto il reporter fluorescente non è espresso. - Ha lasciato le cellule Incubare per 24 h e preparare le celle per citometria a flusso come descritto al punto 2.3.3. Qualsiasi singolo-cellule vitali basate sulla dimensione in avanti e lateralmente a dispersione del cancello ed analizzare l'espressione del gene reporter fluorescente da citometria a flusso (ad esempio i risultati, vedere Figura 3C). Se la spina dorsale tv contiene un secondo reporter fluorescente con promotore (ad es., GFP), schermo qualsiasi cloni per espressione reporter spina dorsale anche (Vedi punto 1.2.2 e la seguente nota per spiegazione).
Nota: Spina dorsale reporter espressione indica indesiderati integrazione delle sequenze di spina dorsale. - Una volta che i cloni della seconda piastra duplicati sono cresciuti sufficientemente (solitamente 24-48 h dopo la duplicazione della piastra 96 pozzetti), preparare lisati cellulari contenenti gDNA per lo screening di PCR. Centrifugare la piastra per 10 min a 300 x g a RT. attentamente decollare il sopranatante senza disturbare il pellet cellulare.
Nota: Tutti i passaggi per la preparazione di lisati e reazioni di PCR possono essere eseguiti con pipette multicanale. - Lavare le cellule con 100 µ l di PBS di pipettaggio delicato e centrifugazione la piastra per 5 min a 300 x g e a TA. Decollare il PBS e aggiungere 200 µ l di tampone di lisi [200 mM NaCl, 100 mM Tris-HCl a pH 8-8.5, 5 mM EDTA, 0,1% SDS; quindi aggiungere 250 – 1.000 µ g/mL di proteinasi K (liofilizzato in polvere, pesare in fresco)] per pozzo. Mescolare delicatamente pipettando e trasferire la sospensione per una nuova piastra PCR.
- Sigillare la piastra con la pellicola di paraffina e incubare per 1 h a 55 ° C in un termociclatore. Centrifugare a massima velocità per 10 min (3.000 x g) a girare giù detriti cellulari e trasferire il surnatante in un nuovo piatto PCR.
Nota: Lisati cellulari in piastre possono essere memorizzati in questa fase a 4 ° C fino al successivo utilizzo. - Preparare una piastra PCR a 96 pozzetti con 110 µ l di dH2O e aggiungere 10 µ l di lysate delle cellule (01:12 diluizione). Lisati cellulari potrebbero essere viscoso e difficile da dispensare. Utilizzare almeno consigli di dispensare 20-µ l.
- Inattivare proteinasi K mediante incubazione per 10 min a 99 ° C in un termociclatore. Successivamente è possibile utilizzare i lisati cellulari inattivato e diluito per lo screening di PCR.
- Disegnare primers per PCR (P5 e P6) basato sulla sequenza di reporter selezionate per amplificare 500 – 800 bp della sequenza reporter di screening. Per controllo positivo PCR, utilizzare primers P7 e P8 che amplificano 630 bp di un locus genomico wild-type, non mirati (geneNUP188 ) (Figura 3 c e tabella 5). Progettare una terza coppia di primer che amplifica 500 – 600 bp del backbone tv come un controllo per l'integrazione non specifico delle sequenze di tv spina dorsale (backbone PCR).
- Per lo screening, controllo e spina dorsale PCR, utilizzare un commerciale mastermix PCR (Vedi tabelle 6 e 7 per PCR ingredienti e condizioni in bicicletta). Utilizzare 2 µ l di diluito e inattivato lisato preparata al punto 3.2.8 come modello ed eseguire PCR per 38 a 40 cicli di amplificazione di PCR in formato da 96 pozzetti.
- Analizzare 5 µ l di prodotti di PCR su un gel di agarosio/TAE 1,5%.
Nota: Per controllo PCR, una banda specifica di 630 bp dovrebbero essere osservati per ogni campione, confermando che la qualità dei lisati cellulari è adeguata per la PCR. Una banda specifica in PCR (500 – 800 bp a seconda del disegno dell'iniettore) di screening indica integrazione della sequenza reporter. Una banda specifica per la spina dorsale PCR (500 – 600 bp, a seconda del disegno dell'iniettore) indica indesiderati integrazione delle sequenze di spina dorsale tv (ad esempio i risultati, vedere Figura 3C). - Combinare i risultati di citometria a flusso (punto 3.2.3) e screening basato su PCR (passo 3.2.12). Selezionare cloni che mostrano le dimensioni corrette dei prodotti di PCR nello screening PCR e controllo positivo PCR ed espressione del reporter fluorescente dopo induzione con PMA-Iono in citometria a flusso. Escludere i cloni che mostrano qualsiasi prodotto di PCR nella spina dorsale PCR o espressione di tv spina dorsale-codificato proteina fluorescente, che indica non specifica integrazione di tv spina dorsale sequenze.
- Espandere gradualmente cloni selezionati dalla piastra 96 pozzetti stock in formati ben più grandi (48/24/12/6-pozzetti) fino ad ottenere un formato di boccetta T75 cultura cella aggiungendo mezzo fresco ogni 2 o 3 giorni. Mantenere una densità di cella tra 1 x 105 e 1 x 106 cellule/mL.
- Assicurarsi di preparare scorte di cella di cloni durante l'espansione: contare le celle, centrifugare a 300 x g per 5 min a RT, scartare il surnatante e sospendere le cellule delicatamente in FCS + 10% DMSO a 5 x 106 cellule/mL. Aliquota di fiale criogeniche e contenitore uso un cryo-congelamento per congelare le cellule a 80 ° C a 1 ° C/min. Per l'archiviazione a lungo termine, è possibile trasferirli al liquido N2.
Nota: Si consiglia di mantenere un matraccio di cultura cellulare T75 (, circa 1 x 107 cellule) durante l'espansione in preparazione di gDNA per la verifica di targeting mediante Southern blotting (Vedi sezione 3.4).
- Verifica dei siti di integrazione mediante PCR/sequenziamento in cloni selezionati
Nota: giunzioni int. 5' e 3' dei cloni selezionati sono PCR amplificato e inviato a Sanger sequenziamento per verificare correggere il targeting a livello di sequenza del DNA.- Preparare gDNA dei cloni selezionati e Jurkat selvaggio-tipo celle utilizzando un kit di estrazione commerciale gDNA.
- Utilizzare coppie di primer obbligatorio l'estremità 5' del reporter e a Monte del 5' HA 5' Junction int. (primer P1 e P2) e l'estremità 3' del reporter e a valle di 3' HA 3' Junction int. (primer P3 e P4) come descritto al punto 2.4. Utilizzare primer P1 e P4 per amplificare il sito di integrazione mirata sull'allele senza reporter integrante (Figura 4a).
- Preparare le reazioni di PCR con 100-200 ng di gDNA come modello ed eseguire PCR usando una polimerasi con attività di proofreading (Vedi tabelle 1 e 2 per PCR ingredienti e condizioni in bicicletta).
Nota: Se si osservano senza bande, considera alterando le condizioni PCR in bicicletta aumentando il numero di cicli o alterando il PCR buffer (ad esempio, con l'aggiunta di DMSO o gli importi aumentati di Mg2 +), o modificando la polimerasi. - Analizzare 5 µ l di prodotti di PCR su un gel di agarosio/TAE 1,5%. Se si osservano la banda corrette dimensioni, purificare il restante prodotto di PCR utilizzando un kit commerciale e sottoporlo a Sanger sequenziamento. Verificare sequenze 5' giunzione int., 3' int. svincolo e mirato sito dell'allele senza reporter integrante allineandoli con la sequenza prevista.
Nota: L'allele omologo dove il reporter non ha integrato probabilmente mostrerà cambiamenti Cas9-mediati nel sito di destinazione, ad esempio del nucleotide inserimenti o eliminazioni (Figura 4a). - Per i cloni che mostrano corretto int. sequenze di giunzione dopo l'allineamento, eseguire una PCR amplificando il reporter tutta mirato e sottoporlo a Sanger sequenziamento per verificare la corretta sequenza dell'integrante.
- Analisi del sud della macchia per la verifica di targeting in cloni selezionati
Nota: Analisi del sud della macchia di cloni selezionati è richiesto per verificare la corretta destinazione ed escludere gli eventi di ricombinazione mediata Cas9 che potrebbero essersi verificato presso il sito di integrazione mirata.- Sviluppare una strategia per gDNA appropriata digestione e il disegno prima di iniziare l'esperimento della sonda.
- Selezionare un enzima di restrizione per restrizione gDNA che genera appropriati frammenti di lunghezza di 2 a 10 kb presso il sito di destinazione. Alcuni enzimi di restrizione, ad esempio Asp718, BamHI, BglI, BglII, EcoRV, HindIII, Ncoio, PstI, PvuII, Scaho, StuI, e SST Ho stato usato con successo per la digestione gDNA ad alto peso molecolare.
- Due diverse sonde meridionale di design: una sonda reporter-specific e sonda genomica. La sonda del reporter specifico ibridizza con una sequenza all'interno del reporter (cioè sonda di, tdTomato-specific). La genomica sonda ibridizza con una regione genomica vicino a (ma non sovrapposte) uno HA.
- Scegliere la sonda genomica in modo che il frammento generato dalla digestione gDNA che verrà rilevata dall'associazione sonda genomica differisce in lunghezza (più di 2 kb) da alleli mirate e non mirati (Figura 4b). Una lunghezza di sonda di 400 a 1.000 bp è raccomandato.
- Progettare gli iniettori di PCR per amplificare le due sonde necessarie. Amplificare la sonda reporter specifico dal modello di tv utilizzando una DNA polimerasi ad alta fedeltà (Vedi tabelle 3 e 4 per PCR ingredienti e condizioni in bicicletta).
- Amplificare la sonda genomica dal selvaggio-tipo Jurkat gDNA preparato con un kit di estrazione commerciale gDNA utilizzando una DNA polimerasi con attività di proofreading (Vedi tabelle 1 e 2 per PCR ingredienti e condizioni in bicicletta). Purificare i prodotti di PCR su un gel di agarosio/TAE ed estrarre i frammenti utilizzando un kit di estrazione del gel commerciale secondo le istruzioni del produttore.
- Estrarre gDNA ad alto peso molecolare da 1 x 107 cellule della cellule Jurkat wild-type e cloni selezionati dal passaggio 3.2.14.
- Agglomerare le cellule mediante centrifugazione a 300 x g per 5 min a RT, lavarlo una volta con PBS e sospendere il pellet in 4 mL di tampone di lisi [200 mM NaCL, 100 mM Tris-HCl a pH 8, 5mm EDTA, 0,1% SDS; quindi aggiungere proteinasi di 250 – 1.000 µ g/mL K (polvere liofilizzata pesano appena)]. Incubare a 55 ° C, agitando a 350 rpm in un thermomixer da tavolo Cod.
- Aggiungere 4 mL di isopropanolo e miscelare capovolgendo 10 a 20 volte. Il gDNA dovrebbe diventare visibile come precipitato bianco. Il gDNA precipitato sulla punta di una pipetta di vetro fine di cursore, lavare da emergenti in 750 µ l 70% EtOH, e lasciate asciugare a RT (5-10 min).
- Versato il precipitato in una provetta di reazione di 1,5 mL contenente 500 µ l di tampone TE 1x (10 mM Tris-HCl pH 8.0, 1 mM EDTA) e lasciate per sciogliere o/n a 4 ° C, agitando a 350 giri/min. Qualsiasi pipettaggio di gDNA da questa fase dovrebbe essere fatto con wide-bore consigli per evitare di tosatura.
Nota: Preparazione di alto peso molecolare gDNA è essenziale per l'analisi del sud della macchia, e kit di preparazione gDNA commercialmente disponibili non sono adatti.
- Digest (due volte) 15 µ g di gDNA dei cloni selezionati e cellule Jurkat wild-type con selezionati degli enzimi di limitazione (Vedi punto 3.4.1.1) in una reazione di 60 µ l con 6 µ l di enzima (20 unità / µ l): in primo luogo, aggiungere DNA, buffer di digestione e ddH2O, incubare o/n a 37 ° C , quindi aggiungete l'enzima e incubare o/n a temperatura di digestione degli enzimi specifici. 15 ug di gDNA digerito è richiesto per ogni sonda meridionale.
- Utilizzare 7 µ l di raccolta di limitazione di 60 µ l per analitiche elettroforesi su un gel di agarosio/TAE di 1%. Una sbavatura indica la digestione completa e buona qualità del DNA per l'analisi del sud della macchia.
- Precipiti il digest di restrizione rimanenti aggiungendo 01:10 3M acetato di sodio e 2 volumi 100% EtOH, quindi incubare per 1 h a-80 ° C e centrifugare per 30 min a 15.600 x g a 4 ° C.
- Scartare il surnatante e lavare la pallina con 70% EtOH. Centrifugare per 15 min a 15.600 x g a 4 ° C, eliminare il supernatante, lasciate che il pellet asciugare brevemente a RT e sciogliere in 20 µ l di ddH2O.
- Eseguire 1% gel macchiante di agarosio/TAE, caricamento 20 uL di gDNA digerito per corsia. Esegua il gel per 2 h a 60 V, 400 mA.
Nota: La percentuale di agarosio gel e corsa tempo/tensione può essere regolata in base alle dimensioni del frammento previsto per il rilevamento del sud della macchia calcolato nel passaggio 3.4.1.1. La seguente procedura di analisi del sud della macchia è descritte in dettaglio in un protocollo supplementare (passaggi da 1 a 18). Questi passaggi è composto da: lavaggio del gel macchiante, macchiare su una membrana di nylon, generazione di sonda radioattiva, sonda di ibridazione e sviluppo della pellicola di autoradiograph. Confrontare il disegno a strisce ottenuta dopo lo sviluppo di autoradiograph nel passaggio 18 (protocollo supplementare) con il modello previsto secondo la strategia del Sud (ad esempio risultati, Vedi Figura 4b).
- Sviluppare una strategia per gDNA appropriata digestione e il disegno prima di iniziare l'esperimento della sonda.
- Analisi degli eventi fuori bersaglio
Nota: Poiché ingegneria genoma CRISPR-Cas9-mediata può generare effetti fuori bersaglio, PCR-amplificare i dieci più alto in classifica in silico-predetto siti fuori bersaglio nella selezione di cloni e sottopongono a Sanger sequenziamento.- Utilizzare CCTop16 (http://crispr.cos.uni-heidelberg.de) per generare un elenco dei dieci più alto in classifica in silico predetto sequenze fuori bersaglio.
- Inserire la sequenza di gRNA tra cui PAM come utilizzato per il targeting come la sequenza di query. Selezionare "NGG" come PAM e "Genoma umano" come riferimento per la stima di fuori bersaglio.
- Impostare la massimale totale mancate corrispondenze a "4" e destinazione sito lunghezza alla lunghezza del gRNA senza PAM. Il file di output fornirà un elenco dettagliato dei siti off-target genomici per gRNA rispettivo.
- In silico estrarre la sequenza genomic 500 bp a Monte e a valle di ciascuna delle dieci colpi fuori bersaglio più alto in classifica a utilizzando UCSC Genome Browser (http://genome.edu.ucsc.edu) e la posizione del bersaglio colpito dall'elenco dei risultati di CCTop.
- Per ogni i siti di destinazione devono essere analizzate, design un primer PCR coppia che amplifica un frammento di 600-700 bp in lunghezza compreso il predetto sito fuori bersaglio.
- Estrarre gDNA dal cloni selezionati e Jurkat selvaggio-tipo celle utilizzando un kit di estrazione commerciale gDNA. Per ogni sito fuori bersaglio, eseguire una PCR utilizzando una DNA polimerasi con attività di proofreading (Vedi tabelle 1 e 2 per ciclismo condizioni e ingredienti PCR) su wild-type e il rispettivo gDNA clone-derivato.
- Analizzare 5 µ l di prodotti di PCR su un gel di agarosio/TAE 1,5%. Se si osservano la banda corrette dimensioni, purificare il restante prodotto di PCR utilizzando un kit di purificazione di PCR commerciale e sottoporlo a Sanger sequenziamento. Confrontare sequenze dei siti fuori bersaglio in cellule Jurkat e i cloni mirati.
- Utilizzare CCTop16 (http://crispr.cos.uni-heidelberg.de) per generare un elenco dei dieci più alto in classifica in silico predetto sequenze fuori bersaglio.
Access restricted. Please log in or start a trial to view this content.
Risultati
In questo esperimento rappresentanza abbiamo scelto di indirizzare un reporter di HIV-1-derivati minimo composta un LTR, la sequenza tdTomato-codificazione e la sequenza di polyA-segnale a due loci in introne 5 del gene BACH217. I loci per il targeting sono stati scelti secondo la vicinanza ai siti pubblicati integrazione ricorrenti trovato in diversi studi su cellule T primarie da pazienti HIV-infettati2,4...
Access restricted. Please log in or start a trial to view this content.
Discussione
Qui, descriviamo un protocollo per generare modelli di HIV-1-derivati Jurkat reporter con siti di integrazione proviral selezionate l'applicazione di ingegneria basati su CRISPR-Cas9 genoma.
Alcuni punti del protocollo richiedono attenzione durante la fase di pianificazione. In primo luogo, il luogo di destinazione dovrebbe essere scelto con attenzione, come alcuni loci potrebbero essere più facile bersaglio di altri (ad es., a seconda dello stato della cromatina della regione e la d...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Ringraziamo Britta Weseloh e Bettina Abel per assistenza tecnica. Ringraziamo anche Arne Düsedau e Jana Hennesen (piattaforma tecnologica flusso cytometry, Heinrich Pette Institut) per il supporto tecnico.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| pX330-U6-Chimeric_BB-cBh-hSpCas9 | Addgene | 42230 | vector for expression of SpCas9 and gRNA |
| pMK | GeneArt | mammalian expression vector for cloning | |
| cDNA3.1 | Invitrogen | V79020 | mammalian expression vector for cloning |
| BbsI | New England Biolabs | R0539S | restriction enzyme |
| NEBuilder Hifi DNA Assembly Cloning Kit | New England Biolabs | E5520S | Assembly cloning kit used for target vector generation |
| TaqPlus Precision PCR System | Agilent Technologies | 600210 | DNA polymerase with proofreading activity used for amplification of homology arms (step 1.2.2.2), verification of integration site and reporter sequence (step 3.3.3 and 3.3.5), generation of genomic probe for Southern blot (step 3.4.1.5) and analysis of off-target events (step 3.5.4) |
| 96-well tissue culture plate (round-bottom) | TPP | 92097 | tissue culture plates for dilution plating |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530 L | DNA polymerase used for detection of targeting events (step 2.4.2) and generation ofreporter-specific probe for Southern blot (step 3.4.1.4) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D9170 | dimethyl sulfoxide as PCR additive |
| Magnesium Chloride (MgCl2) Solution | New England Biolabs | B9021S | MgCl2 solution as PCR additive |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | dNTP mixture with 10 mM of each nt for PCR reactions |
| 5PRIME HotMasterMix | 5PRIME | 2200400 | ready-to-use PCR mix used for screening PCR (step 3.2.11) |
| QIAamp DNA blood mini kit | Qiagen | 51106 | DNA isolation and purification kit |
| QIAquick PCR Purification Kit | Qiagen | 28106 | PCR Purification Kit |
| RPMI 1640 without glutamine | Lonza | BE12-167F | cell culture medium |
| Fetal Bovine Serum South Africa Charge | PAN Biotech | P123002 | cell culture medium supplement |
| L-glutamine | Biochrom | K 0282 | cell culture medium supplement |
| Penicillin/Streptomycin 10.000 U/mL/ 10.000 µg/mL | Biochrom | A 2212 | cell culture medium supplement |
| Gibco Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | 31985062 | cell culture medium with reduced serum concentration optimized for transfection |
| TransIT-Jurkat | Mirus Bio | MIR2125 | transfection reagent |
| phorbol 12-myristate 13-acetate | Sigma-Aldrich | P8139-1MG | cell culture reagent |
| Ionomycin | Sigma-Aldrich | I0634-1MG | cell culture reagent |
| Syringe-driven filter unit, PES membrane, 0,22 µm | Millex | SLGP033RB | filter unit for sterile filtration |
| Heracell 150i incubator | Thermo Fisher Scientific | 51026280 | tissue culture incubator |
| Amershan Hybond-N+ | GE Healthcare | RPN1520B | positively charged nylon membrane for DNA and RNA blotting |
| Stratalinker 1800 | Stratagene | 400072 | UV crosslinker |
| High Prime | Roche | 11585592001 | kit for labeling of DNA with radioactive dCTP using random oligonucleotides as primers |
| illustra ProbeQuant G-50 Micro Columns | GE Healthcare | 28-9034-08 | chromatography spin-columns for purification of labeled DNA |
Riferimenti
- Hughes, S. H., Coffin, J. M. What Integration Sites Tell Us about HIV Persistence. Cell Host and Microbe. 19 (5), 588-598 (2016).
- Marini, B., Kertesz-Farkas, A., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 521 (7551), 227-231 (2015).
- Cesana, D., Santoni de Sio, F. R., et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nature Communications. 8 (1), 498(2017).
- Cohn, L. B., Silva, I. T., et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell. 160 (3), 420-432 (2015).
- Han, Y., Lassen, K., et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 78 (12), 6122-6133 (2004).
- Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. The Journal of Infectious Diseases. 195 (5), 716-725 (2007).
- Wagner, T. A., Mclaughlin, S., et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 345 (6196), 570-573 (2014).
- Maldarelli, F., Wu, X., et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 345 (6193), 179-183 (2014).
- Jordan, A., Bisgrove, D., Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 22 (8), 1868-1877 (2003).
- Mack, K. D., Jin, X., et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. Journal of Acquired Immune Deficiency Syndromes. 33 (3), 308-320 (2003).
- Byrne, S. M., Ortiz, L., Mali, P., Aach, J., Church, G. M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Research. 43 (3), 1-12 (2014).
- ZhangLab. CRISPR Genome Engineering Toolbox: Target Sequence Cloning Protocol. Addgene website. , Available from: https://www.addgene.org/static/cms/filer_public/e6/5a/e65a9ef8-c8ac-4f88-98da-3b7d7960394c/zhang-lab-general-cloning-protocol.pdf (2013).
- Moser, F. Addgene. Gibson Assembly Protocol. Addgene website. , Available from: https://www.addgene.org/protocols/gibson-assembly/ (2009).
- Addgene Plasmid Cloning by PCR. Addgene website. , Available from: https://www.addgene.org/protocols/pcr-cloning/ (2014).
- Addgene Plasmid Cloning by Restriction Enzyme Digest (aka Subcloning). Addgene website. , Available from: https://www.addgene.org/protocols/subcloning/ (2013).
- Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., Mateo, J. L. CCTop: An intuitive, flexible and reliable CRISPR-Cas9 target prediction tool. Public Library of Science (PLoS) ONE. 10 (4), (2015).
- Lange, U. C., Bialek, J. K., Walther, T., Hauber, J. Pinpointing recurrent proviral integration sites in new models for latent HIV-1 infection. Virus Research. 249, (2018).
- Bialek, J. K., Dunay, G. A., et al. Targeted HIV-1 Latency Reversal Using CRISPR-Cas9-Derived Transcriptional Activator Systems. PloS ONE. 11 (6), e0158294(2016).
- Lee, C. M., Davis, T. H., Bao, G. Examination of CRISPR-Cas9 design tools and the effect of target site accessibility on Cas9 activity. Experimental Physiology. 103 (4), 456-460 (2018).
- Jensen, K. T., Fløe, L., et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Letters. 591 (13), 1892-1901 (2017).
- Simonetti, F. R., Sobolewski, M. D., et al. Clonally expanded CD4 + T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 113 (7), 1883-1888 (2016).
- Chen, H. C., Martinez, J. P., Zorita, E., Meyerhans, A., Filion, G. J. Position effects influence HIV latency reversal. Nature Structural and Molecular Biology. 24 (1), 47-54 (2017).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati