
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Baseados em CRISPR-Cas9 genoma engenharia para gerar modelos Jurkat repórter para a infecção de HIV-1 com Sites selecionados Proviral integração
Neste Artigo
Resumo
Nós apresentamos um fluxo de trabalho de engenharia do genoma para a geração de novos modelos in vitro para a infecção de HIV-1 que recapitular integração proviral em locais seleccionados genômicas. Segmentação dos repórteres HIV-derivado é facilitada pela manipulação de genoma CRISPR-Cas9-mediada, site-specific. Protocolos detalhados para geração de clone de célula única, triagem e verificação de direcionamento correta são fornecidos.
Resumo
Vírus de imunodeficiência humana (HIV) integra o DNA proviral não aleatoriamente no genoma da célula de acolhimento em sites recorrentes e genômicas hotspots. Aqui apresentamos um protocolo detalhado para a geração de novos modelos in vitro para a infecção pelo HIV com sites de integração genômica escolhido usando tecnologia de engenharia baseados em CRISPR-Cas9 genoma. Com este método, uma sequência de repórter de escolha pode ser integrada em um locus genômico escolhido, alvo, refletindo sites integração clinicamente relevantes.
No protocolo, o design de um repórter de HIV-derivado e escolhendo de uma sequência de site e gRNA de destino são descritos. Um vetor de direcionamento com braços de homologia é construído e transfectado em células Jurkat T. A sequência de repórter é direcionada para o site selecionado genômico por recombinação homóloga, facilitada por uma quebra de dobro-costa Cas9-mediada no local de destino. Clones de célula única são gerados e examinados para segmentação de eventos por citometria de fluxo e PCR. Os clones selecionados são então expandidos, e direcionamento correto é verificado pela PCR, sequenciamento e mancha do Sul. Eventos fora do alvo potencial de engenharia CRISPR-Cas9-mediada do genoma são analisados.
Usando este protocolo, sistemas de cultura de células de novela essa infecção de HIV modelo em sites de integração clinicamente relevantes pode ser gerada. Embora a geração de clones de célula única e verificação de integração de sequência correto repórter são demorados, as linhas clonais resultantes são ferramentas poderosas para analisar funcionalmente a escolha do local de integração proviral.
Introdução
Integração do DNA proviral no genoma hospedeiro após infecção é um passo crítico no ciclo de vida do vírus de imunodeficiência humana (HIV). Após a integração, o HIV persiste, estabelecendo a latência em subconjuntos de células T CD4 + long-lived tais como as células T CD4 + de memória. Integração do HIV parece ser não-aleatória1,2. Um número de hotspots genômicas com DNA proviral recorrentemente integrado foi detectado em vários estudos através do sequenciamento de sites de integração em indivíduos infectados agudamente e cronicamente2,3,4 ,5,6,7,8. Curiosamente, em alguns destes sites de integração, o mesmo locus foi detectado em uma grande fração de células infectadas, levando à ideia de que integração em locais recorrentes pode afetar positivamente a expansão clonal1.
Para avançar a nossa compreensão da importância de sites de integração recorrentes, escolha de local de integração proviral deve ser explorada. No entanto, vários aspectos técnicos dificultam a integração de HIV estuda site escolha e as consequências. Amplamente utilizados modelos de cultura celular de latência do HIV, como linhas de célula JLat não refletem integração recorrentes clinicamente relevantes sites9. Estudos em células derivado de paciente primários, por um lado, permitem a descrição da paisagem local de integração de sequenciação mas não permitem análises funcionais. A nosso conhecimento, nenhum modelo experimental adequado está disponível para analisar funcionalmente sites selecionados de integração clinicamente relevantes.
Aqui nós apresentamos um fluxo de trabalho detalhado para gerar novos modelos para a infecção pelo HIV, utilizando tecnologia de engenharia baseados em CRISPR-Cas9 genoma. O fluxo de trabalho aqui descrito pode ser usado para gerar linhas de células de repórter derivado de células T que modelam a infecção pelo HIV, carregando um repórter proviral integrado genomically em um site de integração escolhido. Assim, estão servindo como novas ferramentas para explorar como o site de integração proviral pode afetar a biologia do HIV, e como o pró-vírus responde às estratégias de tratamento diferentes (por exemplo,, inducibility por agentes de inversão de latência). Nosso método usa as vantagens de engenharia baseados em CRISPR-Cas9 genoma, no qual a integração do repórter sequência por recombinação homóloga é facilitada por uma quebra Cas9 induzida por nuclease dobro-costa no local de destino. Sites de destino para a integração são escolhidos de acordo com a proximidade aos locais descritos integração recorrentes de estudos em indivíduos infectados pelo HIV e a presença de motivos de PAM apropriados para engenharia de genoma Cas9-mediada.
Em nossos resultados exemplares, enfocamos o locus de gene BACH2, quais códigos para o regulador transcricional BTB e homologia de CNC 2. Em indivíduos cronicamente infectadas com o HIV em terapia anti-retroviral, BACH2 é um dos loci mostrando o enriquecimento de integrado HIV-1 sequências3,6,7,8,10. Nós escolhemos um repórter HIV-derivado mínimo consistindo de HIV-1-derivado longo terminal repetição (LTR), a sequência de código tdTomato e hormônio de crescimento bovino (BGH) sinal de poliadenilação (PA), que podemos ter como alvo a dois locais específicos em BACH2 intron 5. O protocolo apresentado é otimizado para células Jurkat, uma humana CD4 + linha celular suspensão derivado de células T, mas outras células linhas podem ser utilizadas e o protocolo adaptado em conformidade. Apresentamos um fluxo de trabalho detalhado para a seleção do local de destino, a construção do vetor de alvo com braços de homologia, CRISPR Cas9-mediada por direcionamento do repórter no local escolhido genômico, geração e seleção de linhas clonais e abrangentes verificação linha de celular do repórter recém-gerado, alvo.
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. estratégia de segmentação para genoma engenharia e direcionamento de desenho vetorial (tv)
Nota: O primeiro passo da engenharia do genoma envolve a seleção e geração das ferramentas necessárias para CRISPR Cas9-mediada por segmentação. Selecção de um locus de sítio de integração genômica, escolha do tipo de célula para o direcionamento e design de um repórter de HIV-derivado para integração devem preceder esta etapa. Este protocolo descreve como alvo de um repórter de mínimo de HIV-LTR_tdTomato_BGH-PA em células Jurkat-alvo. Um gráfico de fluxo de fluxo de trabalho para CRISPR Cas9-baseado em segmentação, geração, triagem e verificação das linhas clonais é retratado na Figura 1. A estratégia de segmentação descrita usa o S. pyogenes Cas9 (SpCas9) para gerar quebras de dsDNA gRNA-dirigido em um local de integração selecionado. O repórter então é direcionado para o locus genômico escolhido através de recombinação homóloga, fornecendo um não-linearizada direcionamento vetorial (tv) que contém a sequência de repórter ladeada por chamada 5' e 3' homologia braços (HA)11.
-
Escolha do locus alvo, gRNA e direcionamento de desenho vetorial
- Escolha o locus thegenomic ser alvo baseado na questão científica individual. Utilização de listas publicadas dos sítios de integração recorrentes de HIV encontradas em pacientes em diferentes estudos2,3,4,5,6,7,8 como um orientação. In silico extrair a sequência genômica do locus genômico desejado ser alvo (sequência completa do gene) ou pelo menos 5 kb de sequência genômica usando UCSC genome browser (http:// genome.edu.ucsc.edu).
- Escolha a guia RNAs (gRNAs) de 20 nt para o direcionamento do locus genômico escolhido usando o E-CRISP webtool (http://www.e-crisp.org).
- Selecione "Homo sapiens GRCh38" como o organismo. Tensão de entrada 2.000 da sequência genômica cobrindo o desejado locus genômico extraído na etapa 1.1.1.
- Inicie uma pesquisa de gRNA usando as configurações do aplicativo médio (qualquer PAM, qualquer 5' base, fora-alvos toleram incompatibilidades, e consoles de CPG/intrões são excluídos). Com gRNA possíveis projetos irá aparecer uma lista, classificação do mais alto para Baixar Partituras para especificidade e eficiência.
- Selecione uma gRNA que de preferência mostra uma pontuação elevada especificidade e eficiência e é o mais próximo possível para o locus genômico desejado para ser direcionado.
Nota: Um compromisso entre design de gRNA específico e eficiente e proximidade com o locus genômico desejado não foi encontrado.
- A sequência de gRNA escolhido contra o genoma de referência usando o navegador de explosão NCBI (https://blast.ncbi.nlm.nih.gov) para verificar se há singularidade de sítio de ligação a gRNA de explosão.
- Selecione "humano", como o genoma. A sequência de gRNA como a sequência de consulta de entrada. Selecione "sequências altamente similares" (Megarajada) como o programa. Certifique-se de que a sequência de gRNA é exclusiva. Se não, escolhi uma gRNA diferente da etapa 1.1.2.3 e explosão novamente.
- Depois de gRNA sequência é escolhida, selecione em silico 1.000 bp upstream e downstream de gRNA sequência sequência genômica extraída na etapa 1.1.1 como 5' e 3' HA nesse sentido.
Nota: o gRNAs deve ser homólogas para o locus do local escolhido integração genômica e adjacente a um motivo de protospacer adjacentes (PAM; por exemplo,, NGG para SpCas9) (Figura 2a). A tv contém a sequência de repórter que é 5' e 3' ladeado por tem. Tem capa 1000 bp montante e a jusante do gRNA sequência11. A sequência completa de gRNA não deve ser incluída no HA. Uma sobreposição de até 5 nt é aceitável (Figura 2a).
-
Geração de gRNA e direcionamento de vetores
Nota: Para os regimes de vector, ver Figura 2b.- Para gerar um vetor para expressão de SpCas9 e gRNA, use o pX330-U6-Chimeric_BB-cBh-hSpCas9 como a espinha dorsal do qual tanto SpCas9 como o único guia do RNA (sgRNA) podem ser simultaneamente expressa. Para clonar a sequência de gRNA na espinha dorsal, use a BbsI restrição sites12.
- Para gerar a tv, escolha um alto-cópia do plasmídeo como espinha dorsal (por exemplo, pMK, ou cDNA3.1).
- Primeiro, montar o repórter (neste protocolo: LTR_tdTomato_BGH-PA) para a espinha dorsal de construção por Gibson Assembly fechando13 usando um conjunto comercial clonagem kit e introduzir 5' e 3' flanqueando sites de restrição (por exemplo,, 5' PacI e 3' SmaI) para restrição posterior digestão clonagem da tem.
- Amplificar 1000 bp dos fragmentos HA escolhido na etapa 1.1.4 do DNA genômico (gDNA) o tipo de células a ser alvo (neste protocolo: células Jurkat) usando uma DNA polimerase com atividade de revisão (ver tabelas 1 e 2 para ingredientes PCR e ciclismo de condições). Em seguida, introduza o repórter flanqueando locais da limitação nas extremidades 5' e 3' de cada HA (PacI em 5' HA em ambas as extremidades, SmaI na 3' HA em ambas as extremidades).
- Sequencialmente o clone tem a espinha dorsal da construção já contendo o repórter (gerado na etapa 1.2.2.1) pela enzima de restrição clonagem14,15. Primeiro, clonar em 5' HA então usando sites de restrição PacI, clone em 3' HA usando sites de restrição SmaI.
Nota: Se a espinha dorsal de tv contém um repórter fluorescente adicional, integração indesejada espinha dorsal pode ser avaliada por citometria de fluxo (consulte as etapas 3.2.2 e 3.2.3). Se a espinha dorsal de tv não contém nenhum repórter fluorescente, integração de espinha dorsal deve ser avaliada usando PCR (consulte a etapa 3.2.8).

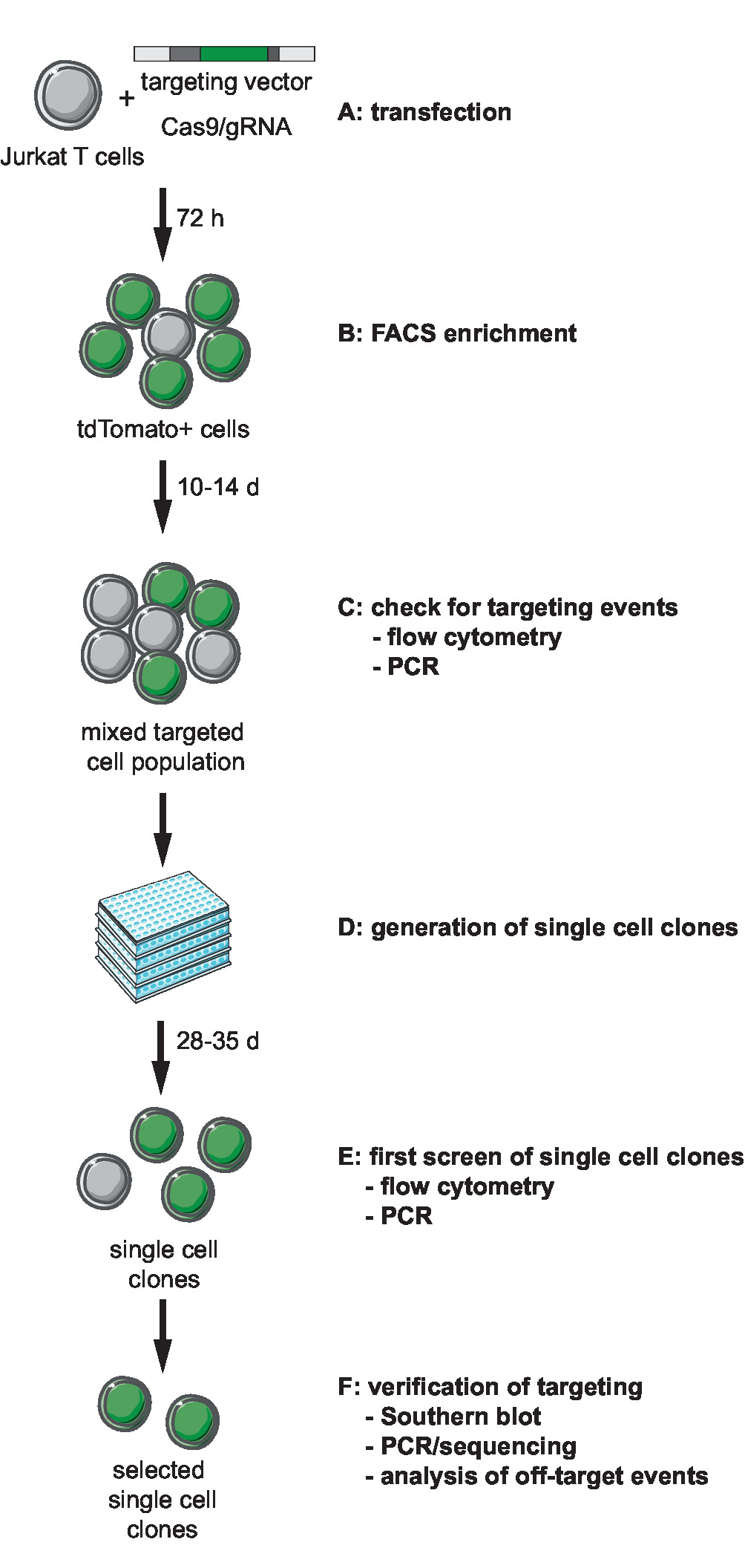
Figura 1: linhas de fluxo de trabalho para CRISPR Cas9-mediada por segmentação, geração e seleção de repórter clonal com site de integração definido. (A) gerar o vetor alvo e transduce células Jurkat T com o vetor de alvo e Cas9/gRNA do plasmídeo de expressão. (B) Enrich o transfeccao de post de 72 h de células transfectadas por FACS. (C) Deixe as células crescem por 10 a 14 dias e confirmam a ocorrência de segmentação de eventos por PCR e citometria de fluxo. (D) gerar clones de célula única limitando clones de diluição e deixe crescerem por 3 semanas. (E) os clones para o direcionamento correto por PCR de tela e fluxo cytometry em formato de 96 poços. Expanda os clones selecionados. (F) verificar correto direcionamento em clones selecionados por Southern blot, PCR e sequenciamento e análise de eventos fora do alvo da atividade de endonuclease Cas9. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: segmentação estratégia e vector design. (um) gRNA e escolha de armas de homologia. 20 nt gRNA é homólogo no site escolhido destino genômica e adjacente a uma homologia de Pam braços são complementares para 1.000 bp - up e a jusante da gRNA e não devem incluir a sequência de gRNA. (b) os esquemas de direcionamento de vetor e o vetor de gRNA/Cas9. O vetor de segmentação consiste em sequência de repórter escolhido é 5' e 3' ladeado pelos braços homologia. O vetor de gRNA/Cas9 baseia-se a espinha dorsal pX330-U6-Chimeric_BB-cBh-hSpCas9. (c) esquema de direcionamento por recombinação homóloga. Vetor alvo e guideRNA/Cas9 vector são transfectadas em células Jurkat. Cas9 Medeia uma dupla vertente pausa no genoma alvo (indicado pelo *) e facilita a recombinação homóloga e a integração de sequência de repórter no locus de genoma alvo. Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. CRISPR-Cas9-direcionamento com base em células Jurkat
-
Transfecção de células Jurkat
- 24 h antes de transfeccao, placa 1,25 x 106 Jurkat T células em 2,5 mL de RPMI 1640 suplementado com 10% (v/v) de soro fetal bezerro (FCS) e 4 mM L-glutamina [referido como "Antibióticos de w.o. RPMI (AB)"] por poço de uma placa de cultura de células de 6-poços. Para uma única experiência de direcionamento, preparar uma completa 6 placa (ou seja,, 6 poços cada um com 2,5 mL de suspensão de células).
- No dia seguinte, co transfect as células com circular tv e pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA usando um reagente de transfeccao específico para células Jurkat.
- Adicione 2 µ g de tv circular e 2 µ g de pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA por alvéolo a 250 µ l de comercial meio RPMI com concentração reduzida de soro otimizada para transfeccao (RPMI com redução de 50% de soro) em um tubo de reação e misture bem.
- Adicionar 12 µ l de reagente de transfeccao lentamente ao meio de DNA sem tocar a parede do tubo e agite. Deixe a mistura incubar durante 15 min e adicionar gota a gota para um poço de células. Incube as celulas em 37 ° C e 5% de CO2.
Nota: a preparação da reação de transfeccao pode ser ampliada. Nenhuma mudança média é necessária depois do transfection.
-
Enriquecimento de células transfectadas por fluorescência-ativado da pilha (FACS) de classificação
- 72 h pós-transfeccao, piscina as células transfectadas, contá-los e prepare-se para enriquecimento por FACS. Coletar as células em um tubo cônico de 50 mL, centrifugação a 300 x g e temperatura (RT) para 4 min, lavar as células uma vez em PBS, centrifugue novamente, suspender a pelota em uma quantidade adequada de buffer de FACS (PBS suplementado com FCS 1% + 1 mM EDTA) 1 x 10 7 células/mL e finalmente transferir para um tubo de FACS.
- Submeter as células a FACS e classificar aqueles que expressam o fluorescente repórter da tv (por exemplo,, tdTomato neste protocolo). Colete as células RPMI 1640 suplementado com 10% de FCS, 4 mM L-glutamina e 50 penicilina U/mL e estreptomicina (referido como "RPMI w / AB").
- Após a triagem de FACS, lavar as células uma vez adicionando 20 mL de RPMI w / AB para células classificadas e centrifugar a 300 x g durante 4 min em RT. resuspenda o pellet de células em uma quantidade adequada de RPMI w / AB e placa as células em um poço de uma placa de cultura de células com a volume apropriado de acordo com o número de célula post-FACS.
Nota: Cultura classificar as células em um pequeno volume (por exemplo,, 24-bem), como níveis consideráveis de morte celular tem sido observados na primeira semana pós-segmentação (até 80-90%). - Expanda a população de mistura de células alvo até uma densidade de 1 x 106 células/mL em um frasco de cultura de células de 75 cm ². Isto vai demorar em torno de 10 a 14 dias post-FACS classificação.
-
Confirmação de segmentação de eventos por citometria de fluxo na população mista célula alvo
- Após 10 a 14 dias de expansão (quando células chegaram a uma densidade de 1 x 106 células/mL em balão de cultura de células 75 cm2 ), dois poços de placa com 1 x 106 células do misto direcionados a população de células em 1 mL de RPMI w / AB em uma cultura celular de 12-poços placa.
- Induzir o HIV LTR do repórter (geração de tv é descrita na etapa 1.2.2.1.) em um dos poços, adicionando 50 ng/mL phorbol myristate 12 13-acetato (PMA) e 1 µM Ionomycin (designado PMA-Iono). Use células no segundo bem como o controle de não-induzida. Cultura o induzido e as células não induzida por 24 h.
- Tomar 0,5 mL de suspensão de células de células não-induzido e induzidas (cada um), lave-os vez em PBS e suspender cada um em 200 µ l de tampão de FACS.
- Analise a 100.000 células por citometria de fluxo. Portão as single-células viáveis com base no tamanho de dispersão para a frente e lateralmente e analisar a expressão do gene repórter fluorescentes.
Nota: Neste passo (10 – 14 dias post-FACS classificação), expressão transiente de repórter fluorescente por transfecção já não deve ser detectável. Expressão fluorescente neste ponto do tempo indica integração genômica de sequência de repórter.
-
Deteção de segmentação de eventos por PCR em DNA genômico da população mista célula alvo
Nota: Para detectar eventos direcionamentos através de PCR, projeto dois pares de primer específico para a junção de integração (int.) 5' e 3' int. junção. Para 5' int. junção do PCR, o primer para a frente deve ligar a montante da 5' HA e o primer reverso na LTR do repórter (primeiras demão P1 e P2 na Figura 3a). O par de primer para a junção de int. 3' PCR deve abranger desde o PA do repórter a 100 – 200 bp a jusante de 3' HA (primeiras demão P3 e P4 na Figura 3a). As primeiras demão P1 e P4 servirá também para a amplificação do alelo na população alvo misto não específico. Para um esquema, consulte Figura 3a.- Prepare gDNA de 2 mL de suspensão de células da população mista célula alvo da etapa 2.2.4. Use um kit de extração gDNA de acordo com o protocolo do fabricante. Então prepare o gDNA de células não-alvo como um controle.
- Executar a junção int. PCRs (primers P1/P2 e P3/P4 para 5' e 3' int. junction, respectivamente) e alelo não específico do PCR (primeiras demão P1/P4) usando uma alta fidelidade DNA polymerase (ver tabelas 3 e 4 para condições de ciclismo e ingredientes PCR) . Analise a 5 µ l de produtos do PCR em um gel de agarose/TAE de 1,5%.
Nota: Se a população-alvo mista contém células que sofreram engenharia de genoma, um produto PCR específico deve ser observado que não é detectável em gDNA de células não-alvo (controle negativo). Para o não-alvo alelo PCR, deve-se observar um produto do mesmo tamanho para ambos os alvo e não alvo células (controle positivo para genômicas cartilhas P1 e P4). Se não há bandas são observadas, considere alterar condições de ciclagem do PCR, aumentando o número de ciclos ou alterando o buffer PCR (por exemplo através da adição de DMSO ou maiores quantidades de Mg2 +), ou alterando o polymerase.
3. geração de linhas Clonal e triagem para o direcionamento correto
Nota: Após a confirmação dos eventos direcionamentos na população alvo mistura de células por citometria de fluxo e PCR (seções 2.2-2.4), gerar clones de célula única (duração: 28 a 35 dias) e tela para integração correta da sequência de repórter.
- Geração de clones de célula única através de chapeamento de diluição
- Preparar meio condicionado Jurkat antecipadamente: Tire RPMI w / médio AB de saudáveis, não tratadas células Jurkat T crescido a 1 x 106 células/mL, centrifugar durante 5 min à x ge filtrar o sobrenadante usando uma unidade de filtro de seringa 0,22 µm.
Nota: Manter o meio condicionado a 4 ° C para armazenamento de curto prazo ou a-20 ° C para o armazenamento de mais de 1 semana. Prepare-se 20 a 30 mL de meio condicionado antes de chapeamento de diluição. - Contar as células alvo da etapa 2.2.4 após 10 a 14 dias de expansão e diluí-los em RPMI w / AB a uma concentração de 1 x 105 células/mL. Pegue 100 µ l de solução de 1 x 105 células/mL e diluir com 9,9 mL de meio para atingir uma concentração de 1.000 células/mL. Tomar 1 mL de solução de 1.000 células/mL e diluir com 9 mL de meio para atingir uma concentração de 100 células/mL.
- Placa de 96 poços placas contendo 1 célula por alvéolo e 2 células por poço. Para 1 célula por alvéolo, tomar 1 mL de solução de 100 células/mL e misture suavemente com 5 mL de meio condicionado e 4 mL de meio fresco em um reservatório de reagente estéril.
- Para 2 células por poço, tomar 2 mL de solução de 100 células/mL e misture suavemente com 5 mL de meio condicionado e 3 mL de meio fresco. Placa de 96 poços fundo redondo placas com 100 µ l da diluição respectiva célula por bem, usando uma pipeta multicanal.
Nota: 5 a 10 placas de 96 poços por segmentação de construção são suficientes para obter clones para rastreio. - Empilhe as placas de 96 poços, cobrir cada pilha com uma placa de 6 contendo 3 mL de PBS em cada poço e incubar as placas a 37 ° C numa incubadora de cultura celular umidificado com 5% de CO2 por 3 semanas.
Nota: Não alterar o meio de cultura celular durante este tempo. Não abra a incubadora mais de uma vez ou duas vezes por semana. Os melhores resultados são observados em incubadoras com reservatório de água aberta. - Após 3 semanas de incubação, confirmar visualmente a presença de colônias crescidas usando microscopia de luz (ampliação de 4x) e marcar os poços com colônias crescidas, então eles são visíveis como pontos no fundo dos poços.
- Prepare uma placa de 96 poços fundo redondo com 100 µ l de RPMI w / AB por bem. Resuspenda suavemente as células de um marcado bem pipetando. Transferir 100 µ l de suspensão de células em um poço da placa de 96 poços novo já contendo 100 µ l de RPMI w / AB e, em seguida, misture suavemente pipetando. Transferi 100 µ l da suspensão celular para um vazio de segundo placa de 96 poços fundo redondo para duplicar a placa.
- Continue com todos os poços marcados com colônias crescidas. Preencha todos os poços em branco com 200 µ l de RPMI w / médio de AB. Incubar as placas a 37° C e 5% CO2.
Nota: Dentre estas placas servirá para expansão de clones de célula única ("placa de estoque") e o outro como um prato"duplicado" para a seleção.
- Preparar meio condicionado Jurkat antecipadamente: Tire RPMI w / médio AB de saudáveis, não tratadas células Jurkat T crescido a 1 x 106 células/mL, centrifugar durante 5 min à x ge filtrar o sobrenadante usando uma unidade de filtro de seringa 0,22 µm.
- Seleção de clones de célula única por citometria de fluxo e PCR
Nota: Enquanto os clones de célula única estão expandindo, utilize a placa duplicada da etapa 3.1.8 para clones de célula única tela para presença de sequência de repórter pelo PCR (3.2.4–3.2.12 passos) e expressão de repórter fluorescente por citometria de fluxo (3.2.2–3.2.3 passos) (Figura 3C).- Deixe a placa duplicada incubar por 24 a 48 h e duplique a placa novamente. Para fazer isso, adicione 100 µ l de RPMI w / AB a cada poço, misture suavemente pipetando e transferir 100 µ l de uma nova placa de 96 poços fundo redondo usando uma pipeta multicanal. Use um prato para triagem de citometria de fluxo e a outra para rastreio baseados em PCR.
- Para rastreio de citometria de fluxo, estimulam as células com PMA-Iono. Preparar um mastermix de 0,1 µ l de Ionomocin (1 mM estoque), 0,1 µ l de PMA (50 µ g / µ l, estoque), e 4,8 µ l de RPMI w / AB por número de poços, em seguida, adicione 5 µ l de mastermix por bem.
Nota: Indução é necessária identificar com sucesso clones, onde pode ser a LTR transcriptionally silencioso e, portanto, o repórter fluorescente não é expresso. - Deixe células incubar por 24 h e preparar células por citometria de fluxo, conforme descrito na etapa 2.3.3. Portão qualquer single-células viáveis com base no tamanho de dispersão para a frente e lateralmente e analisar a expressão do gene repórter fluorescente por citometria de fluxo (por exemplo, resultados, ver Figura 3C). Se a espinha dorsal de tv contém segundo repórter fluorescente com promotor (por exemplo,, GFP), qualquer clones para expressão de repórter de espinha dorsal da tela também (ver passo 1.2.2 e a seguinte nota para explicação).
Nota: Backbone repórter expressão indica indesejada integração das sequências de espinha dorsal. - Uma vez que os clones na segunda placa duplicado têm crescido suficientemente (normalmente de 24 a 48 h após a duplicação da placa de 96 poços), preparar lisados celulares contendo gDNA para rastreio de PCR. Centrifugar a placa durante 10 minutos a 300 x g em RT. cuidadosamente Tire o sobrenadante sem perturbar o centrifugado.
Nota: Todos os passos para a preparação de lisados e reações de PCR podem ser realizados com pipetas multicanais. - Lavam-se células com 100 µ l de PBS por pipetagem suave e centrifugação a placa durante 5 min à 300 x g em RT Tire a PBS e adicionar 200 µ l de tampão de Lise [200 mM NaCl, pH 100 mM Tris-HCl 8-8,5, 5 mM EDTA, 0,1% SDS; em seguida, adicione 250 – 1.000 µ g/mL de proteinase K (liofilizado em pó, pesar em fresco)] por bem. Misture suavemente pipetando e a suspensão de transferência para uma nova placa PCR.
- Selar a placa com a película de parafina e incubar durante 1 h a 55 ° C, em um thermocycler. Centrifugar a velocidade máxima durante 10 min (3.000 x g) a girar para baixo os restos da célula e transferir o sobrenadante para um novo prato PCR.
Nota: Lisados celulares em placas podem ser armazenados nesta fase a 4 ° C até utilização posterior. - Preparar um prato PCR de 96 poços com 110 µ l de dH2O e adicionar 10 µ l de lisado celular (01:12 diluição). Lisados celulares podem ser viscoso e difícil de pipeta. Use pelo menos pontas de pipeta de 20 µ l.
- Inactivar proteinase K por incubação por 10 min a 99 ° C em um thermocycler. Posteriormente, use os lysates célula inativada e diluídos para rastreio de PCR.
- Desenho de primers para PCR (P5 e P6) com base na sequência de repórter escolhido para amplificar a bp 500 – 800 da sequência de repórter de triagem. Para o controlo positivo do PCR, use primers P7 e P8 que amplificam 630 bp de um locus genômico tipo selvagem, não específico (geneNUP188 ) (Figura 3 c e tabela 5). Desenha um terceiro par de primer que amplifica bp 500 – 600 do backbone tv como um controle para integração inespecífica de sequências de tv espinha dorsal (backbone de PCR).
- Para triagem, controle e espinha dorsal do PCR, use um comercial do PCR mastermix (ver tabelas 6 e 7 para condições de ciclismo e ingredientes PCR). Use 2 µ l do diluído e inactivados lisado preparado na etapa 3.2.8 como um modelo e executar PCR para 38 a 40 ciclos de amplificação por PCR em formato de 96 poços.
- Analise a 5 µ l de produtos do PCR em um gel de agarose/TAE de 1,5%.
Nota: Para controle do PCR, uma banda específica de 630 bp deve ser observado para cada amostra, confirmando que a qualidade de lisados celulares é adequada para o PCR. Uma banda específica na triagem do PCR (bp 500 – 800 dependendo do projeto da primeira demão) indica a integração da sequência de repórter. Uma banda específica para a espinha dorsal do PCR (bp 500 – 600, dependendo do projeto da primeira demão) indica indesejada integração das sequências de espinha dorsal de tv (por exemplo, resultados, ver Figura 3C). - Combine os resultados de citometria de fluxo (etapa 3.2.3) e triagem baseados em PCR (etapa 3.2.12). Selecione clones que mostram tamanhos corretos dos produtos PCR no rastreio do PCR e controle positivo PCR e expressão de repórter fluorescente após indução com PMA-Iono em citometria de fluxo. Exclua os clones que mostram qualquer produto do PCR na espinha dorsal do PCR ou expressão de televisão codificados em espinha dorsal proteína fluorescente, indicando integração inespecífica de sequência de espinha dorsal de tv.
- Gradualmente expanda clones selecionados da placa de 96 poços estoque para formatos bem maiores (48/24/12/6-bem) até alcançar um formato de balão de cultura de células T75 adicionando meio fresco a cada 2 a 3 dias. Manter uma densidade celular entre 1 x 105 e 1 x 106 células/mL.
- Certifique-se de preparar a existências de célula de clones durante a expansão: contar as células, centrifugar a 300 x g durante 5 min à RT, desprezar o sobrenadante e suspender as células suavemente no FCS + 10% de DMSO em 5 x 106 células/mL. Alíquota em frascos criogênicos e uso um congelamento cryo-recipiente para congelar as células a 80 ° C, em 1 ° C/min. Para armazenamento a longo prazo, transferi-los para líquido N2.
Nota: é aconselhável manter um frasco de cultura celular T75 (ou seja,, em torno de 1 x 107 células) durante a expansão em preparação de gDNA para verificação de definião mancha do Sul (ver secção 3.4).
- Verificação de sites de integração por PCR/sequenciamento em clones selecionados
Nota: junções de int. 5' e 3' dos clones selecionados são PCR amplificado e enviado para Sanger sequenciamento para verificar corrigir direcionamento no nível de sequência do DNA.- Prepare gDNA de clones selecionados e Jurkat selvagem-tipo células usando um kit de extração gDNA comercial.
- Use vinculação a extremidade 5' do repórter e o montante de 5' HA para 5' junção int. (primeiras demão P1 e P2) e a extremidade 3' do repórter e a jusante da 3' HA para 3' junção int. (primeiras demão P3 e P4), conforme descrito na etapa 2.4 de pares da primeira demão. Use as primeiras demão P1 e P4 para amplificar o site alvo integração o alelo sem repórter integrantes (figura 4a).
- Reações de PCR com 100-200 ng de gDNA como um modelo de preparar e realizar PCR usando uma polimerase com atividade de revisão (ver tabelas 1 e 2 para condições de ciclismo e ingredientes PCR).
Nota: Se não há bandas são observadas, considere alterar as condições de ciclagem do PCR, aumentando o número de ciclos ou alterando o buffer PCR (por exemplo, pela adição de DMSO ou maiores quantidades de Mg2 +), ou alterando o polymerase. - Analise a 5 µ l de produtos do PCR em um gel de agarose/TAE de 1,5%. Se a banda correcta tamanhos são observados, purificar o restante produto do PCR usando um kit comercial e submetê-la a Sanger sequenciamento. Verificar sequências de junção de int. 5' e 3' junção int. local pretendido do alelo sem repórter integrantes alinhando-os com sequências esperadas.
Nota: o alelo homólogo, onde o repórter não tem integrado provavelmente mostrará Cas9 mediada por alterações no local de destino, tais como nucleotídeos inserção e deleção (figura 4a). - Para os clones que mostram sequências de junção de int. correto após o alinhamento, executar um PCR amplificando o repórter todo alvo e submeta a Sanger sequenciamento para verificar a sequência correta do integrante.
- Borrão do Sul análise para verificação de direcionamento em clones selecionados
Nota: Análise de Southern blot dos clones selecionados é necessário para verificar o direcionamento correto e excluir os eventos de recombinação Cas9-mediada que possam ter ocorrido no local de integração orientada.- Desenvolver uma estratégia para a digestão apropriada gDNA e sonda o projeto antes de começar o experimento.
- Selecione uma enzima de restrição para restrição gDNA que gera fragmentos apropriados de comprimento de 2 a 10 kb, no local de destino. Determinadas enzimas de restrição, tais como Asp718, BamHI, Bgleu, BglII, EcoRV, HindIII, Ncoeu, Psteu, PvuII, Scaeu, Stu, e SST Eu tenho sido usado com sucesso para digerir gDNA de alto peso molecular.
- Desenha duas sondas Sul diferentes: um repórter específico sonda e sonda genômica. A sonda de repórter específicos se para uma sequência dentro o repórter (ou seja, sonda de, tdTomato-específico). A sonda genômica se para uma região genômica perto (mas não se sobrepõem) um HA.
- Escolha a sonda genômica para que o fragmento gerado pela digestão gDNA que será detectado pela ligação de genômica sonda difere de comprimento (mais de 2 kb) dos alelos visados e não específico (figura 4b). Um comprimento de sonda de 400 a 1.000 bp é recomendado.
- Projeto do PCR primers para amplificar as duas sondas necessárias. Amplificar a sonda repórter específicos do modelo de tv usando uma alta fidelidade DNA polymerase (ver tabelas 3 e 4 para condições de ciclismo e ingredientes PCR).
- Amplificar a sonda genômica do selvagem-tipo Jurkat gDNA preparado com um kit de extração comercial gDNA usando uma DNA polimerase com atividade de revisão (ver tabelas 1 e 2 para condições de ciclismo e ingredientes PCR). Purificar os produtos PCR em um gel de agarose/TAE e extrair os fragmentos usando um kit de extração do gel de comerciais de acordo com as instruções do fabricante.
- Extrai gDNA de alto peso molecular de 1 x 107 células do selvagem-tipo Jurkat pilhas e clones selecionados da etapa 3.2.14.
- Pelotas as células por centrifugação a 300 x g durante 5 min à RT, lave uma vez com PBS e suspender o sedimento em 4 mL de tampão de Lise [200 mM NaCL, pH 100 mM Tris-HCl 8, 5 mM EDTA, 0,1% SDS; em seguida, adicione da protease µ g/mL de 250 – 1.000 K (pó liofilizado pesar fresco)]. Incube o/n a 55 ° C, agitando a 350 rpm em um thermomixer do tabletop.
- Adicionar 4 mL de isopropanol e misture por inversão de 10 a 20 vezes. O gDNA deve tornar-se visível como precipitado branco. Spool o precipitado gDNA para a ponta fina de uma pipeta de vidro, lave por emergentes em 750 µ l de 70% EtOH e deixar seco no RT (5 a 10 min).
- Derramou o precipitado para um tubo de reação de 1,5 mL contendo 500 µ l de tampão de TE 1 x (10 mM Tris-HCl pH 8,0, 1 mM EDTA) e deixar para dissolver o/n a 4 ° C, agitando a 350 rpm. Qualquer pipetagem de gDNA desta etapa deve ser feito com furo largo dicas para evitar distorção.
Nota: Preparação de gDNA de alto peso molecular é essencial para a análise de Southern blot, e kits de preparação gDNA comercialmente disponíveis não são adequados.
- Digest (duas vezes) 15 µ g de gDNA de clones selecionados e selvagem-tipo Jurkat células com enzima de restrição selecionada (consulte a etapa 3.4.1.1) em uma reação de 60 µ l com 6 µ l da enzima (20 unidades / µ l): em primeiro lugar, adicionar DNA, tampão de digestão e ddH2O, incubam o/n a 37 ° C , em seguida, adicionar a enzima e incubar o/n a temperatura de digestão enzimática específica. 15 µ g de gDNA digerido é necessário por sul da sonda.
- Use 7 µ l do Resumo de restrição 60 µ l para a electroforese do gel de analítica em um gel de agarose/TAE 1%. Um esfregaço indica a digestão completa e boa qualidade de DNA para análise de Southern blot.
- Precipitar o restante digest de restrição adicionando 01:10 de acetato de sódio 3M e 2 volumes 100% EtOH, então incubar durante 1 h a-80 ° C e centrifugação por 30 min a 15.600 x g a 4 ° C.
- Desprezar o sobrenadante e lavar o sedimento com 70% EtOH. Centrifugar por 15 min a 15.600 x g a 4 ° C, desprezar o sobrenadante, deixar o sedimento secar brevemente a RT e dissolver em 20 µ l de DDQ2O.
- Execute 1% gel do agarose/TAE mancha, carregando 20 uL de gDNA digerido por faixa. Funcione o gel por 2 h a 60 V, 400 mA.
Nota: a porcentagem de agarose gel e execução tempo/tensão pode ser ajustada de acordo com o tamanho do fragmento esperado para a deteção de Southern blot, calculado na etapa 3.4.1.1. As seguintes etapas de análise de Southern blot são descritas em detalhes em um protocolo complementar (etapas 1 a 18). Estes passos são compostos por: lavagem do gel mancha, mancha em uma membrana de nylon, geração de sonda radioativa, hibridização da sonda e desenvolvimento do filme autoradiograph. Comparar o padrão de bandas obtido após o desenvolvimento de autoradiograph na etapa 18 (protocolo suplementar) com o padrão esperado de acordo com a estratégia do Sul (por exemplo, resultados, veja figura 4b).
- Desenvolver uma estratégia para a digestão apropriada gDNA e sonda o projeto antes de começar o experimento.
- Análise de eventos fora do alvo
Nota: Desde engenharia genoma CRISPR-Cas9-mediado pode gerar efeitos fora do alvo, PCR-amplificar os dez mais altos do ranking em silico-previu sites fora do alvo em selecionado clones e sujeitá-los a Sanger sequenciamento.- Usar CCTop16 (http://crispr.cos.uni-heidelberg.de) para gerar uma lista dos dez mais altos do ranking em silico previu sequências fora do alvo.
- A sequência de gRNA incluindo PAM como usado para o direcionamento como a sequência de consulta de entrada. Selecione "NGG" como PAM e "Genoma humano" como referência para a previsão de fora do alvo.
- Conjunto de incompatibilidades totais máximas para "4" e alvo site comprimento para o comprimento da gRNA sem PAM. O arquivo de saída irá fornecer uma lista classificada de genômicas sites fora do alvo para gRNA respectivo.
- In silico extrair a sequência genómica 500 bp montante e a jusante de cada um dos dez mais altos do ranking fora do alvo hits usando UCSC Genome Browser (http://genome.edu.ucsc.edu) e a posição do fora-alvo atingido CCTop lista de resultados.
- Para cada um, fora os sites de destino para análise, projeto de uma cartilha PCR par que amplifica um fragmento de 600 a 700 bp em comprimento, incluindo o site fora do alvo previsto.
- Extraia o gDNA dos clones selecionados e Jurkat selvagem-tipo pilhas usando um kit de extração gDNA comercial. Para cada site de fora do alvo, realizar uma PCR usando uma DNA polimerase com atividade de revisão (ver tabelas 1 e 2 para condições de ciclismo e ingredientes PCR) no tipo selvagem e o respectivo gDNA clone-derivado.
- Analise a 5 µ l de produtos do PCR em um gel de agarose/TAE de 1,5%. Se a banda correcta tamanhos são observados, purificar o restante produto do PCR usando um kit de purificação de PCR comercial e submetê-la a Sanger sequenciamento. Compare sequências dos sites fora do alvo em células Jurkat e clones do alvo.
- Usar CCTop16 (http://crispr.cos.uni-heidelberg.de) para gerar uma lista dos dez mais altos do ranking em silico previu sequências fora do alvo.
Access restricted. Please log in or start a trial to view this content.
Resultados
Neste experimento representativos, optámos por um repórter de HIV-1-derivado mínimo consistindo de um LTR, sequência tdTomato-codificação e sequência de polyA-sinal para dois loci no intron 5 do gene BACH217-alvo. Os Locus para o direcionamento foram escolhidos de acordo com a proximidade de sites publicados integração recorrentes encontrados em diferentes estudos primários células T de pacientes infectados pelo HIV2,
Access restricted. Please log in or start a trial to view this content.
Discussão
Aqui, descrevemos um protocolo para gerar modelos de HIV-1-derivado Jurkat repórter com sites de integração proviral escolhido aplicando engenharia CRISPR-Cas9-baseado do genoma.
Vários pontos do protocolo requerem atenção cuidadosa durante a fase de planejamento. Primeiro, o locus ser alvo deve ser escolhido com cuidado, como alguns loci pode ser mais fácil para o alvo do que outros (por exemplo,, dependendo do estado da cromatina da região e o destino de sequências em si). ...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Agradecemos a Britta Weseloh e Bettina Abel para assistência técnica. Agradecemos também Arne Düsedau e Jana Hennesen (fluxo cytometry plataforma tecnológica, Heinrich Pette Institut) para suporte técnico.
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| pX330-U6-Chimeric_BB-cBh-hSpCas9 | Addgene | 42230 | vector for expression of SpCas9 and gRNA |
| pMK | GeneArt | mammalian expression vector for cloning | |
| cDNA3.1 | Invitrogen | V79020 | mammalian expression vector for cloning |
| BbsI | New England Biolabs | R0539S | restriction enzyme |
| NEBuilder Hifi DNA Assembly Cloning Kit | New England Biolabs | E5520S | Assembly cloning kit used for target vector generation |
| TaqPlus Precision PCR System | Agilent Technologies | 600210 | DNA polymerase with proofreading activity used for amplification of homology arms (step 1.2.2.2), verification of integration site and reporter sequence (step 3.3.3 and 3.3.5), generation of genomic probe for Southern blot (step 3.4.1.5) and analysis of off-target events (step 3.5.4) |
| 96-well tissue culture plate (round-bottom) | TPP | 92097 | tissue culture plates for dilution plating |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530 L | DNA polymerase used for detection of targeting events (step 2.4.2) and generation ofreporter-specific probe for Southern blot (step 3.4.1.4) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D9170 | dimethyl sulfoxide as PCR additive |
| Magnesium Chloride (MgCl2) Solution | New England Biolabs | B9021S | MgCl2 solution as PCR additive |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | dNTP mixture with 10 mM of each nt for PCR reactions |
| 5PRIME HotMasterMix | 5PRIME | 2200400 | ready-to-use PCR mix used for screening PCR (step 3.2.11) |
| QIAamp DNA blood mini kit | Qiagen | 51106 | DNA isolation and purification kit |
| QIAquick PCR Purification Kit | Qiagen | 28106 | PCR Purification Kit |
| RPMI 1640 without glutamine | Lonza | BE12-167F | cell culture medium |
| Fetal Bovine Serum South Africa Charge | PAN Biotech | P123002 | cell culture medium supplement |
| L-glutamine | Biochrom | K 0282 | cell culture medium supplement |
| Penicillin/Streptomycin 10.000 U/mL/ 10.000 µg/mL | Biochrom | A 2212 | cell culture medium supplement |
| Gibco Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | 31985062 | cell culture medium with reduced serum concentration optimized for transfection |
| TransIT-Jurkat | Mirus Bio | MIR2125 | transfection reagent |
| phorbol 12-myristate 13-acetate | Sigma-Aldrich | P8139-1MG | cell culture reagent |
| Ionomycin | Sigma-Aldrich | I0634-1MG | cell culture reagent |
| Syringe-driven filter unit, PES membrane, 0,22 µm | Millex | SLGP033RB | filter unit for sterile filtration |
| Heracell 150i incubator | Thermo Fisher Scientific | 51026280 | tissue culture incubator |
| Amershan Hybond-N+ | GE Healthcare | RPN1520B | positively charged nylon membrane for DNA and RNA blotting |
| Stratalinker 1800 | Stratagene | 400072 | UV crosslinker |
| High Prime | Roche | 11585592001 | kit for labeling of DNA with radioactive dCTP using random oligonucleotides as primers |
| illustra ProbeQuant G-50 Micro Columns | GE Healthcare | 28-9034-08 | chromatography spin-columns for purification of labeled DNA |
Referências
- Hughes, S. H., Coffin, J. M. What Integration Sites Tell Us about HIV Persistence. Cell Host and Microbe. 19 (5), 588-598 (2016).
- Marini, B., Kertesz-Farkas, A., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 521 (7551), 227-231 (2015).
- Cesana, D., Santoni de Sio, F. R., et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nature Communications. 8 (1), 498(2017).
- Cohn, L. B., Silva, I. T., et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell. 160 (3), 420-432 (2015).
- Han, Y., Lassen, K., et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 78 (12), 6122-6133 (2004).
- Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. The Journal of Infectious Diseases. 195 (5), 716-725 (2007).
- Wagner, T. A., Mclaughlin, S., et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 345 (6196), 570-573 (2014).
- Maldarelli, F., Wu, X., et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 345 (6193), 179-183 (2014).
- Jordan, A., Bisgrove, D., Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 22 (8), 1868-1877 (2003).
- Mack, K. D., Jin, X., et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. Journal of Acquired Immune Deficiency Syndromes. 33 (3), 308-320 (2003).
- Byrne, S. M., Ortiz, L., Mali, P., Aach, J., Church, G. M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Research. 43 (3), 1-12 (2014).
- ZhangLab. CRISPR Genome Engineering Toolbox: Target Sequence Cloning Protocol. Addgene website. , Available from: https://www.addgene.org/static/cms/filer_public/e6/5a/e65a9ef8-c8ac-4f88-98da-3b7d7960394c/zhang-lab-general-cloning-protocol.pdf (2013).
- Moser, F. Addgene. Gibson Assembly Protocol. Addgene website. , Available from: https://www.addgene.org/protocols/gibson-assembly/ (2009).
- Addgene Plasmid Cloning by PCR. Addgene website. , Available from: https://www.addgene.org/protocols/pcr-cloning/ (2014).
- Addgene Plasmid Cloning by Restriction Enzyme Digest (aka Subcloning). Addgene website. , Available from: https://www.addgene.org/protocols/subcloning/ (2013).
- Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., Mateo, J. L. CCTop: An intuitive, flexible and reliable CRISPR-Cas9 target prediction tool. Public Library of Science (PLoS) ONE. 10 (4), (2015).
- Lange, U. C., Bialek, J. K., Walther, T., Hauber, J. Pinpointing recurrent proviral integration sites in new models for latent HIV-1 infection. Virus Research. 249, (2018).
- Bialek, J. K., Dunay, G. A., et al. Targeted HIV-1 Latency Reversal Using CRISPR-Cas9-Derived Transcriptional Activator Systems. PloS ONE. 11 (6), e0158294(2016).
- Lee, C. M., Davis, T. H., Bao, G. Examination of CRISPR-Cas9 design tools and the effect of target site accessibility on Cas9 activity. Experimental Physiology. 103 (4), 456-460 (2018).
- Jensen, K. T., Fløe, L., et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Letters. 591 (13), 1892-1901 (2017).
- Simonetti, F. R., Sobolewski, M. D., et al. Clonally expanded CD4 + T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 113 (7), 1883-1888 (2016).
- Chen, H. C., Martinez, J. P., Zorita, E., Meyerhans, A., Filion, G. J. Position effects influence HIV latency reversal. Nature Structural and Molecular Biology. 24 (1), 47-54 (2017).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados