
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Fluoreszenz Biomembrane Kraft Sonde: Concurrent Quantifizierung von Rezeptor-Ligand-Kinetik und Binding-induzierte intrazelluläre Signaling auf Einzelzell
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
We describe a technique for concurrently measuring force-regulated single receptor-ligand binding kinetics and real-time imaging of calcium signaling in a single T lymphocyte.
Zusammenfassung
Membranrezeptor-Liganden-Wechselwirkungen vermitteln viele zelluläre Funktionen. Bindungskinetik und nachgeschalteten Signal durch diese molekularen Wechselwirkungen ausgelöst werden wahrscheinlich durch die mechanische Umgebung, in der Bindung und Signalisierung erfolgen betroffen. Eine neuere Studie hat gezeigt, dass eine mechanische Kraft kann die Antigen-Erkennung durch regulieren und Auslösen der T-Zellrezeptor (TCR). Dies wurde durch eine neue Technologie, die wir entwickelt und bezeichnet Fluoreszenz Biomembran Kraftsonde (fBFP), die Einzelmolekülkraftspektroskopie mit Fluoreszenzmikroskopie kombiniert möglich. Mit seinem ultra-soft menschlichen roten Blutkörperchen als sensitive Kraftsensor, ein High-Speed-Kamera und Echtzeit-Bildgebung Tracking-Techniken ist von ~ 1 pN (10 -12 N), ~ 3 nm die fBFP und ~ 0,5 ms in Kraft, räumlicher und zeitlicher Auflösung. Mit der fBFP, kann man genau einzigen Rezeptor-Ligand-Bindungskinetik unter Kraftregelung und gleichzeitig image Bindung ausgelöste intrazelluläre cal messencium Signalisierung auf einer einzelnen lebenden Zellen. Diese neue Technologie kann verwendet werden, um andere Membranrezeptor-Liganden-Wechselwirkung und Signalisierung in anderen Zellen unter mechanischer Regulierung untersuchen.
Einleitung
Zell-zu-Zell und Zell-extrazellulären Matrix (ECM) Haftung wird durch die Bindung zwischen Zelloberflächenrezeptoren, ECM-Proteine und / oder Lipide 1 vermittelt. Bindung erlaubt den Zellen funktionalen Strukturen 1 zu bilden, ebenso wie zu erkennen, zu kommunizieren und zu reagieren, um die Umwelt 1-3. Anders als lösliche Proteine (zB Cytokine und Wachstumsfaktoren), die binden aus einem dreidimensionalen (3D) Fluidphase auf die Zelloberflächenrezeptoren Zelladhäsionsrezeptoren bilden Bindungen mit ihren Liganden über einen schmalen Spalt junctional zu zwei einander gegenüberliegenden Oberflächen, die Molekular beschränken brücken Diffusion in einem zweidimensionalen (2D) Schnittstelle 4-7. Im Gegensatz zu 3D-Kinetik, die üblicherweise durch herkömmliche Bindungstests (zB Oberflächenplasmonresonanz oder SPR) gemessen werden, 2D-Kinetik mit spezialisierten Techniken wie Rasterkraftmikroskopie (AFM) 8-10 quantifiziert werden, Durchflusskammer 11,12, Mikro 13,14, optischePinzette 15 und Biomembran Kraftsonde (BFP) 16-21.
Mehr als lediglich die Bereitstellung physikalische Verknüpfung für die zelluläre Kohäsion, sind Adhäsionsmoleküle, eine Hauptkomponente des Signal-Maschine, damit die Zelle mit ihrer Umgebung zu kommunizieren. Hat eine zunehmende Interesse für das Verständnis, wie Ligand Eingriff Adhäsionsmoleküle initiiert intrazellulären Signalisierung und wie die anfängliche Signal innerhalb der Zelle transduziert. Intuitiv Eigenschaften des Rezeptor-Ligand-Bindung kann die Signale induziert auswirken. Es ist jedoch schwierig, mechanistische Beziehungen zwischen der extrazellulären Interaktion und intrazelluläre Signalereignisse sezieren Verwendung traditioneller Ensemble von biochemischen Assays wegen ihrer vielen Einschränkungen, beispielsweise eine schlechte zeitliche Auflösung und das völlige Fehlen von räumlicher Auflösung. Bestehende Verfahren, die sowohl biophysikalischer (2D-Rezeptor-Liganden-Bindung Kinetik) zu ermöglichen und biochemische (Signalisierung) Beobachtungen über Live-Zellen umfassen Substrate von abstimmbaren Steifigkeit 22, Elastomer Säule Arrays 23 und Strömungsraum / mikrofluidischen Vorrichtungen mit Fluoreszenzfähigkeit 24-26 integriert. Jedoch haben Auslesungen Signalisierung und Rezeptor-Ligandenbindungs separat (häufig durch verschiedene Verfahren) erhalten werden, was es erschwert, zeitlichen und räumlichen Beziehungen der Bindungseigenschaften mit Signalisierungsereignisse zu sezieren.
Konventionelle BFP ist eine ultrasensitive Kraftspektroskopie mit hoher Raum-Zeit-Auflösung 17. Es verwendet eine flexible roten Blutkörperchen (RBC) als Kraftsensor, eine Messung der Einzelmolekül 2D Kinetik, mechanische Eigenschaften und Konformationsänderungen 14,16,19-21,27-29. Ein Fluoreszenz-Bildgebung basiert BFP (fBFP) korreliert die Rezeptor-Ligand-Bindungskinetik mit der Bindung ausgelöste Zellsignalisierung bei Einzelmolekülskala. Mit diesem Setup in situ Zellsignalisierung Tätigkeiten im Zusammenhang mit der Oberfläche mechanical Stimulation wurde in T-Zellen, 27 beobachtet. Die fBFP ist vielseitig und kann für die Untersuchung der Zellhaftung und Signalisierung durch andere Moleküle in anderen Zellen vermittelt werden.
Protokoll
Dieses Protokoll folgt den Richtlinien und wurde von der menschlichen Forschung Ethik-Kommission des Georgia Institute of Technology genehmigt.
1. Menschen RBCs Isolation, Biotinylierung und Osmolarität Adjustment
Hinweis: Schritt 1.1 sollte von einem ausgebildeten Arzt, wie eine Krankenschwester durchgeführt werden, mit einem Institutional Review Board genehmigten Protokoll.
- Erhalten 8-10 ul (ein Tropfen) Blut von Finger stechen und fügen Sie 1 ml der Carbonat / Bicarbonat-Puffer (Tabelle 1 und 2). Vorsichtig vortexen oder einer Pipette die Mischung und Zentrifuge für 1 min bei 900 x g. Überstand verwerfen und waschen Sie einmal.
- In einem kleinen Becherglas wiegen 3,5-4 mg Biotin-NHS-PEG3500-Linker (Tabelle 1). Löst ihn in Carbonat / Bicarbonat-Puffer, um eine Endkonzentration von 3 mg / ml zu ergeben.
- Mischungs 171 ul / Bicarbonat-Puffer, 10 ul RBC Packung und 1049 ulBiotin-PEG3500-NHS Linker Lösung, Inkubation bei Raumtemperatur 30 min. Waschen Sie die RBC einmal mit Carbonat / Bicarbonat-Puffer und dann zweimal mit N2-5% Puffer (Tabelle 1 und 2).
- In der Zwischenzeit setzen Sie die Linker-Flasche mit gelockerten Kappe in einem Glas Vakuumexsikkator mit Trockenmittel in den Boden und Vakuum für 5 Minuten gefüllt, und füllen Sie den Exsikkator mit Argon. Ziehen Sie den Deckel und nehmen Sie die Flasche aus. Verschließen Sie die Flasche mit Kunststoffparaffinfilm (Tabelle 1), legen Sie sie in einen Behälter mit Trockenmittel auf der Unterseite und lagern in -20 ° C gefüllt.
Hinweis: die Schritte, die die Verwendung von Biotin-PEG3500-NHS Linker beinhalten, einschließlich 1,2-1,4 (außer der Inkubation und Waschen in 1,3), müssen so schnell wie möglich durchgeführt werden. - Verdünnter Nystatin in N2-5% Puffer bis zu einer Endkonzentration von 40 ug / ml zu ergeben. Mischen Sie 5 ul biotinyliertes RBC mit 71,4 ul Nystatin (Tabelle 1) Lösung, Inkubation für 1 h bei 0 &# 176; C. Zweimal waschen mit N2-5% Puffer und Speicher mit N2-5% Puffer + 0,5% BSA (Tabelle 1) im Kühlschrank (4 ° C).
2. Glasperlen Silanisierung
- Reinigung der Partikeloberfläche
- Wiegen 50 mg Glaskügelchen Pulver und resuspendieren sie in 500 & mgr; l DI-Wasser.
- Mischungs 0,5 ml 30% H 2 O 2 (Tabelle 1) mit 9,5 ml DI-Wasser in einem 50 ml Becherglas, dann 2 ml konzentrierte NH 4 OH (Tabelle 1) und bringt diese Lösung in einem Kessel auf eine heiße Platte .
- In der Glasperlen in die kochende Lösung und weiterhin für weitere 5 Minuten kochen lassen. Vorsichtig schwenken die Lösung alle min.
- Nach dem Kochen zu übertragen ~ 5 ml dieser heißen Perlensuspension in ein 15 ml Mikrozentrifugenröhrchen geben und mit RT DI-Wasser. Zentrifuge bei 3.500 xg für 5 min, zu entfernen und den Überstand verwerfen.
- Übertragen Sie weitere 5 ml heißem Beadsuspension und zu den gewaschenen Perlen, Auffüllen mit DI-Wasser, gut mischen und zentrifugieren erneut. Diesen Vorgang wiederholen, bis etwa 50 ml DI-Wasser verwendet wird, die insgesamt 4 bis 5 mal Waschen wird.
- Übertragen Sie die Bead-Suspension in eine 1 ml-Fläschchen. Wiederholen Waschen der Perlen mit Methanol (Tabelle 1) durch Zentrifugation bei 17.000 xg für 5 Minuten 3 Mal und schließlich resuspendieren der Kügelchen in 1 ml 100% igem Methanol.
- Wulstoberfläche Thiolierung
- In ein 50 ml Zentrifugenglas 45,6 ml Methanol, 0,4 ml Essigsäure (Tabelle 1), 1,85 ml entionisiertes Wasser, 1,15 ml 3-Mercaptopropyltrimethoxysilan (MPTMS) (Tabelle 1) und 1 ml Kügelchen enthaltenden Suspension in 2.1 hergestellten dann bei RT inkubiert 3 Stunden.
- Nach der Reaktion entfernen Sie alle Reaktionspartner durch einmaliges Waschen mit frischem Methanol und resuspendieren die Perlen in 500 ul Methanol. Teilen Sie gleichmäßig Diese konzentrierte Glasperlensuspension in einen Satz von 20 trockene und saubere Glasgefäße mitSchraubverschlüsse. Abzudampfen das Methanol unter Verwendung einer Strahl aus trockenem Argon und langsam rotieren die Fläschchen, um so eine dünne Schicht von trockenen Perlen auf den Seiten jedes Fläschchen zu machen.
- Legen Sie die Durchstechflaschen mit Perlen in einem vorgewärmten Trockenschrank bei 120 ° C für 5 Minuten und dann herausnehmen und schnell setzen Sie die Kappe (n) lose auf. Legen Sie die Fläschchen in einem Glas Vakuum-Exsikkator mit Trockenmittel in den Boden gefüllt und Vakuum Exsikkator mit einer Vakuumpumpe, bis gekühlt.
- Spülen Sie die gesaugt Exsikkator mit trockenem Argon in den Exsikkator zu normalem Atmosphärendruck zu bringen. Entfernen Sie den Deckel und Exsikkator schnell nachziehen die Kappe (n) auf dem Fläschchen. Verschließen Sie die Fläschchen mit Kunststoffparaffinfilm und speichern Sie sie bei Raumtemperatur in einem trockenen dunklen Aufbewahrungsbox.
- Nach den sofortigen Einsatz, nehmen Sie eine Flasche mit Trockenperlen und einmal mit Phosphatpuffer (Tabellen 1 und 2), re-suspendieren in 50 ul Phosphatpuffer und bei 4 ° C zu waschen. Diese konzentrierte Wulst Vorbereitung wird Bezug genommen werdenals "MPTMS Perlen" in den folgenden Schritten.
Hinweis: Bei sachgerechter Lagerung könnte MPTMS Perlen Funktions für bis zu drei Monate bleiben.
3. Bead Funktionalisierung
- Kovalent Beschichtung Proteine auf Beads
- Nehmen Sie ein bestimmtes Volumen (beispielsweise 2,5 & mgr; l) des Proteins Lager und mit gleichen Volumen von Carbonat / Bicarbonat-Puffer, um Lösung 1 machen mischen.
Hinweis: Das Volumen hängt von der Ausgangskonzentration und der gewünschten endgültigen Ort Dichte des Proteins auf der Oberfläche Kügelchen. - In einem kleinen Becherglas wiegen 2-3 mg MAL-PEG3500-NHS-Linker (Tabelle 1) zu und löse es mit Carbonat / Bicarbonat-Puffer, um eine endgültige Konzentration von 0,231 mg / ml zu erreichen.
- Mischungslösung 1 mit einem gleichen Volumen des in 3.1.2 hergestellten Lösung Linker. Inkubieren der Mischung bei RT für 30 min zur Lösung 2 zu machen.
- In der Zwischenzeit setzen Sie die Linker-Flasche mit gelockerten Kappe in einem Glas Vakuumexsikkator filled mit Trockenmittel in den Boden und Vakuum für 5 Minuten, und füllen Sie den Exsikkator mit Argon. Ziehen Sie den Deckel und nehmen Sie die Flasche aus. Verschließen Sie die Flasche mit Kunststoffparaffinfilm, legen Sie sie in einen Behälter mit Trockenmittel auf der Unterseite und lagern in -20 ° C gefüllt.
Hinweis: die Schritte, die die Verwendung von MAL-PEG3500-NHS Linker beinhalten, einschließlich 3.1.2-3.1.4 (außer für die Inkubation in 3.1.3), müssen so schnell wie möglich durchgeführt werden. - Mischen Sie 5 ul MPTMS Perlen mit Lösung 2 und fügen Phosphatpuffer (Tabelle 1), um ein Endvolumen von 250 ul zu machen.
- Die Perlen Inkubieren über Nacht bei RT, wäscht 3 mal mit Phosphatpuffer und resuspendieren in 100 ul Phosphatpuffer und bei 4 ° C.
- Nehmen Sie ein bestimmtes Volumen (beispielsweise 2,5 & mgr; l) des Proteins Lager und mit gleichen Volumen von Carbonat / Bicarbonat-Puffer, um Lösung 1 machen mischen.
- Vorbereiten Protein / Streptavidin (SA) Beschichtete Beads
- Folgen Protokoll 3.1.1-3.1.4.
- Mischen 5 ul MPTMS Perlen mit Lösung 2 und 5 & mgr; l 4 mg / ml Streptavidin-maleimid (SA-MAL) (Tabelle 1) Lösung und fügen Sie anschließend Phosphatpuffer, um ein Endvolumen von 250 ul zu machen.
- Die Perlen Inkubieren über Nacht bei RT, wäscht 3 mal mit Phosphatpuffer und schließlich resuspendieren in 100 ul Phosphatpuffer und bei 4 ° C.
- Beschichtung Streptavidin auf Glasperlen
- Mischen Sie 5 ul MPTMS Perlen mit 5 ul von 4 mg / ml SA-Lösung und fügen Sie 140 ul Phosphatpuffer.
- Die Perlen Inkubieren über Nacht bei RT, wäscht 3 mal mit Phosphatpuffer und resuspendieren in 50 ul Phosphatpuffer und bei 4 ° C.
- Beschichtung der SA Coated Beads mit einem biotinylierten Protein
- Mischen 5 ul SA-beschichteten Kügelchen mit dem Protein (Volumen je nach der gewünschten Beschichtungsdicke) und fügen Phosphat-Puffer, um das Endvolumen auf 100 ul zu sein.
- Inkubieren der Mischung über Nacht bei 4 ° C oder 3 h bei RT, wäscht 3 mal mit Phosphatpuffer und resuspendieren in 50 ul phosphate puffern und bei 4 ° C.
4. Zellpräparation
Hinweis: Um die Zellen zu reinigen, folgen Standardzelle Reinigungsprotokolle entsprechend der Art der Zellen, in Verwendung, beispielsweise T-Zellen, 27 oder bestimmte Zelllinien 21,29.
- Für fBFP Experimenten sobald die Zellsuspension hergestellt wird, hinzuzufügen Fura2-AM (Tabelle 1), gelöst in DMSO in die Zellsuspension zu einer Endkonzentration von 2 & mgr; M zu erreichen, Inkubation für 30 min bei RT und dann noch einmal zu waschen. Bewahren Sie diese fluoreszierend geladen Zellsuspension in dunklen bis zur Verwendung.
5. Vorbereitung für Mikropipetten und eine Zellkammer
- Vorbereiten Mikropipetten
- Geschnitten lange Kapillare Glasrohre (Tabelle 1) mit einem Glasschneider in kurze Stücke von etwa 3 cm in der Länge. Berg ein Stück auf die Mikropipette Abzieher (Tabelle 1), klicken Sie auf "Pull" aberTonne, so dass die Mitte der Kapillare wird von der Maschine aufgeheizt werden und die Kapillare wird an den beiden Enden gezogen, um zwei Kapillaren mit scharfen Spitzen (raw Mikropipetten) zu machen.
Hinweis: Indem man dem Produktrichtlinie, die gewünschte Morphologie des Ausgangs Pipette 6-8 mm Konus und 0,1-0,5 & mgr; m Spitze. - Montieren Sie eine rohe Pipette auf den Pipettenhalter des Mikroschmiede (Tabelle 1). Wärme, um die Glaskugel auf die Schmiede zu schmelzen. Legen Sie die Spitze des rohen Pipette in der Glaskugel. Abkühlung der Glaskugel und ziehen Sie die raw-Pipette, um es von außen zu brechen und hinterlassen ihre Spitze in der Kugel. Wiederholen Sie diesen Vorgang, bis der gewünschte Spitzenöffnung erhalten wird.
Anmerkung: Beispiele einer Mikropipettenspitze Innendurchmesser: 2,0-2,4 & mgr; m für eine RBC, ~ 1,5 & mgr; m für einen Wulst ~ 2-4 & mgr; m für eine T-Zelle und -7 um eine Hybridomzelle.
- Geschnitten lange Kapillare Glasrohre (Tabelle 1) mit einem Glasschneider in kurze Stücke von etwa 3 cm in der Länge. Berg ein Stück auf die Mikropipette Abzieher (Tabelle 1), klicken Sie auf "Pull" aberTonne, so dass die Mitte der Kapillare wird von der Maschine aufgeheizt werden und die Kapillare wird an den beiden Enden gezogen, um zwei Kapillaren mit scharfen Spitzen (raw Mikropipetten) zu machen.
- Der Aufbau einer Zellkammer
Hinweis: Der Zellraum wird auf der Basis eines Heim-mad gebaute Kammerhalterung, die aus zwei Metallstücken Quadrate (Kupfer / Aluminium) und einem Griff, der sie miteinander verbindet (Figur 1D) besteht. - Schneiden eine 40 mm x 22 mm x 0,2 mm Deckglas mit einem Glasschneider in zwei 40 mm x 11 mm x 0,2 mm große Stücke (Deckglas 1 und 2). Kleber Deck 1 durch Fett auf die Oberseite des Kammerhalterung in der Weise, dass sie die beiden Metallquadraten überbrückt und in ähnlicher Kleber Deckglas 2 an der Unterseite, die eine Parallel-Deckzellraum (1D) bildet.
- Verwende eine Pipette, um 200 ul Versuchspuffer zwischen den beiden Deckplättchen injizieren. Sicherstellen, dass der Puffer wird an beiden Deckgläsern. Vorsichtig drehen und schütteln Sie die Kammer, damit der Puffer berühren beide Enden der Kammer.
- Mineralöl sorgfältig zu injizieren in beide Seiten der Kammer flankieren den experimentellen Pufferzone, wodurch die Puffer von der Open-Air-Abdichtung. Injizieren Suspensionen Sondenkügelchen (zB pMHC-beschichteten Perlen), RBCs und Zielvorgaben(Zum Beispiel T-Zellen) in den oberen, mittleren und unteren Bereich der Pufferzone auf.
6. BFP Experiment

Abbildung 1: fBFP Baugruppe (A) Eine Übersicht Bild des fBFP Hardware-System.. (B) eine schematische Zeichnung des fBFP Hardwaresystem. (C) Das Doppelnockensystem "DC2" (orange), auf die die Hochgeschwindigkeitskamera (blau) und ein Fluoreszenz-Kamera (weiß) montiert wurden. (D) Der Mikroskoptisch, der einen Experimentierraum und drei Mikromanipulationssysteme anpasst. (E und F) Mikroskopische Aufnahmen von BFP-Einstellung in einer Versuchskammer. (E) Mikropipetten Anordnung, die die Sonde Pipette (links), Zielpipette (oben rechts) und Helfer-Pipette (untere rechts). (F) Probe Perlenplatzierung. Eine Sonde Raupe wurde von einem Helfer Pipette manipuliert und zu einem RBC Spitze, um eine Kraft zu bilden Sonde angebracht ist. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
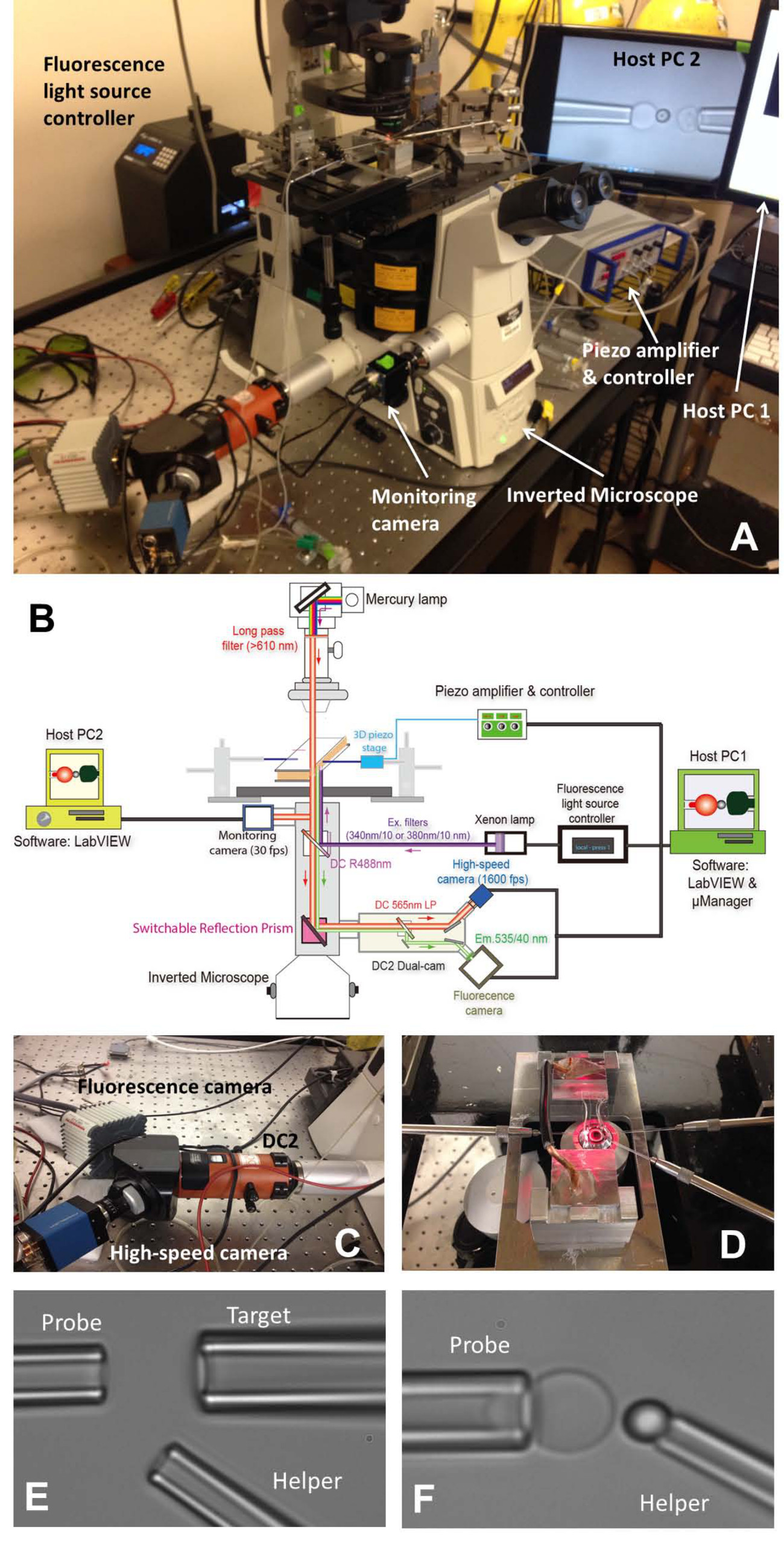{kind=link}
- Einschalten des Mikroskops (Tabelle 1) und der Lichtquelle. Setzen Sie die Kammer auf die Hauptmikroskoptisch (1A, D).
- Installieren Sie alle drei Mikropipetten von BFP (1D. Links: Sonde, um eine RBC, richtig zu greifen: Ziel, um eine Zelle oder ein Kügelchen, unten rechts greifen: Helfer, um eine Perle zu greifen).
- Verwenden Sie ein Mikro-Injektor (Tabelle 1) zu einer Mikropipette mit experimentellen Puffer verfüllen. Nehmen Sie den Pipettenhalter (Tabelle 1) und halten Sie sie zu einem niedrigeren Ort, um Wasser tropft von der Spitze zu ermöglichen. Legen Sie schnell die Mikropipette in die Halterung Spitze und sicherstellen, dass keine Luftblase wird in die Mikropipettewährend des Einsetzens. Ziehen Sie die Halteschraube.
- Hängen Sie jede Pipettenhalter auf dem entsprechenden Mikromanipulator. Schieben Sie die Mikropipette zu der Kammer, so daß ihre Spitzen in die Kammer Pufferbereich. Stellen Sie die Position der Mikropipette und finden sie unter dem Mikroskop Feld.
- Bewegen Sie sich die Kammer Halter Bühne, um die Kolonien aus drei Elementen (die Erythrozyten, die Ziele und die Sonde Perlen) finden one-by-one. Stellen Sie die Position des entsprechenden Mikropipette, indem Sie die Knöpfe der Manipulatoren, um die Spitze der Mikropipette Ansatz einer Zellen / Perlen lassen. Absaugen Zellen / Perlen durch Einstellen des Drucks innerhalb des entsprechenden Mikropipette. Alle drei Mikropipetten werden ihre entsprechenden Elemente zu erfassen.
- Bewegen Sie sich die Kammer Halter Bühne, um einen offenen Raum von den Kolonien der eingespritzte Elemente, bei denen das Experiment durchgeführt werden, zu finden. Schalten Sie das Mikroskop visuellen Modus, um das Bild auf dem Layout-Datei visualisierenuter-Programm auf dem Computerbildschirm. Verschieben Sie alle drei Elemente auf Pipettenspitzen in Sichtfeld des Programms.
- Ausrichten der Sonde Wulst und RBC und vorsichtig manövrieren die Sonde Wulst zum Scheitelpunkt der RBC kurz auftreffen den Wulst auf den RBC und sanft zurückzuziehen. Stellen Sie den Druck der Hilfsmikropipette vorsichtig blasen den Wulst entfernt, so dass es auf die RBC Scheitel (1F) links geklebt werden. Entfernen Sie den Helfer Mikropipette und richten Sie das Ziel und Sonde Wulst (2A).

Abb. 2: BFP System und seine Testzyklus (A) Video-Aufnahme zeigt eine Kraft-Sonde (links) und ein Ziel-T-Zellen (rechts) durch ihre jeweiligen pipettes.The stationären Kraft-Sonde abgesaugt besteht aus einem geschwollenen RBC und ein angeschlossenes Liganden-tragenden Kügelchen. Die Rezeptor-tragendenT-Zelle (Ziel) mit einem Piezotranslator an der Sonde gegenüberliegende ausgerichtet ist. Die ROI wird grün angezeigt. Der Rand-Tracker ist in einem blauen Linie dargestellt. Das Insert zeigt die Ligand (pMHC, Wulstseite) und Rezeptor (TCR, T-Zellen-Seite) Paar an den beiden gegenüberliegenden Oberflächen in dem in orange markierten Bereich. (B) Das Intensitätsprofil der Wulstrand in (A). Die ROI-Bereich in der x-Richtung wird als x-Achse (in Pixelzahl) und der Lichtintensität (in Grauwert) von Binning 30 Pixel entlang der y-Richtung gemittelt aufgetragen. (C) Die Auslenkung der RBC und die Position des Wulstes und dem Target (T-Zelle) in einem Testzyklus der Kraftspann Assay. Die vertikalen und horizontalen gestrichelten Linien zeigen den Nullkraft-Position des RBC Apex und dem zeitlichen Verlauf auf. Die Linie Rand tracker des RBC Verformung wird in blau in jeder Tafel gezeigt. Die gleichen Schritte werden jedoch weniger Haftung Frequenz angenommenAssay und Temperaturschwankungen Assays (das den Schritt der "dissoziieren" fehlt) (welches die Schritte "clamp" und "dissoziieren" fehlt).
- Auf dem Programm, in dem Sichtfeld Fenster mit den Werkzeugen im Programm, um die jeweiligen Radien der Sonde Mikropipette (R p) zu messen, die RBC (R 0), die kreisförmige Kontaktfläche zwischen der RBC und Sonde Wulst (R c) , das die Abschätzung der Federkonstante der RBC (k) durch die folgende Gleichung 17,30 ermöglicht,

wo Δ p der Druck, bei Aspiration Sonde Pipettenspitze.
Hinweis: Aus dem Hookeschen Gesetz, dass die Bindungskraft, F, kann durch das Produkt der Federkonstante und Verschiebung der Sonde Wulst (d), dh F = d quantifiziert werden (Abbildung 2C). - Geben Sie die gewünschte RBC Federkonstante in das Programm (Bitte zu Protokoll Abschnitt 6.6 verwiesen. Die Federkonstante wird in der Regel bei 0,25 oder 0,3 pN / nm für die Kraftklemme Assay und Haftung Frequenz-Assay und 0,1 pN / nm für die thermische Fluktuation Test eingestellt) , die eine erforderliche Ansaugdruck in der Einheit Zentimeter Wasser zurück. Stellen Sie die Höhe des Wassertanks, die an der Sonde Pipette verbindet, bis die erforderliche Absaugen Druck erreicht ist.
- Zeichnen Sie eine horizontale Linie über die RBC Spitze, die eine Kurve in der benachbarten Fenster, das die Helligkeit (Grauwert) von jedem Pixel entlang dieser Linie ergeben wird. Ziehen Sie die Schwellenlinie bei etwa der Hälfte der Tiefe der Kurve (2A, B) sein.
Hinweis: Der Minimalpunkt auf der Helligkeitskurve unterhalb der Schwellenlinie zeigt die Position des Wulstes Grenze, also nur ein lokales Minimum erlaubt. Wenn zwei oder mehrere lokale Minima gibt es derzeit,zeigt das Bild nicht optimal ist (wahrscheinlich aufgrund nicht scharf das Bild, oder eine schlechte Ausrichtung zwischen der Sonde und dem Wulst RBC). - Wählen Sie den gewünschten Modus Experiment: Temperaturschwankungen Assay Haftung Frequenz-Assay oder Klemmkraft-Test. Stellen Sie die Parameter wie gewünscht (zB Aufprallkraft = 15 pN, Laderate = 1.000 pN / s, Kontaktzeit = 1 s, Schließkraft = 20 pN (für Klemmkraft-Test), etc).
- Klicken Sie auf "Start", die das Programm, um die Zielpipette voran zu treiben, das Ziel in und aus dem Kontakt mit der Sonde (siehe Repräsentative Ergebnisse Abschnitt) ermöglicht. Datenerfassung werden parallel, die die Position der Sonde Wulst in Echtzeit aufzeichnet geführt werden. Beenden Sie das Programm, indem Sie auf die Schaltfläche "Stop-Experiment", zu welchem Zeitpunkt ein Fenster öffnet sich, damit das Speichern der erfassten Daten.
7. Fluoreszenz BFP (fBFP) Experiment
- Um die fluore verwendenscence Funktion der BFP Systems den Anregungslichtquelle (Tabelle 1) und dem Fluoreszenzkamera (Tabelle 1), die durch ein separates Programm (Tabelle 1) gesteuert werden. Auf dem Programm, wählen Sie die Parameter für die Fluoreszenz-Bildgebung, einschließlich Verstärkung, Belichtung, Anregungskanäle (in diesem Fall 340 nm und 380 nm-Licht), etc. Folgen alle Zubereitungen in BFP Versuchsprotokolls, einschließlich Ausrichten der Sonde und des Ziels, das zur Visualisierung der Zielzelle Live Fluoreszenzbild von 340 nm oder 380 nm Anregungslicht angeregt ermöglichen.
- Verwenden Sie die Schneidewerkzeug grob Abschnitt der Bereich, in dem die Zelle während der gesamten Aufzeichnungsdauer bleiben.
Hinweis: Durch die Verwendung des Ansatzes kontaktRückzugZyklus wird die Zelle vorwärts und rückwärts bewegt, wiederholt, damit die geschnittene Fläche wesentlich größer ist als die Zelle selbst. - Klicken Sie auf "Record", um die 340 nm eine Zulassungsd 380 nm-Licht, um abwechselnd erregt die intrazelluläre Fluoreszenzfarbstoff (Fura2) und ein Paar von entsprechenden Fluoreszenzbilder abwechselnd etwa einmal in jeder Sekunde aufgezeichnet werden. Gleichzeitig klicken Sie auf "Start" in das Programm, um die BFP Experiment zur Analyse molekularer Interaktionen und der Fluoreszenz-Imaging Experiment, um intrazellulären Calcium-Signalisierung überwachen zu beginnen. Das System wird eine Rohdatendatei für die Rezeptor-Ligand-Bindung (siehe 6A unten) und eine Serie von Fluoreszenzbildern in TIFF-Format für die Calciumsignale erzeugen.
8. Datenanalyse
- BFP Data Analysis
- Datenanalyse für die Haftung Frequenz-Test
- Sequentiell zu inspizieren jedes Zyklus "Kraft-Zeit" -Signal und einfach aufzeichnen, die Zyklen enthalten, einen Haft Veranstaltung und welche nicht, und zusammenfassen, um eine durchschnittliche Haftfrequenz ergeben.
- Sammeln Sie die Bruchkraft jeder Haftung Veranstaltung, dieist der Spitzenwert der linear rampenförmige Kraft, bevor Bindungsbruch. Nach dem Sammeln einer ausreichenden Menge an Bruchkräfte in einem Bereich von Rampenraten, leiten die Bruchkraftverteilung an jeder Rampenrate aus dem das kraftabhängige off-Rate von Rezeptor-Ligand-Dissoziation mit dynamischen Kraftspektroskopie-Analyse 18,31 abgeleitet.
- Datenanalyse für die thermische Fluktuation Test
- Sequentiell inspizieren die Klemmphasensignal eines jeden Zyklus, die wahrscheinlich enthält Fachbindung Assoziation und Dissoziation Veranstaltungen. Verwenden Sie die Spannphase Temperaturschwankungen Ebene (die durchschnittliche Standardabweichung von einer gleitenden Intervall von 70 aufeinanderfolgenden Zeitpunkten der Wulst-Position) in der "Kraft-Zeit" -Signal als Leitfaden, um die Bindung Assoziation und Dissoziation Ereignissen zu unterscheiden, da eine Bindung Bildung entspricht einer Verringerung der Temperaturschwankungen.
- Bestimmen Sie das Intervall vom Zeitpunkt des Bindungsdissoziationsenergie (wenn die thermal Schwankung wechselt in die normale Ebene) zu dem Zeitpunkt des nächsten Bindungsbildung als Wartezeit, und bezeichnen die Dauer der Bindung von ihrer Zuordnung zu Dissoziation Bindung Lebensdauer, die sowohl während der Datenprüfung erfasst werden. Berechnen Sie die durchschnittliche Wartezeit und durchschnittliche Bindungslebenszeit, die jeweils während der Kehrwert des on-Rate und der off-Rate unter Nullkraft 16,30.
- Datenanalyse für Kraftspanntest
- Nehmen Parameter aller Lebens Veranstaltungen, darunter die durchschnittliche Kraft und Rentenlaufzeit mit der Sequenznummer als auch die Startzeit und die Endzeit, die erlauben es, eine kumulierte Lebensdauerkurve ziehen wird (zum Beispiel 6C, gelbe Kurve).
- Sammeln Sie eine ausreichende Menge an Lebensereignisse unter einer Reihe von Kräften. Gruppe sie in unterschiedlichen Kraft Bins, die eine durchschnittliche Lebensdauer in jeder Kraft bin produzieren wird, und zusammen ergeben eine "mittlere Lebensdauer vs. Kraft "Kurve (Abbildung 4).
- Datenanalyse für die Haftung Frequenz-Test
- Calcium Fluoreszenz Imaging Data Analysis
- Einzustellen, die Schwellenintensität, bis die Fluoreszenzbilder zeigen eine deutliche Kontur der Zelle in beiden 340 nm und 380 nm-Kanäle ohne Hintergrundrauschen (5A, B). Dann überprüfen die intrazelluläre Ca 2+ Signalrahmen durch Rahmen mit einer Pseudofarbe, die den Intensitätspegel (6B), die abgeleitet ist von der Intensität Verhältnis von 340 nm / 380 nm, um den "normalisierten Ca 2+ Intensität vs erzeugen . Zeit "-Kurve (6C). Mithilfe der Pseudofarbfluoreszenzbilder, um einen Film, der die Fluoreszenzniveau zweite zeigt von Sekunde zu erzeugen.
Ergebnisse
Die BFP Technik wurde von der Evans Labor 1995 17 voran. Diese picoforce Werkzeug wurde ausgiebig auf Wechselwirkungen von Proteinen auf Oberflächen immobilisiert zu messen, um so zweidimensionale Kinetik von Adhäsionsmolekülen zu analysieren, um die Interaktion mit ihren Liganden 16,19,20 verwendet, 30, um das Molekular Elastizität 21,29 zu messen und zu bestimmen, Protein Konformationsänderungen 21. Für eine fBFP, ein zusätzlicher Satz von Epifluoreszenz bezogene Einr...
Diskussion
Eine erfolgreiche fBFP Experiment bringt einige kritische Überlegungen. Erstens, für die Kraftberechnung zu zuverlässig sein, die Mikropipette, die RBC und die Sonde Perle sollte so nah wie möglich koaxial ausgerichtet werden. Die Projektion der RBC in der Pipette über eine Sonde Pipetten Durchmesser sein, so dass die Reibung zwischen dem RBC und dem Pipetten vernachlässigbar ist. Für eine typische menschliche RBC ist die optimale Pipetten Durchmesser von 2,0-2,4 um, die eine beste Anpassung der Gleichung 1 ...
Offenlegungen
The authors have nothing to disclose.
Danksagungen
Research related to this paper and the development of the fBFP technology in the Zhu lab were supported by NIH grants AI044902, AI077343, AI038282, HL093723, HL091020, GM096187, and TW008753. We thank Evan Evans for inventing this empowering experimental tool, and members of the Evans lab, Andrew Leung, Koji Kinoshita, Wesley Wong, and Ken Halvorsen, for helping us to build the BFP. We also thank other Zhu lab members, Fang Kong, Chenghao Ge and Kaitao Li, for their helps in the instrumentation development.
Materialien
| Name | Company | Catalog Number | Comments |
| Sodium Phosphate Monobasic Monohydrate (NaH2PO4 • H2O) | Sigma-Aldrich | S9638 | Phosphate buffer preparation |
| Anhy. Sodium Phosphate Dibasic (Na2HPO4) | Sigma-Aldrich | S7907 | Phosphate buffer preparation |
| Sodium Carbonate (Na2CO3) | Sigma-Aldrich | S2127 | Carbonate/bicarbonate buffer preparation |
| Sodium Bicarbonate (NaHCO3) | Sigma-Aldrich | S5761 | Carbonate/bicarbonate buffer preparation |
| Sodium chloride (NaCl) | Sigma-Aldrich | S7653 | N2-5% buffer preparation |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 | N2-5% buffer preparation |
| Potassium phosphate monobasic (KH2PO4) | Sigma-Aldrich | P5655 | N2-5% buffer preparation |
| Sucrose | Sigma-Aldrich | S0389 | N2-5% buffer preparation |
| MAL-PEG3500-NHS | JenKem | A5002-1 | Bead functionalization |
| Biotin-PEG3500-NHS | JenKem | A5026-1 | RBC biotinylation |
| Nystatin | Sigma-Aldrich | N6261 | RBC osmolarity adjustment |
| Ammonium Hydroxide (NH4OH) | Sigma-Aldrich | A-6899 | Glass bead silanization |
| Methanol | BDH | 67-56-1 | Glass bead silanization |
| 30% Hydrogen Peroxide (H2O2) | J. T. Barker | Jan-86 | Glass bead silanization |
| Acetic Acid (Glacial) | Sigma-Aldrich | ARK2183 | Glass bead silanization |
| 3-Mercaptopropyltrimethoxysilane (MPTMS) | Uct Specialties, llc | 4420-74-0 | Glass bead functionalization |
| Borosilicate Glass beads | Distrilab Particle Technology | 9002 | Glass bead functionalization |
| Streptavidin−Maleimide | Sigma-Aldrich | S9415 | Glass bead functionalization |
| BSA | Sigma-Aldrich | A0336 | Ligand functionalizing |
| Fura2-AM | Life Technologies | F-1201 | Intracellular calcium fluorescence dye loading |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D2650 | Intracellular calcium fluorescence dye loading |
| Quantibrite PE Beads | BD Biosciences | 340495 | Density quantification |
| Flow Cytometer | BD Biosciences | BD LSR II | Density quantification |
| Capillary Tube 0.7-1.0 mm x 30 inches | Kimble Chase | 46485-1 | Micropipette making |
| Flaming/Brown Micropipette Puller | sutter instrument | P-97 | Micropipette making |
| Pipette microforce | Narishige | MF-900 | Micropipette making |
| Mineral Oil | Fisher Scientific | BP2629-1 | Chamber assembly |
| Microscope Cover Glass | Fisher Scientific | 12-544-G | Chamber assembly |
| Micro-injector | World Precision Instruments | MF34G-5 | Chamber assembly |
| 1 ml syringe | BD | 309602 | chamber assembly |
| Micropipette holder | Narishige | HI-7 | Chamber assembly |
| Home-designed mechanical parts and adaptors fabrications using CNC machining. | Biophysics Instrument | All parts are customized according to the CAD designs. | BFP system |
| Microscope (TiE inverted) | Nikon | MEA53100 | BFP system |
| Objective CFI Plan Fluor 40x (NA 0.75, WD 0.72 mm, Spg) | Nikon | MRH00401 | BFP system |
| Camera, GE680, 640 x 480, GigE, 1/3" CCD, mono | Graftek Imaging | 02-2020C | BFP system |
| Prosilica GC1290 - ICX445, 1/3", C-Mount, 1280 x 960, Mono., CCD, 12 Bit ADC | Graftek Imaging | 02-2185A | BFP system |
| Manual submicron probehead with high resolution remote control | Karl Suss | PH400 | BFP system |
| Anti-vibration table (5’ x 3’) | TMC | 77049089 | BFP system |
| 3D manual translational stage | Newport | 462-XYZ-M | |
| SolidWorks 3D CAD software | SOLIDWORKS Corp. | Version 2012 SP5 | BFP system |
| LabVIEW software | National Instruments | Version 2009 | BFP system, BFP program |
| 3D piezo translational stage | Physik Instrumente | M-105.3P | BFP system |
| Linear piezo accuator | Physik Instrumente | P-753.1CD | BFP system |
| Micromanager software | Version 1.4 | fBFP system, fluorescence imaging program | |
| Dual Cam (DC-2) | Photometrics | 77054724 | fBFP system |
| Dual Cam emission filter (T565LPXR) | Photometrics | 77054725 | fBFP system |
| Fluorescence Camera | Hamamatsu | ORCA-R2 C10600-10B | fBFP system |
| Plastic paraffin film (Parafilm) | Bemis Company, Inc | PM996 | bottle sealing |
| Carbonate/bicarbonate buffer (pH 8.5) | 8.4 g/L sodium carbonate (Na2CO3), 10.6 g/L sodium bicarbonate (NaHCO3) | ||
| Phosphate buffer (pH 6.5-6.8) | 27.6 g/L NaPhosphate monobasic (NaH2PO4 • H2O), 28.4 g/L Anhy. NaPhosphate dibasic (Na2HPO4) | ||
| N2-5% buffer (pH 7.2) | 20.77 g/L potassium chloride (KCl), 2.38 g/L sodium chloride (NaCl), 0.13 g/L potassium phosphate monobasic (KH2PO4), 0.71 g/L anhy. sodium phosphate dibasic (Na2HPO4), 9.70 g/L sucrose |
Referenzen
- Aplin, A. E., Howe, A., Alahari, S. K., Juliano, R. L. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacological reviews. 50, 197-263 (1998).
- Davis, M. M., Bjorkman, P. J. T-cell antigen receptor genes and T-cell recognition. Nature. 334, 395-402 (1988).
- Dado, D., Sagi, M., Levenberg, S., Zemel, A. Mechanical control of stem cell differentiation. Regenerative medicine. 7, 101-116 (2012).
- Edwards, L. J., Zarnitsyna, V. I., Hood, J. D., Evavold, B. D., Zhu, C. Insights into T cell recognition of antigen: significance of two-dimensional kinetic parameters. Frontiers in immunology. 3, 86 (2012).
- Zhu, C., Jiang, N., Huang, J., Zarnitsyna, V. I., Evavold, B. D. Insights from in situ analysis of TCR-pMHC recognition: response of an interaction network. Immunological reviews. 251, 49-64 (2013).
- Huang, J., Meyer, C., Zhu, C. T. T cell antigen recognition at the cell membrane. Molecular immunology. 52, 155-164 (2012).
- Zarnitsyna, V., Zhu, C. T. T cell triggering: insights from 2D kinetics analysis of molecular interactions. Physical biology. 9, 045005 (2012).
- Binnig, G., Quate, C. F., Gerber, C. Atomic Force Microscope. Physical Review Letters. 56, 930-933 (1986).
- Marshall, B. T., et al. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 423, 190-193 (2003).
- Kong, F., Garcia, A. J., Mould, A. P., Humphries, M. J., Zhu, C. Demonstration of catch bonds between an integrin and its ligand. The Journal of cell biology. 185, 1275-1284 (2009).
- Yago, T., et al. Catch bonds govern adhesion through L-selectin at threshold shear. The Journal of cell biology. 166, 913-923 (2004).
- Yago, T., et al. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. The Journal of clinical investigation. 118, 3195-3207 (2008).
- Chesla, S. E., Selvaraj, P., Zhu, C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophysical journal. 75, 1553-1572 (1998).
- Huang, J., et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature. 464, 932-936 (2010).
- Heinrich, V., Wong, W. P., Halvorsen, K., Evans, E. Imaging biomolecular interactions by fast three-dimensional tracking of laser-confined carrier particles. Langmuir : the ACS journal of surfaces and colloids. 24, 1194-1203 (2008).
- Chen, W., Evans, E. A., McEver, R. P., Zhu, C. Monitoring receptor-ligand interactions between surfaces by thermal fluctuations. Biophysical journal. 94, 694-701 (2008).
- Evans, E., Ritchie, K., Merkel, R. Sensitive force technique to probe molecular adhesion and structural linkages at biological interfaces. Biophysical. 68, 2580-2587 (1995).
- Evans, E., Leung, A., Heinrich, V., Zhu, C. Mechanical switching and coupling between two dissociation pathways in a P-selectin adhesion bond. Proceedings of the National Academy of Sciences of the United States of America. 101, 11281-11286 (2004).
- Ju, L., Dong, J. -. f., Cruz, M. A., Zhu, C. The N-terminal Flanking Region of the A1 Domain Regulates the Force-dependent Binding of von Willebrand Factor to Platelet Glycoprotein Ib. Journal of Biological Chemistry. 288, (2013).
- Chen, W., Lou, J., Zhu, C. Forcing switch from short- to intermediate- and long-lived states of the alphaA domain generates LFA-1/ICAM-1 catch bonds. The Journal of biological chemistry. 285, 35967-35978 (2010).
- Chen, W., Lou, J., Evans, E. A., Zhu, C. Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. The Journal of cell biology. 199, 497-512 (2012).
- Judokusumo, E., Tabdanov, E., Kumari, S., Dustin, M. L., Kam, L. C. Mechanosensing in T lymphocyte activation. Biophysical journal. 102, L5-L7 (2012).
- Bashour, K. T., et al. CD28 and CD3 have complementary roles in T-cell traction forces. Proceedings of the National Academy of Sciences of the United States of America. 111, 2241-2246 (2014).
- Nesbitt, W. S., et al. Distinct glycoprotein Ib/V/IX and integrin alpha IIbbeta 3-dependent calcium signals cooperatively regulate platelet adhesion under flow. The Journal of biological chemistry. 277, 2965-2972 (2002).
- Mazzucato, M., Pradella, P., Cozzi, M. R., De Marco, L., Ruggeri, Z. M. Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Ibalpha mechanoreceptor. Blood. 100, 2793-2800 (2002).
- Lefort, C. T., Ley, K. Neutrophil arrest by LFA-1 activation. Frontiers in immunology. 3, 157 (2012).
- Liu, B., Chen, W., Evavold, B. D., Zhu, C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell. 157, 357-368 (2014).
- Lou, J., et al. Flow-enhanced adhesion regulated by a selectin interdomain hinge. The Journal of cell biology. 174, 1107-1117 (2006).
- Fiore, V. F., Ju, L., Chen, Y., Zhu, C., Barker, T. H. Dynamic catch of a Thy-1-alpha5beta1+syndecan-4 trimolecular complex. Nature communications. 5, 4886 (2014).
- Chen, W., Zarnitsyna, V. I., Sarangapani, K. K., Huang, J., Zhu, C. Measuring Receptor-Ligand Binding Kinetics on Cell Surfaces: From Adhesion Frequency to Thermal Fluctuation Methods. Cellular and molecular bioengineering. 1, 276-288 (2008).
- Marshall, B. T., Sarangapani, K. K., Lou, J., McEver, R. P., Zhu, C. Force history dependence of receptor-ligand dissociation. Biophysical. 88, 1458-1466 (2005).
- Xiang, X., et al. Structural basis and kinetics of force-induced conformational changes of an alphaA domain-containing integrin. PloS one. 6, e27946 (2011).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten