Method Article
Eine einfache Fraktionierte Extraktionsmethode für die umfassende Analyse von Metaboliten, Lipiden und Proteinen aus einer einzigen Probe
In diesem Artikel
Zusammenfassung
Ein Protokoll zur umfassenden Extraktion von Lipiden, Metaboliten und Proteinen aus biologischen Geweben mit einer Probe wird vorgestellt.
Zusammenfassung
Das Verständnis komplexer biologischer Systeme erfordert die Messung, Analyse und Integration mehrerer Verbindungsklassen der lebenden Zelle, die in der Regel durch transkriptomische, proteomische, metabolomische und lipidomische Messungen bestimmt wird. In diesem Protokoll stellen wir ein einfaches Verfahren zur reproduzierbaren Extraktion von Metaboliten, Lipiden und Proteinen aus biologischen Geweben mit einem einzigen Aliquot pro Probe vor. Das Extraktionsverfahren basiert auf einem Methyl- tert -butylether: Methanol: Wassersystem für flüssige: flüssige Partitionierung von hydrophoben und polaren Metaboliten in zwei unmischbare Phasen zusammen mit dem Ausfällen von Proteinen und anderen Makromolekülen als festem Pellet. Diese Methode liefert daher drei verschiedene Fraktionen der spezifischen molekularen Zusammensetzung, die vollständig mit den üblichen Hochleistungs-Omics-Technologien wie Flüssigchromatographie (LC) oder Gaschromatographie (GC), die an Massenspektrometer gekoppelt sind, kompatibel sind. Obwohl die Methode init warFür die Analyse verschiedener Pflanzengewebeproben entwickelt, hat es sich als voll kompatibel für die Extraktion und Analyse von biologischen Proben aus so vielfältigen Systemen wie Algen, Insekten und Säugetiergeweben und Zellkulturen erwiesen.
Einleitung
Die Systembiologie, die in der Mitte des letzten Jahrhunderts 1 entstand und durch die umfangreiche Analyse der genomischen und transkriptomischen Datensätze 2 , 3 vorangebracht wurde, hat sich zu einem neuen und unentbehrlichen Ansatz für die Analyse komplexer biologischer Systeme entwickelt 4 , 5 . Das Hauptziel der Systembiologie ist es, die Komponenteninteraktionen und -abhängigkeiten in biologischen Systemen zu entschlüsseln und die Verbindung zwischen Genotypen, deren Realisierung, molekulare Transformationen und daraus resultierende Phänotypen zu überbrücken. Dementsprechend ist die Integration von umfassenden Datensätzen, die durch die vielfältigen analytischen Ansätze, nämlich Genomik, Transkriptomik, Metabolomik, Lipidomik und Proteomik und deren Berechnungsanalyse, hergestellt werden, eine Voraussetzung für die Beschreibung und das Verständnis komplexer biologischer Systeme.
Bas Über die enorme chemische Vielfalt und Komplexität der biologischen Komponenten in jedem lebenden System beruht die Produktion von großen und umfassenden "Omics" -Datensätzen stark auf die Qualität der angewandten Extraktionsmethode 9 . Neben der Qualität der Extraktionsmethode ist die Wirtschaftlichkeit der Methode wichtig; Dies bedeutet, dass es wünschenswert wäre, so viel molekulare Informationen von so wenig Stichprobeneingabe wie möglich zu erhalten. Oft können die Probenmengen begrenzt werden, und daher ist es höchst wünschenswert, eine Extraktionsmethode zu verwenden, die so viele Molekülklassen aus einer einzigen Extraktion einer gegebenen Probe ableiten kann. Dies bedeutet, dass anstelle der Verwendung mehrerer spezialisierter Extraktionsmethoden für die Extraktion verschiedener Compoundklassen aus verschiedenen Probenaliquoten derselben Probe ein sequentielles Extraktionsverfahren angewendet wird, das die molekularen Bestandteile eines einzelnen Aliquots in verschiedene molekulare Fraktionen fraktioniert.
_content "> Die übliche Methode, die für diese fraktionierten Extraktionsverfahren angewendet wird, basiert auf der Zwei-Phasen-Lipid-Extraktionsmethode von Folch et al. , Die 1957 entwickelt wurde. 10. Diese Methode basiert auf einer Chloroform: Methanol / Wasser-Partitionierung von polaren und hydrophoben Metaboliten und war Beabsichtigt, die Proben für eine qualitativ hochwertige Lipidanalyse aufzuräumen und zu dekomplexieren. Mit der Entwicklung der Multi-Omics-Systembiologie wurde die Folch-Methode weiter schrittweise verbessert, indem sie für die Probenpartitionierung von Proteinen und polaren Metaboliten und Lipiden verwendet wurde Gas- und Flüssigchromatographie-basierte Metabolomik und Lipidomik von polaren und hydrophoben Verbindungen, zusätzlich zu Flüssigchromatographie-basierten Proteomik 11 , 12 , 13 , 14. Leider sind alle diese Methoden auf eine Chloroform-basierte Extraktionsmethode angewiesen, die nicht nur Führt zu den unerwünschten foRmation des Proteinpellets als Zwischenphase zwischen der Polar- und der Lipidphase, die aber auch ein unerwünschtes Lösungsmittel aus der Perspektive der Grünchemie 15 , 16 ist . Jedoch überwindet das Lösungsmittel Methyl- tert -butylether (MTBE) beide dieser vorgenannten Probleme und ist ein geeigneter Ersatz für Chloroform. Basierend auf diesen Anforderungen haben wir uns entschlossen, eine MTBE: methanol: wasserbasierte Extraktionsmethode zu etablieren, die alle vorgenannten Spezifikationen erfüllt und somit als idealer Ausgangspunkt für eine umfassende Multimedienanalyse dient 16 .Dieses Protokoll führt den Anwender Schritt für Schritt durch den einfachen, schnellen und reproduzierbaren Arbeitsablauf der Probenvorbereitung, einschließlich der Fehlersuche bei häufig beobachteten Problemen. Weiterhin werden wir kurz beispielhafte analytische Daten aus der Ultra-Performance-Flüssigkeitschromatographie-Massenspektrometrie (UPLC-MS) -bas vorstellenEd Lipidomik, Metabolomik und Proteomik Profilierung von Pflanzengewebe Proben. Obwohl die angegebenen Beispiele aus 50 mg einer Arabidopsis-Thaliana-Blattgewebeprobe stammen, wurde dieses Protokoll für mehrere andere biologische Proben und Gewebe verwendet, darunter Algen 17 , 18 , Insekten 19 und Säugetierzellen, Organe und Gewebe 20 , 21 , 22 Der Umfang des präsentierten Extraktionsprotokolls soll eine klare und detaillierte Beschreibung der Vorbehandlung der Probenahme und des Extraktionsverfahrens selbst ermöglichen. Obwohl wir drei kurze Beispiele für die analytische Anwendung vorstellen, können detaillierte Informationen über die vor- und nachanalytische Datenverarbeitung aus unseren früheren Publikationen 16 , 23 , 24 , 25 , 26 .
Protokoll
Achtung : Methanol (MeOH) und Methyl- tert -butylether (MTBE), die während der Extraktion verwendet werden, sind entflammbar und können bei längerer Exposition und / oder Kontakt-, Atem-, Augen- oder Hautreizung auftreten. Bitte behandeln Sie diese sorgfältig nur in einer Dunstabzugshaube und verwenden Sie bei der Extraktion die entsprechenden Sicherheitsmaßnahmen (Laborkittel, Schutzbrille, Handschuhe usw. ). Flüssiger Stickstoff und Trockeneis, die in mehreren Schritten dieses Protokolls verwendet werden, können schwere Verbrennungen durch längeren Hautkontakt verursachen. Bitte behandeln Sie sie sorgfältig, indem Sie Schutzhandschuhe und Gläser tragen. Benutzer können verschiedene Chemikalien, Reagenzien oder interne Standards für die Probenanalyse verwenden, von denen einige giftig sein können. Bitte prüfen Sie die relevanten Sicherheitsdatenblätter für alle verwendeten Materialien.
1. Sammlung und Ernte von biologischen Proben
- Bereiten Sie markierte Ernterohre vor.
HINWEIS: Hier ernten Sie biologische Proben in etikettierten, 2 mL, rund-unten, sichere locK Mikrozentrifugenröhrchen mit zwei 5 mm Durchmesser, Metallkugeln für den Gewebehomogenisator. - Einen gefüllten flüssigen Stickstoff-Dewar vorbereiten.
- Ernte die biologische Probe und schnappt das Gewebe in flüssigem Stickstoff ein. Führen Sie diesen Schritt so schnell wie möglich innerhalb von wenigen Sekunden durch, um metabolische Veränderungen zu vermeiden, die durch Verwundung verursacht werden.
Anmerkung: Für Demonstrationszwecke verwenden Sie Rosettenblätter von 30 Tage alten Wildtyp Arabidopsis thaliana (Col-0), die auf dem Boden unter langen Tagesbedingungen gewachsen sind. - Halten Sie die geernteten Proben auf Trockeneis für kurzfristige Pausen oder lagern Sie sie bei -80 ° C für längere Zeiträume.
2. Schleifen und Gewebeunterbrechung
- Vorbereitung der Röhrchenhalter des Gewebehomogenisators in flüssigem Stickstoff für mindestens 10 min. Wenn ein Gewebehomogenisator nicht vorhanden ist, verwenden Sie saubere und vorgekühlte Mörser und Stößel.
- Nehmen Sie die Proben aus dem flüssigen Stickstoff, Trockeneis oder -80 ° C Gefrierschrank und legen Sie sie in den vorgekühltenRohrhalter.
- Schnell die Röhrchenhalter in den Gewebehomogenisator stellen.
- Das biologische Material zu einem feinen und homogenen Pulver schleifen. Verwenden Sie 20 Hz für 1 min für Blätter.
Hinweis; Die Homogenisierungszeit und -geschwindigkeit kann je nach Gewebe variiert werden, stellen Sie sicher, dass die Probe zu einem feinen Pulver homogenisiert wird und dass dieses Pulver bei jedem Schritt der Homogenisierung eingefroren wird. - Nehmen Sie die biologischen Proben aus den Röhrenhaltern und halten Sie sie bis zur weiteren Extraktion gefroren.
3. Wiegen von Geweben
- Verwenden Sie eine Analysenwaage mit ausreichender Genauigkeit für die benötigten Probenmengen.
- Bereiten Sie ein beschriftetes 2-mL-Rundboden-Safe-Lock-Mikrozentrifugenröhrchen vor.
- Die Röhrchen und Spatel in flüssigem Stickstoff vorkühlen.
- Aliquotieren Sie die erforderliche Menge an Gewebepulver in das 2 ml-Sicherheitsschloss-Mikrozentrifugenröhrchen.
Achtung: Vermeiden Sie das Auftauchen des Pflanzenmaterials durch Minimierung der Zeit, die auf w genommen wirdAcht die Proben. - Geben Sie die Aliquotten sofort nach dem Wiegen auf flüssigen Stickstoff zurück.
- Aufzeichnung für jede Probe das genaue Gewicht. Verwenden Sie 10-50 mg ± 10% für die meisten pflanzlichen Gewebe.
- Lagern Sie die aliquotierten Proben bei -80 ° C bis zur weiteren Extraktion.
4. Reagenz-Setup
- Verwenden Sie eine Extraktionsmischung aus Methyl- tert -butylether (MTBE) / Methanol (MeOH).
- Zur Herstellung von 100 ml Extraktionslösungsmittel werden 75 ml MTBE auf 25 ml MeOH gegeben, um eine Mischung aus MTBE: MeOH (3: 1, vol / vol) herzustellen.
- Hinzufügen von internen Standards für die Nachanalyse Normalisierung nach analytischen Bedürfnissen. In der Regel werden 50 μl 1,2-Diheptadecanoyl- sn- glycero-3-phosphocholin (1 mg / ml in Chloroform) als interner Standard für die UPLC-MS-basierte Lipidanalyse zugegeben, während 50 μl 13 C Sorbit ( 1 mg / ml in Wasser) als interne Standards für die GC-MS-basierte Analyse der primärenMetaboliten Interne Standards für die UPLC-MS-basierte Metabolitanalyse sind 50 μl Corticosteron (1 mg / ml in Methanol) und 25 μl Ampicillin (1 mg / ml in Methanol).
- Übertragen Sie das Lösungsmittel auf eine saubere Glasflasche, die mit MTBE: MeOH-Mischung gespült wurde.
- Die Extraktionsmischung bis zu 1 Woche bei 4 ° C aufbewahren.
HINWEIS: Die Extraktionsmischung längere Zeit nicht aufbewahren, um reproduzierbare Ergebnisse zu erhalten.
- Um die Phasentrennung zu induzieren, verwenden Sie Wasser (H 2 O) / Methanol (MeOH).
- Zur Herstellung von 100 ml H & sub2 ; O: MeOH werden 75 ml H & sub2 ; O zu 25 ml MeOH gegeben, um H & sub2 ; O: MeOH (3: 1, vol / vol) zu ergeben.
- Übertragen Sie das Lösungsmittel auf eine saubere Glasflasche, die mit H 2 O: MeOH-Mischung gespült wurde. Dieses Lösungsmittel kann mehrere Wochen bei Raumtemperatur gelagert werden.
5. Extraktion von Proben
- Vorbereitung der Extraktion m(MTBE: MeOH, 3: 1, vol / vol) bis -20 ° C mit einem Flüssigkeitskühlsystem oder einem Gefrierschrank von -20 ° C.
- Nehmen Sie die aliquotierten Proben einzeln heraus und geben Sie 1 ml des vorgekühlten Extraktionsgemisches zu jedem Probenröhrchen hinzu. Achtung: Führen Sie diesen Schritt schnell durch die niedrige Viskosität von MTBE durch.
- Mischen Sie sofort auf einen Vortex-Mischer, bis das Gewebe innerhalb der Extraktionsmischung gut homogenisiert ist.
HINWEIS: Dieser Schritt ist sehr wichtig, da es erforderlich ist, die Proteine auszufällen und ihre enzymatischen Aktivitäten zu inaktivieren. - Inkubieren Sie alle Proben auf einem Orbitalschüttler bei 100 U / min für 45 min bei 4 ° C.
- Sonden Sie die Proben für 15 min in einem eisgekühlten Beschallungsbad.
6. Fraktionierung durch Phasentrennung
- Füge 650 μl H 2 O: MeOH (3: 1, vol / vol) zu jedem Probenröhrchen hinzu.
- Mischen Sie gut durch Vortexen für 1 min.
- Die Proben mit einer Geschwindigkeit von 20.000 x g für 5 min bei 4 ° C zentrifugieren.
HINWEIS: Nach diesem Schritt gibt es zwei unmischbare flüssige Phasen mit einem festen Pellet im Boden des Röhrchens.
Achtung: Die Rohre sorgfältig behandeln, um das Mischen der beiden flüssigen Phasen zu vermeiden und das ausgefällte Pellet zu vermeiden.
7. Aliquotierung von polaren und hydrophoben Fraktionen
- Übertragen Sie 500 μl des Lösungsmittels aus der oberen, lipidhaltigen Phase in ein markiertes 1,5 mL Mikrozentrifugenröhrchen.
HINWEIS: Die aliquotierten Lipidproben können direkt zur sofortigen UPLC-MS-Analyse konzentriert werden (Schritt 8.1) oder mehrere Wochen bei -80 ° C gelagert werden. - Sobald die 500 μl aus der Probe entfernt wurden, entfernen Sie die verbleibende Lipidphase mit einer 200 μl Pipette.
- Übertragen Sie 400 μl des Lösungsmittels aus der unteren Phase (polare und halbpolare Metaboliten) in ein markiertes 1,5 mL Mikrozentrifugenröhrchen. Die aliquotierten Polarproben können direkt zur sofortigen UPLC-MS-Analyse konzentriert werden (Schritt 8.2) oder gelagert werdenR mehrere Wochen bei -80 ° C.
- Nehmen Sie ein zusätzliches Aliquot von 200 μl, um eine zusätzliche Analyse durchzuführen, z. B. Gaschromatographie-basierte Metabolitanalyse, wie es kostbar beschrieben ist.
- Entfernen Sie den Rest der wässrigen Phase durch Abpumpen des überschüssigen Volumens.
- Waschen des erhaltenen Proteins, Stärke, Zellwandpellets mit 500 μl Methanol durch gründliches Vortexen für 1 min.
- Die Proben mit einer Geschwindigkeit von 10.000 x g für 5 min bei 4 ° C zentrifugieren.
- Führen Sie die Proteinextraktion und den Verdau (Schritt 11) oder die Stärke / Zellwandanalyse wie zuvor beschrieben durch.
HINWEIS: Wenn nicht sofort verwendet, können diese Pellets für mehrere Wochen bei -80 ° C gelagert werden.
8. Konzentration und Lagerung von Fraktionen
- Verdampfe das Lösungsmittel aus Lipidproben (aus Schritt 7.1) entweder in einem Vakuumkonzentrator ohne Erhitzen (für 1-2 Stunden) oder vorzugsweise ein NitrDurchflussverdampfer zur Vermeidung oxidativer Modifikationen der Lipide.
HINWEIS: Getrocknete Proben sollten sofort analysiert werden. Zur Aufbewahrung von Proben in MTBE-Lösung, idealerweise in Glasfläschchen (Schritt 7.1). - Verdampfe das Lösungsmittel aus den wässrigen Proben (ab Schritt 7.3 oder 7.4) über Nacht in einem Vakuumkonzentrator ohne Erwärmung. HINWEIS: Die getrockneten Proben können für mehrere Wochen bei -80 ° C vor der Analyse gelagert werden.
9. Analyse der Lipide mit UPLC-MS 24
- Die getrockneten Lipidfraktionen (aus Schritt 8.1) in 400 μl Acetonitril: 2-Propanol (7: 3, vol / vol) erneut suspendieren.
- Übertragen Sie genügend Flüssigkeit in Glasfläschchen und Kappe fest.
- Setzen Sie die Glasfläschchen in einen gekühlten Autosampler (4 ° C).
- 2 μl pro Probe injizieren und die Lipide auf einer Umkehrphase (RP) C 8 -Säule, die bei 60 ° C gehalten wird, unter Verwendung eines UPLC-Systems, das mit einer Flussrate von 400 & mgr; l / min läuft, trennen.
- Benutze das HandyPhasen, die in Tabelle 1 für die chromatographische Trennung beschrieben sind.
- Erfassen Sie die Massenspektren im positiven und negativen Ionisationsmodus mit einem geeigneten MS-Instrument, das den Massenbereich zwischen 150 und 1.500 m / z abdeckt.
10. Analyse von polaren und semipolaren Metaboliten mit UPLC-MS 25 .
- Die polare Phase (ab Schritt 8.2) in 200 μl UPLC-Klasse Methanol: Wasser (1: 1, vol / vol) erneut suspendieren.
- Übertragen Sie genügend Flüssigkeit in Glasfläschchen und Kappe fest.
- Setzen Sie die Glasfläschchen in einen gekühlten Autosampler (4 ° C).
- 2 & mgr; l von jeder Probe injizieren und die Metaboliten auf einer RP C 18 -Säule, die bei 40 ° C gehalten wird, unter Verwendung eines UPLC-Systems, das mit einer Flussrate von 400 & mgr; l / min läuft, trennen.
- Verwenden Sie die mobilen Phasen für die chromatographische Trennung mit den in Tabelle 2 angegebenen Parametern.
- Erwerben Sie vollständige Scan-Massenspektren im positiven und negativen IonisationsmodusEin geeignetes Massenspektrometer mit einem Massenbereich zwischen 50 und 1.500 m / z.
11. Protein Extraktion, Verdauung und Analyse 16
- Das gewaschene Protein / Stärke / Zellwandpellet (ab Schritt 7.8) in 200 μl des Proteinextraktionspuffers der Wahl erneut suspendieren. Anmerkung: Wir verwenden Harnstoff / Thioharnstoffpuffer (5 M Harnstoff, 2 mM Thioharnstoff, 15 mM DTT, 2% CHAPS und Protease und Phosphataseinhibitoren) 27 .
- Sonden Sie die Proben für 10 min in einem eisgekühlten Schallbad.
- Inkubieren Sie die Proben für 30 min auf einem Orbitalschüttler (100 U / min) bei Raumtemperatur.
- Zentrifugieren der gelösten Proteine bei 10.000 x g für 5 min.
- Sammle den Proteinüberstand in einem neuen Röhrchen.
- Bestimmen Sie die Proteinkonzentration aus dem gesammelten Überstand 28 .
- 50 μg Protein in-Lösung mit einem Protokoll der Wahl verdauen. Verwenden Sie typischerweise die Trypsin / Lys-C-MischungNg zur Bedienungsanleitung.
- Nach der Verdauung die Entsalzung der Peptide vor der Massenspektrometrie unter Verwendung von C 18 -Stufentests durchführen und die verdauten Peptide 29 eluieren.
- Konzentrieren Sie die Proben in nahezu Trockenheit in einem Vakuumkonzentrator ohne Erwärmung.
- Die Proben in einem geeigneten Beladungspuffer (z. B. 5% Acetonitril, 0,5% Ameisensäure) erneut suspendieren und die Peptidmischungen durch LC-MS / MS unter Verwendung eines hochauflösenden Massenspektrometers, das mit einem Nano-LC-System verbunden ist, analysieren.
HINWEIS: In dem beispielhaften Proteomik-Datensatz, der in diesem Protokoll vorgestellt wurde, verwendeten wir einen Gradienten, wie in Tabelle 3 beschrieben. - Stellen Sie das Massenspektrometer mit einer Top-15-Strategie ein, bei der ein voller Scan (FS) bis zu 15 datenabhängige MS / MS-Scans verfolgt wurde, auf folgende Parameter: Der FS lag im Massenbereich von 200 - 2.000 m / z an Eine Auflösung von 70.000 mit einem Zielwert von 3x10 6 Ionen. Erhalten Sie die datenabhängigen MS / MS-Scans von höheremKlinische Dissoziation (HCD). Setzen Sie die Zielwerte für die MS / MS auf 1e 5 Ionen mit einer maximalen Ionenfüllzeit von 50 ms, einem Isolationsfenster von 4,0 m / z, einer normalisierten Kollisionsenergie (NCE) von 30% und einem Unterfüllungsverhältnis von 1%. Messen Sie die MS / MS-Ionen mit einer Auflösung von 17.500 und der dynamische Ausschluss wurde auf 60 s gesetzt.
Ergebnisse
Umfassende multimediale Datensätze sind für das Verständnis komplexer biologischer Systeme von unschätzbarem Wert. Die Strategie für ein erfolgreiches biologisches Experiment beginnt in der Regel von einem sinnvollen experimentellen Design, Experimentaufbau und Performance, gefolgt von Probenahme, Extraktion, analytische Datenerfassung, Rohdatenverarbeitung, statistische Datenanalyse, Identifizierung relevanter Metaboliten und biologischer Dateninterpretation Einschließlich Wegweiser und Visualisierung (Abbildung 1 ).
In dem hier vorgestellten Extraktionsprotokoll konzentrieren wir uns auf die Stichproben-, Handling- und -Extraktionsschritte, die in der detaillierten Workflow-Übersicht in Abbildung 2 dargestellt sind. Zu Demonstrationszwecken wurden 50 mg Arabidopsis-Blattgewebe ausgewählt. Dieses Material wurde geerntet, geschliffen und extrahiert, bevor es drei ausgesetzt wurdeBeispielhafte analytische UPLC-MS-Plattformen, die Daten liefern, die für gezielte und ungezielte lipidomische, metabolomische und proteomische Analysen genutzt werden können. Die Fig. 2 und 6 enthalten zusätzlich repräsentative Bilder, wie unter Standardbedingungen das Extraktionslösungsmittel aussehen sollte. Zusätzlich werden Beispiele von Proben, die übermäßige Mengen an ausgefällten Makromolekülen (Proteine und Stärke) und Proben mit ungeeigneter Probenhomogenisierung enthalten, gezeigt (Abbildung 3 ). Die Fehlersuche für diese beiden gemeinsamen Probleme ist in Abbildung 3 kurz dargestellt, aber es wird auch in unserer früheren Publikation näher erläutert.
Die Fig. 4 und 5 beschreiben Beispiele von drei analytischen Chromatogrammen, die aus dem Lipid, dem Polar / SemI-polare Metaboliten und Proteinanalysen. Lipide, die aus der oberen MTBE-Phase (Abbildung 2 ) entnommen wurden, wurden durch Umkehrphasen- (RP) C 8- Ultra-Performance-Flüssigkeitschromatographie analysiert, die mit einer hochauflösenden Massenspektrometrie gekoppelt war. Lipide können mit positiven und negativen MS-Ionisationsmodi (Abbildung 4 , obere Scheibe) 16 , 24 gewonnen werden .
Polare und halbpolare primäre und sekundäre Metaboliten wurden aus der polaren (Wasser / Methanol) Phase (Abbildung 2 ) durch Umkehrphase (RP) C 18 UPLC-MS 25 analysiert. Das dargestellte Verfahren, das die Umkehrphasenchromatographie verwendet, ist in hohem Maße kompatibel zur Analyse von halbpolaren Metaboliten (nämlich Metaboliten aus dem sekundären Stoffwechsel), die mit positiven und negativen Ionisationsmodi in der MS analysiert werden können( Abbildung 4 , untere Scheibe) 16 . Mehr hydrophile Metaboliten aus dieser Fraktion (Zucker, polare Aminosäuren etc.), die keine gute Retention auf dem umgekehrten Phasenmaterial zeigen, können durch andere analytische Methoden wie GC-MS 16 oder hydrophile Wechselwirkungs-Flüssigkeitschromatographie 30 analysiert werden.
Die Proteine, die aus dem festen Pellet im Boden des Extraktionsröhrchens ( Fig. 2 ) gewonnen wurden, wurden in der Lösung verdaut und unter Verwendung der Schrotflinte LC-MS ( Fig. 5 ) analysiert, während das Protokoll für die Extraktion von Stärke und Zellwand Material kann aus unserem bisher veröffentlichten Protokoll 16 gewonnen werden .
Zusammenfassend zeigten sich mehr als 200 Lipidarten, 50 annähernd halbpolare Metaboliten und mehrere tausend ProteiNs können routinemäßig aus Proben des in unserem Beispiel verwendeten Typs identifiziert werden. Zusätzlich zeigte das Verfahren eine breite Anwendbarkeit unter Verwendung verschiedener Gewebe, Organe und Zellkulturmaterial ( 6 ).

Abbildung 1: Allgemeiner Workflow für Großmaßstab Ungezielte Omics-Analyse. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2 : Probenvorbereitung und Extraktions-Workflow zur Analyse von Lipiden, Metaboliten und Proteinen aus einem einzigen Aliquot einer biologischen Probe.Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 3 : Beispielhafte Beispiele für häufig beobachtete Probleme mit zweiphasigen Partitionierungs-Extraktionsprotokollen. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 4 : Repräsentative Chromatogramme von Lipid- und Semipolmetaboliten aus Arabidopsis thaliana-Blätterextrakten . Basis-Peak-ChromatogrammS von Lipiden (oberer Scheibe) und halbpolaren Metaboliten (untere Scheibe), die im positiven Ionisationsmodus analysiert wurden 16 . Die Kreisdiagramme in der oberen rechten Ecke jedes Chromatogramms zeigen die Anzahl der identifizierten Lipide und Metaboliten, die verschiedenen chemischen Klassen zugeordnet sind. Chl, Chlorophylle; DAG, Diacylglycerid; DGDG, Digalactosyldiacylglycerin; FA, Fettsäure; LysoPC, Lysophosphatidylcholin; MGDG, Monogalactosyldiacylglycerin; PC, Phosphatidylcholin; PE, Phosphatidylethanolamin; PG, Phosphatidylglycerin; PI, Phosphatidylinositol; PS, Phosphatidylserin; SP, Sphingolipid; SQDG, Sulfoquinovosyldiacylglycerin; TAG, Triacylglycerid. Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

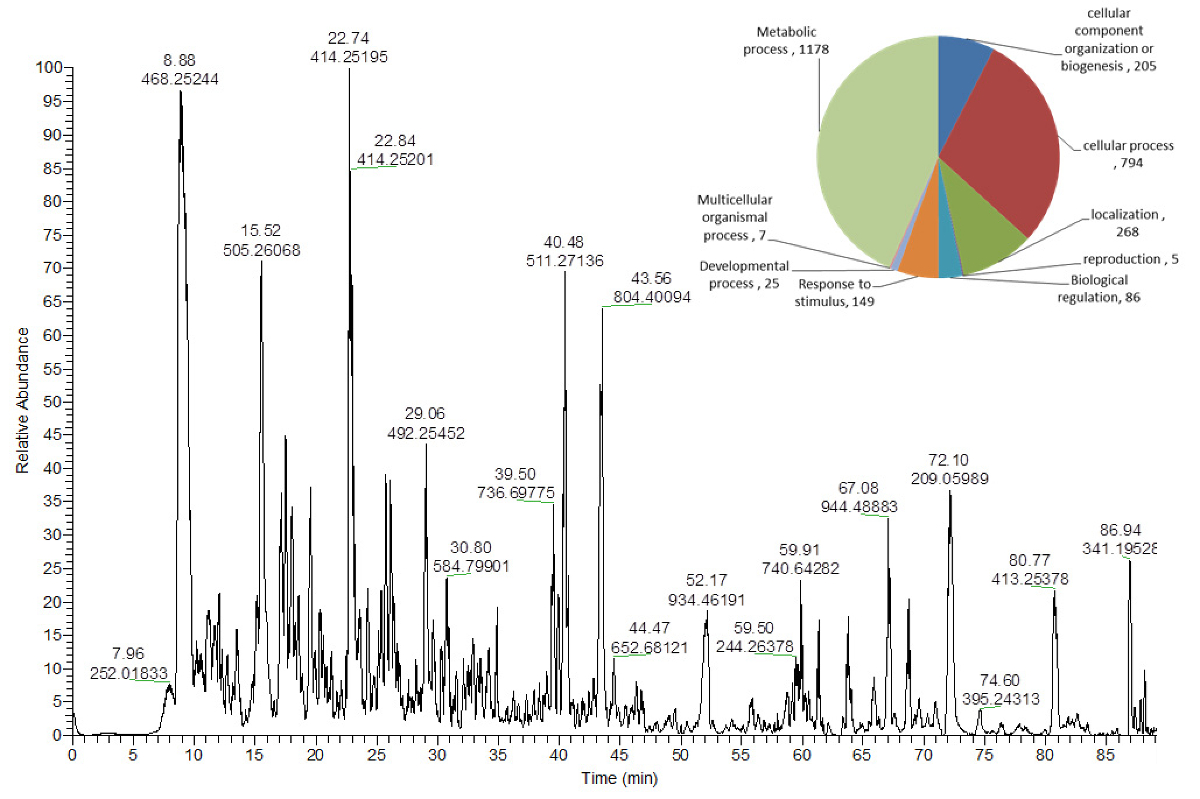
Abbildung 5 Trong>: Repräsentatives Basis-Peak-Chromatogramm von Peptiden aus Arabidopsis thaliana-Blätterextrakten .
Das Kreisdiagramm zeigt in der oberen rechten Ecke die Anzahl der identifizierten Proteine, die verschiedenen biologischen Prozessen zugeordnet sind 16 . Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 6 : Repräsentative Extraktionsbeispiele für verschiedene Gewebetypen aus Wildtyp- Arabidopsis thaliana .
Alle Proben wurden mit 50 mg Frischgewicht aus den angegebenen Geweben extrahiert."> Bitte klicken Sie hier, um eine größere Version dieser Figur zu sehen.
| Zeit (min) | % Puffer A zu Puffer B | |
| 0 bis 1 min | 45% A | Puffer A: 1% 1M NH4-Acetat, 0,1% Essigsäure in UPLC MS-Wasser |
| 1 bis 4 min | Linearer Gradient von 45% A bis 25% A | Puffer B: 1% 1M NH4-Acetat, 0,1% Essigsäure in Acetonitril / Isopropanol 7: 3, (v: v) |
| 4 bis 12 min | Linearer Gradient von 25% A bis 11% A | Durchflussrate 400 μl / min |
| 12 bis 15 min | Linearer Gradient von 11% A bis 0% A | Einspritzvolumen 2 μl |
| 15 bis 19,5 min | Die Säule für 4,5 min mit 0% A waschen | |
| 19,50 bis 19,51 min | Zurück zu 45% A | |
| 19,51 bis 24 min | Equilibrieren mit 45% A |
Tabelle 1: Gradientenparameter für RP-UPLC Trennung von Lipiden RP, umgekehrte Phase. UPLC, Ultra-Performance-Flüssigkeitschromatographie.
| Zeit (min) | % Puffer A zu Puffer B | |
| 0 bis 1 min | 99% A | Puffer A: 0,1% Ameisensäure in UPLC-Grad Wasser |
| 1 bis 11 min | Linearer Gradient von 99% A bis 60% A | Puffer B: 0,1% Ameisensäure in UPLC-Acetonitril |
| 11 bis 13 min | Linearer Gradient von 60% A bis 30% A | Durchflussrate 4001; L / min |
| 13 bis 15 min | Linearer Gradient von 30% A bis 1% A | Einspritzvolumen 2 μl |
| 15 bis 16 min | Die Säule 1 min mit 1% A waschen | |
| 16 bis 17 min | Linearer Gradient von 1% A bis 99% A | |
| 17 bis 20 min | Äquilibrieren für 3 min bei 99% A |
Tabelle 2: Gradientenparameter für die RP-UPLC-Trennung von polaren und semipolaren Metaboliten. RP, umgekehrte Phase. UPLC, Ultra-Performance-Flüssigkeitschromatographie.
| Zeit (min) | % Puffer B zum Puffer A | |
| 0 bis 5 min | Linearer Gradient von 0 bis 10% | Puffer A: 0,1% Ameisensäure in UPLC-Grad Wasser |
| 5 bis 80 min | Linearer Gradient von 10% bis 40% | Puffer B: 0,1% Ameisensäure in 60% Acetonitril der UPLC-Klasse |
| 80 bis 85 min | Linearer Gradient von 40% bis 60% | Durchflussmenge 300 nL / min |
| 85 bis 86 min | Linearer Gradient von 60% bis 95% | Einspritzvolumen 5 μl |
| 86 bis 91 min | Säule für 5 min mit 95% | |
| 91 bis 92 min | Linearer Gradient von 95% bis 0% | |
| 93 bis 110 min | Äquilibrieren Sie die Säule für 17 min bei 0% |
Tabelle 3: Gradientenparameter für die Nano-LC-Trennung von Peptiden LC, Flüssigchromatographie.
Diskussion
In diesem Artikel beschreiben und illustrieren wir ein einfaches und hoch anwendbares Extraktionsprotokoll für eine umfassende lipidomische, metabolomische und proteomische Analyse aus einer einzigen 50 mg Blattprobe. Die Methode wurde bisher in mehreren Studien verwendet, die in verschiedenen Artikeln 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 31 , 32 , 33 , 34 , 35 , 36 , 37 veröffentlicht wurden Und erwies sich, zusätzlich zu seinem geradlinigWorkflow und hohe Anwendbarkeit robust und reproduzierbar.
Die hier bereitgestellten Anwendungen zeigen einige Routineverfahren für das anfängliche Screening einer komplexen biologischen Probe. Diese veranschaulichten metabolomischen und lipidomischen Datensätze können umfangreiche Informationen über die breiten oder spezifischen Veränderungen des Stoffwechsels des analysierten biologischen Systems liefern, während die aus der Analyse von Proteinen gewonnenen Daten Einblicke in die quantitativen (Fülle) und qualitativen (Modifikationen) liefern ) Veränderungen von Enzymen, Strukturproteinen oder Transkriptionsfaktoren (TFs), die zelluläre Funktionen und Maschinen steuern. Dementsprechend haben integrative Omikendaten das Potenzial, erste Informationen über mögliche Veränderungen, die durch genetische oder biotische und / oder abiotische Störungen eines biologischen Systems induziert werden, zu offenbaren, indem molekulare Veränderungen verschiedener Moleküle, die mit spezifischen Stoffwechselwegen oder zellulären Prozessen assoziiert sind, aufgeklärt werden.
Na sicherLangfristig ist es für eine erfolgreiche systembiologische Analyse von grundlegender Bedeutung, die Anzahl der analysierten und kommentierten molekularen Einheiten zu maximieren und so die Überwachung von zellularen Funktionen und Aktivitäten möglichst vollständig zu ermöglichen. Zu diesem Zweck können die erhaltenen Fraktionen zusätzlich zu verschiedenen analytischen Methoden angewendet werden, wobei auf weitere Verbindungen oder Verbindungsklassen geachtet werden (Abbildung 4 ).
Dabei ist zu erwähnen, dass die globale Analysestrategie der gewonnenen Daten durch zwei verschiedene Strategien verfolgt werden kann: Einerseits haben wir die Aufklärung zellulärer Funktionen durch Quantifizierung bekannter Verbindungen hervorgehoben. Auf der anderen Seite sind viele der gemessenen Metaboliten und Lipide noch nicht bekannt oder kommentiert. Diese noch nicht kommentierten zusammengesetzten Messungen enthalten auch viele aussagekräftige Informationen, die durch statistische Methoden zur Klassifizierung oder Diskriminierung genutzt werden können. BZwischen Gruppen oder Behandlungen 20 , 21 , 22 .
Dennoch müssen diese unbekannten Verbindungen, insbesondere diejenigen, die für die Gruppenklassifizierung relevant sind oder als Biomarker dienen, identifiziert werden. Dieser Identifikationsvorgang ist leider recht mühsam und kann ohne zusätzliche analytische Messungen oder Strategien nicht erreicht werden 38 . Wie aus Fig. 4 ersichtlich ist, ist die Anzahl der nicht annotierten Verbindungen ziemlich hoch (eigentlich die überwiegende Mehrheit). Dennoch können diese chromatographischen Peaks, wie oben erwähnt, innerhalb der Datenanalyse gehandhabt werden, und daher können signifikant beeinflusste Entitäten aufgeklärt und weiteren Identifikationsstrategien unterworfen werden.
Zusammenfassend lässt sich feststellen, dass das hier eingeführte Protokoll mehrere Vorteile für die experimentelle Systembiologie sowie für klassische statistische Anwendungen bietetAtions
Zunächst wird, da alle Fraktionen aus einer einzigen Probe extrahiert werden, die Variation zwischen den verschiedenen experimentellen Datensätzen (Lipide, Metaboliten, Proteine) signifikant reduziert, da jeder Datensatz aus dem gleichen Probenaliquot abgeleitet wird. Dies führt eindeutig zu einer erhöhten Vergleichbarkeit der erzielten Ergebnisse.
Zweitens ist das Verfahren leicht skalierbar und macht es daher sehr kompatibel mit kleinen bis großen Probenmengen. Wir verwenden routinemäßig 10-100 mg Gewebeproben, aber erfolgreiche lipidomische Studien wurden auch auf so wenig wie 20 Arabidopsis Samen durchgeführt 31 . Vor allem die Kompatibilität mit kleinen Probenmengen macht dieses Verfahren anwendbar, wenn begrenzte Mengen an biologischen Geweben oder Proben verfügbar sind. Dennoch, auch wenn genügend Probenmaterial vorhanden ist, bietet die hier vorgestellte Methode den Vorteil, diese Proben in einer größeren Anzahl von experimentellen Replikaten anstelle von u zu nutzenSingen sie für verschiedene Extraktionsverfahren. Dies ermöglicht eine bessere und verfeinerte statistische Datenanalyse.
Drittens stellt das Verfahren, da das Verfahren auf einer Flüssig-Flüssig-Fraktionierung von polaren und nichtpolaren Molekülen basiert, im Gegensatz zu einfachen Einphasenextraktionsverfahren ( z. B. Methanol-Extraktionen) einen signifikanten dekomplexierenden Schritt im Verfahren dar. Diese effiziente Probenentkomplexierung führt zu einer partiellen Reinigung der einzelnen Fraktionen aufgrund der Trennung von chemisch interferierenden Molekülen voneinander. Dementsprechend bietet der chemische Partitionierungsprozess nicht nur einen praktischen Vorteil für die systematische Aliquotierung der extrahierten Proben in verschiedene chemische Klassen, sondern verbessert auch die einzelnen analytischen Messungen, da sie kontaminierende Verbindungen aus den verschiedenen Fraktionen entfernt. Klar können wir beobachten, dass vor allem die Lipide, die auf die organische Phase aufgeteilt sind und die sich meist negativ beeinflussenChromatographische Analyse von polaren Verbindungen, wird nahezu vollständig aus der polaren Fraktion fehlen. Das gleiche gilt für die Analyse der hydrophoben Lipide, die an den polaren Verbindungen abgebaut werden. Neben der Reinigung von polaren und unpolaren Verbindungen auseinander entleeren und sammeln wir Proteine und andere Makromoleküle aus der Probe, die nicht nur eine separate Fraktion liefert, die für die Protein-, Stärke- und Zellwandanalyse 16 genutzt werden kann, sondern auch Führt zu einer saubereren Probe innerhalb der einzelnen Fraktionen. Dies ist besonders relevant, da bekannt ist, dass das Vorhandensein großer Makromoleküle zu einer Beschädigung oder zumindest einer kürzeren Lebensdauer der analytischen Säulen führt.
Last but not least wurde die beschriebene MTBE-Extraktionsmethode, die auf dem weniger gefährlichen und günstigeren Chloroform-Ersatzlösungsmittel 15 beruht, bereits in mehreren Studien aus unserer Gruppe gezeigt worden, um weit verbreitet zu seinIcability für verschiedene biologische Proben aus Pflanzen 16 , Algen 17 , 18 , Fliegen 19, aber auch mehrere Säugetiergewebe, Organe oder Zellen 20 , 21 , 22 .
Offenlegungen
Die Autoren haben nichts zu offenbaren
Danksagungen
MS wird von einem vollständigen Promotionsstipendium aus dem GERLS-DAAD-Programm unterstützt. Wir danken Dr. Andrew Wiszniewski für den Nachweis und das Kommentieren des Manuskripts. Wir sind allen Mitgliedern des Giavalisco Labors am Max-Planck-Institut für Molekulare Pflanzenphysiologie, Golm, für ihre Hilfe sehr dankbar.
Materialien
| Name | Company | Catalog Number | Comments |
| Reagents and standards | |||
| Ampicillin | Sigma Aldrich | A9393-5G | Internal standard for metabolites |
| Corticosterone | Sigma Aldrich | 27840-500MG | Internal standard for metabolites, HPLC grade |
| 13C Sorbitol | Sigma Aldrich | 605514 | Internal standard for metabolites, ISOTEC® Stable Isotopes |
| 1,2-diheptadecanoyl-sn-glycero-3-phosphocholine (17:0 PC) | Avanti Polar Lipids | 850360P | Internal standard for lipids |
| Methanol (MeOH) | Biosolve Chemicals | 13684102 | ULC-MS grade |
| Water | Biosolve Chemicals | 23214102 | ULC-MS grade |
| Methyl tert-butyl ether (MTBE) | Biosolve Chemicals | 13890602 | HPLC grade |
| Trypsin/Lys-C mix | Promega | V5072 | Enzymatic digestion of proteins |
| Equipment | |||
| Balance | Sartorius Corporation | 14 557 572 | |
| Tissue grinding mixer mill | Retsch, Mixer Mill MM 300 | 20.746.0001 | |
| Mortar and pestle | Sigma Aldrich | Z247464-1EA | |
| Vortex mixer | Vortex-Genie 2, Model G560 | SI-0236 | |
| Vacuum concentrator | Scan Speed Maxi Vac Alpha Evaporators | 7.008.500.002 | |
| 2 mL Safe-lock microcentrifuge tubes | Eppendorf | 30120094 | Used for sample extarction |
| 1.5 mL Safe-lock microcentrifuge tubes | Eppendorf | 30120086 | Used for fractions |
| Shaker | Eppendorf Thermomixer 5436 | 2050-100-05 | |
| Sonicator | USC 300 TH | 142-0084 | |
| Refrigerated microcentrifuge | Eppendorf, model 5427R | 22620701 | |
| UPLC system | Waters Acquity UPLC system (Waters, Machester, UK) | ||
| MS system | Exactive, Orbitrap-type, MS (Exactive, Thermo-Fisher, Bremen, Germany). | ||
| Reversed Phase (RP) Bridged Ethyl Hybrid (BEH) C8 column (100 mm × 2.1 mm containing 1.7 μm diameter particles) | Waters, Machester, UK | 186002878 | Analysis of lipids |
| RP High Strength Silica (HSS) T3 column (100 mm × 2.1 mm containing 1.8 μm diameter particles) | Waters, Machester, UK | 186003539 | Analysis of metabolites |
| Q ExactivePlus high resolution mass spectrometer connected to an EASY-nLC 1000 system | Thermo-Fisher, Bremen, Germany | Analysis of peptides |
Referenzen
- Hodgkin, A. L., Huxley, A. F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 117 (4), 500-544 (1952).
- Wang, Z., Gerstein, M., Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 10 (1), 57-63 (2009).
- Yang, M. Q., et al. The emerging genomics and systems biology research lead to systems genomics studies. BMC Genomics. 15, 1 (2014).
- Sheth, B. P., Thaker, V. S. Plant systems biology: insights, advances and challenges. Planta. 240 (1), 33-54 (2014).
- Ideker, T., Galitski, T., Hood, L. A new approach to decoding life: Systems biology. Annu Rev Genom Hum G. 2, 343-372 (2001).
- Somvanshi, P. R., Venkatesh, K. V. A conceptual review on systems biology in health and diseases: from biological networks to modern therapeutics. Syst Synth Biol. 8 (1), 99-116 (2014).
- Oliver, S. G., Winson, M. K., Kell, D. B., Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 16 (9), 373-378 (1998).
- Tweeddale, H., Notley-McRobb, L., Ferenci, T. Effect of slow growth on metabolism of Escherichia coli, as revealed by global metabolite pool ("metabolome") analysis. J Bacteriol. 180 (19), 5109-5116 (1998).
- Kim, H. K., Verpoorte, R. Sample preparation for plant metabolomics. Phytochemical analysis : PCA. 21 (1), 4-13 (2010).
- Folch, J., Lees, M., Stanley, G. H. S. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J Biol Chem. 226 (1), 497-509 (1957).
- Weckwerth, W., Wenzel, K., Fiehn, O. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics. 4 (1), 78-83 (2004).
- Wienkoop, S., et al. Integration of metabolomic and proteomic phenotypes: analysis of data covariance dissects starch and RFO metabolism from low and high temperature compensation response in Arabidopsis thaliana. Mol Cell Proteomics. 7 (9), 1725-1736 (2008).
- Valledor, L., et al. A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant J. 79 (1), 173-180 (2014).
- Nakayasu, E. S., et al. MPLEx: a Robust and Universal Protocol for Single-Sample Integrative Proteomic, Metabolomic, and Lipidomic Analyses. mSystems. 1 (3), (2016).
- Alfonsi, K., et al. Green chemistry tools to influence a medicinal chemistry and research chemistry based organisation. Green Chem. 10 (1), 31-36 (2008).
- Salem, M. A., Juppner, J., Bajdzienko, K., Giavalisco, P. Protocol: a fast, comprehensive and reproducible one-step extraction method for the rapid preparation of polar and semi-polar metabolites, lipids, proteins, starch and cell wall polymers from a single sample. Plant Methods. 12, 45 (2016).
- Hemme, D., et al. Systems-wide analysis of acclimation responses to long-term heat stress and recovery in the photosynthetic model organism Chlamydomonas reinhardtii. Plant Cell. 26 (11), 4270-4297 (2014).
- Sharma, D. K., et al. UPLC-MS analysis of Chlamydomonas reinhardtii and Scenedesmus obliquus lipid extracts and their possible metabolic roles. J Appl Phycol. 27 (3), 1149-1159 (2015).
- Delgado, R., Munoz, Y., Pena-Cortes, H., Giavalisco, P., Bacigalupo, J. Diacylglycerol activates the light-dependent channel TRP in the photosensitive microvilli of Drosophila melanogaster photoreceptors. J Neurosci. 34 (19), 6679-6686 (2014).
- Bozek, K., et al. Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron. 85 (4), 695-702 (2015).
- Khrameeva, E. E., et al. Neanderthal ancestry drives evolution of lipid catabolism in contemporary Europeans. Nat Commun. 5, 3584 (2014).
- Bozek, K., et al. Exceptional evolutionary divergence of human muscle and brain metabolomes parallels human cognitive and physical uniqueness. PLoS Biol. 12 (5), 1001871 (2014).
- Krueger, S., Steinhauser, D., Lisec, J., Giavalisco, P. Analysis of subcellular metabolite distributions within Arabidopsis thaliana leaf tissue: a primer for subcellular metabolomics. Methods Mol Biol. 1062, 575-596 (2014).
- Hummel, J., et al. Ultra performance liquid chromatography and high resolution mass spectrometry for the analysis of plant lipids. FrontPlant Sci. 2, 54 (2011).
- Giavalisco, P., et al. Elemental formula annotation of polar and lipophilic metabolites using (13) C, (15) N and (34) S isotope labelling, in combination with high-resolution mass spectrometry. Plant J. 68 (2), 364-376 (2011).
- Caldana, C., et al. Systemic analysis of inducible target of rapamycin mutants reveal a general metabolic switch controlling growth in Arabidopsis thaliana. Plant J. 73 (6), 897-909 (2013).
- Giavalisco, P., et al. Proteome analysis of Arabidopsis thaliana by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionisation-time of flight mass spectrometry. Proteomics. 5 (7), 1902-1913 (2005).
- Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72, 248-254 (1976).
- Rappsilber, J., Mann, M., Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nature protocols. 2 (8), 1896-1906 (2007).
- Spagou, K., et al. Hydrophilic interaction chromatography coupled to MS for metabonomic/metabolomic studies. J Sep Sci. 33 (6-7), 716-727 (2010).
- Hauck, O. K., et al. Uric acid accumulation in an Arabidopsis urate oxidase mutant impairs seedling establishment by blocking peroxisome maintenance. Plant Cell. 26 (7), 3090-3100 (2014).
- Avin-Wittenberg, T., et al. Global analysis of the role of autophagy in cellular metabolism and energy homeostasis in Arabidopsis seedlings under carbon starvation. Plant Cell. 27 (2), 306-322 (2015).
- Watanabe, M., et al. Comprehensive dissection of spatiotemporal metabolic shifts in primary, secondary, and lipid metabolism during developmental senescence in Arabidopsis. Plant physiol. 162 (3), 1290-1310 (2013).
- Lambers, H., et al. Proteaceae from severely phosphorus-impoverished soils extensively replace phospholipids with galactolipids and sulfolipids during leaf development to achieve a high photosynthetic phosphorus-use-efficiency. New Phytol. 196 (4), 1098-1108 (2012).
- Degenkolbe, T., et al. Differential remodeling of the lipidome during cold acclimation in natural accessions of Arabidopsis thaliana. Plant J. 70, 972-982 (2012).
- Bromke, M. A., et al. Liquid chromatography high-resolution mass spectrometry for fatty acid profiling. Plant J. 81 (3), 529-536 (2015).
- Li, N., et al. FAX1, a novel membrane protein mediating plastid fatty acid export. PLoS Biol. 13 (2), 1002053 (2015).
- Kueger, S., Steinhauser, D., Willmitzer, L., Giavalisco, P. High-resolution plant metabolomics: from mass spectral features to metabolites and from whole-cell analysis to subcellular metabolite distributions. Plant J. 70 (1), 39-50 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten