
Method Article
Mikromanipulation Techniken Analyse der morphogenetischen Dynamik und Umsatz des Zytoskeletts Regulatoren
In diesem Artikel
Zusammenfassung
Wir beschreiben, wie Mikro- und Fotomanipulation Techniken wie FRAP und photoaktivierungen ermöglichen die Bestimmung der Motilität Parameter und die räumlich-zeitliche Dynamik der Proteine innerhalb Migration Zellen. Experimentellen Messwerte sind subzelluläre Dynamik und Umsatz der Motilität Regulierungsbehörden oder des zugrunde liegenden Aktin-Zytoskeletts.
Zusammenfassung
Prüfung der räumlich-zeitliche Dynamik der Proteine kann ihre funktionale Bedeutung in verschiedenen Kontexten zeigen. In diesem Artikel ist es diskutiert wie fluoreszierende Erholung nach Immunofluoreszenz (FRAP) und photoaktivierungen-Techniken verwendet werden, um die räumlich-zeitliche Dynamik der Proteine an subzellulären Orten zu studieren. Wir zeigen auch, wie diese Techniken einfach Bestimmung verschiedener Parameter verbunden mit Aktin-Zytoskeletts Regulierung und Zelle Motilität zu ermöglichen. Darüber hinaus die Mikroinjektion von Zellen zusätzlich eine alternative Behandlung (möglicherweise vor oder ergänzend zu die vorgenannten Photomanipulation Techniken) Trigger sofortige Wirkungen der translozierten Proteine auf Zelle bezeichnet man als Morphologie und Funktion. Mikromanipulation wie Protein Einspritzung oder lokale Anwendung von Plasmamembran durchlässig Drogen oder Zellskelett Hemmer dienen als mächtiges Werkzeug, um die unmittelbaren Auswirkungen einer bestimmten Behandlung auf Zelle Verhalten auf die einzelne Zelle und subzellulärer erfassen Ebene. Dies ist hier durch sofortige Induktion der Lamellipodial Zelle Rand Vorsprung durch die Injektion von rekombinanten Rac1 Protein veranschaulicht, wie vor einem Viertel Jahrhundert gegründet. Darüber hinaus bieten wir ein Protokoll für die Bestimmung des Umsatzes der erweiterten grün fluoreszierendes Protein (EGFP)-VASP, ein Aktin-Filament-Polymerase prominent an Lamellipodial Spitzen der B16-F1 Zellen ansammeln beschäftigt FRAP und einschließlich der zugehörigen Daten Analyse und Kurvenanpassung. Wir präsentieren auch Richtlinien für die Schätzung der Preise der Lamellipodial Aktin Netzwerk Polymerisation, beispielhaft durch Zellen, die mit dem Ausdruck EGFP getaggt β-Aktin. Zu guter Letzt sind Anweisungen für wie zu untersuchen, die Preise der Aktin-Monomer-Mobilität innerhalb der Zelle Zytoplasma, gefolgt von Aktin Einbau an Standorten von rapid Filament Assembly, wie die Spitzen der hervorstehenden Lamellipodia, mit photoaktivierungen Ansätze. Keines dieser Protokolle ist auf Komponenten oder Regulatoren des Aktin-Zytoskeletts beschränkt, sondern kann problemlos erweitert werden, um in Analog Mode die raumzeitliche Dynamik und Funktion von Proteinen in verschiedenen verschiedenen subzellulären Strukturen oder funktionelle erkunden Kontexten.
Einleitung
Überwachung der räumlich-zeitliche Dynamik von Proteinen und anderen Molekülen in lebenden Zellen ist ein wesentliches Instrument in vielen Bereichen der Zell- und Molekularbiologie geworden. Erweiterte Fluoreszenz mikroskopiertechniken einschließlich Fluoreszenz Resonanz Energietransfer (FRET) und Bund-Fluoreszenz Lifetime imaging (Bund-FLIM) oder FRAP, Verlust der Fluoreszenz in Immunofluoreszenz (FLIP) und photoaktivierungen sowie viele andere zulassen für die zeitliche und räumliche Verfolgung von Protein-Protein-Wechselwirkungen, Konformationsänderungen, sowie die Bestimmung der Kinetik der Diffusion und Lokalisierung von verschiedenen Proteinen in der Zelle1,2. FRAP und photoaktivierungen Techniken sind vor allem für die Prüfung der Regulierungsbehörden der Aktin-Zytoskelett und Zelle Migration allgemein anwendbar. Diese Techniken können allein oder in Kombination mit zusätzlichen Mikromanipulation Techniken wie Mikroinjektion3angewendet werden und beinhalten die Expression von Proteinen eindringmittel beschriftet. Sie ermöglichen die Abschätzung der Kinetik der Verband Protein Aktin-reiche Strukturen Zellwanderung, z. B. Filopodien oder Lamellipodia, der Umsatz der Proteine in fokalen Adhäsionen4beteiligt oder verzweigte Aktin Netzwerke5. Sie ermöglichen auch die Bestimmung der Lamellipodial-Aktin-Polymerisation-Preise, die Bewertung der Dispersion von Monomeren Aktin im Zytosol, die Rate der subzellulären Actin Monomeren Translokation, dadurch actinfilamente im vorstehenden Lamellipodia6und andere Parameter.
FRAP ist eine Methode zur Visualisierung und Quantifizierung der Mobilität von Proteinen innerhalb einer lebenden Zelle, ursprünglich in den 1970er Jahren von Axelrod7entwickelt. Eine Region of Interest (ROI) innerhalb einer Zelle, bevölkert mit fluoreszent-markierten Proteinen ist ein Laser von hoher Intensität, zu bleichen der Fluorophor Moleküle vorhanden in dieser Region einen bestimmten kurzen Zeitraum hinweg führen vorübergehend ausgesetzt. Ungebleicht, eindringmittel beschriftet Proteine befindet sich außerhalb der ROI beim Bleichen, diffuse und die gebleichte Region abhängig von ihrer raumzeitlichen Dynamik, verursacht die Verschiebung der Photobleached Moleküle im Laufe der Zeit zu infiltrieren. Die Rate der Fluoreszenz Wiederaufnahme in gebleichtem Regionen ist abhängig von verschiedenen Faktoren ab, einschließlich die Größe und die Rate der Verbreitung eines bestimmten Moleküls und natürlich die Fluktuationsrate innerhalb der vermeintlichen gebleichten Struktur verbunden. So lösliche Proteine wird die Erholung der Fluoreszenz in der gebleichten ROI schnell durch Diffusion, während Proteine fest zugeordnete Strukturen wie fokale Adhäsionen zu vermitteln, haben längere Umschlagzeiten, wie deren Fluoreszenz-Erholung wird richten Sie sich sowohl auf die Diffusion der löslichen Fraktion der Protein- und Dissoziation-Verband Kinetik des Bruches Struktur verbunden. Fluoreszenz-Recovery ist in der Regel erworben und quantifiziert, bis das ursprüngliche Niveau der Pre-Bleichmittel Intensität der Fluoreszenz erreicht ist. Allerdings tritt dies nicht auf, wenn ein Teil der ursprünglichen Fluoreszenzintensität gehört zu den sogenannten unbeweglich Fraktion, die nicht durch Diffusion wieder aufgefüllt werden kann oder ist sehr langsam Preisen im Vergleich zu den Großteil der Moleküle bestehend aus mobilen Auffüllen Bruchteil. Zur Bestimmung des Protein-Umsatz, FRAP Kurven erzeugt werden, das Ausmaß der Fluoreszenz-Erholung im Laufe der Zeit darstellt. Aus diesen Kurven Erholung können durchschnittliche zweieinhalbmal Proteingewinnung berechnet werden. Kurve passt der Durchschnittsdaten FRAP, und damit mathematischen Analysen erstellen, ist es auch möglich, abzuleiten, die durchschnittliche Fluktuationsrate der mobilen Fraktion aus einer homogenen Bevölkerung von Molekülen zusammengesetzt ist, ob es besteht mindestens zwei Subpopulationen von Molekülen mit Differential Rate umdrehen. Neben der Schätzung Protein Umsatzraten durch quantitative Ansätze, können verfolgen die Erholung der Photobleached Regionen in Lamellipodia für genaue Quantifizierung von Lamellipodial Motilität Parameter wie retrograde Flow, Vorsprung auch, und Aktin-Polymerisation-Preise. FRAP stellt, ein vielseitiges Werkzeug für die Beurteilung verschiedener Parameter innerhalb der Strukturen lebender Zellen angewendet werden.
Photoaktivierungen ist eine Methode verwendet, um die Verbreitung und die Mobilität der Proteine oder Moleküle, die aus einem bestimmten zellulären Ort verfolgen. Die Technik beschäftigt, zum Beispiel eine Variante des Wildtyp grün fluoreszierendes Protein (GFP), ursprünglich entwickelt von Patterson und Lippincott-Schwartz8, die in einer Art und Weise mutiert ist, mit dem seine Fluoreszenz hoch nach Exposition gegenüber erhöht werden können Ultraviolettes (UV) Licht (rund 400 nm; hier, 405 nm). Wie beschrieben von Patterson Et Al., Wildtyp GFP Chromophore existieren als eine gemischte Bevölkerung von neutralen Phenole und anionischen Phenolates, erzeugen eine große Absorption Peak bei ca. 397 nm und eine kleinere noch 475 nm, beziehungsweise. Bei der Bestrahlung des Proteins mit UV-Licht erfährt die Bevölkerung Ladungszustand, Verschiebung in Richtung der anionischen Form. Wenn begeistert von 488 nm, das Photoconverted/photoaktiviert-Protein stellt eine Erhöhung der 3-fold in Fluoreszenz, nicht ausreichend in der Praxis für die Unterscheidung zwischen aktiviert und nicht GFP aufgrund der hohen intrinsischen Hintergrundfluoreszenz aktivierte. Jedoch wurde eine Abnahme der hintergrundintensität erreicht, durch die Einführung einer einzigen Aminosäure Mutation in der GFP-Sequenz (Histidin Substitution an Position 203). Das daraus resultierende T203H mutierte, auch bekannt als Photoactivatable-GLP (PA-GFP) zeichnet sich durch einen erheblichen Rückgang der Extinktion der kleine Spitze, die bei Bestrahlung mit UV-Licht fast 100fach erhöht wird, wenn anschließend mit 488 nm Licht angeregt. Überexpression von PA-GFP-markierten Proteinen ist eine weit verbreitete Ansatz, der die Bestimmung der Diffusion und Beweglichkeit der Moleküle in den Zellen ermöglicht. Wir haben zuvor PA-GFP-Tags Aktin um festzustellen, die Rate der Dispersion von Actin-Monomere vom cytosolischen Regionen angewendet ermöglicht nicht nur die Erforschung ihrer Mobilität innerhalb der Zytosol, sondern auch ihre Aufnahme Rate in die hervorstehenden Lamellipodial Aktin Netzwerk6. Neuere Literatur beschreibt auch neuartige, Foto-Cabrio Proteine, die im Prinzip in einer analogen Weise verwendet werden können, sondern beherbergen den möglichen Vorteil bereits vor der Foto-Konvertierung sichtbar. Beispiele für diese Gruppe von fluoreszierenden Proteinen sind Dendra2 und mEos29,10,11,12.
In diesem Artikel erklären wir die Methodik der microinjecting Zellen mit Proteinen. Wir erklären weiter, wie diese Technik mit FRAP, durch Immunofluoreszenz Proteine Aktin-Zytoskelett-Verordnung und Motilität, beteiligt kombiniert werden kann und wie FRAP Kurven und Halbzeit der Verwertung von mobilen Fraktionen abgeleitet werden können. Darüber hinaus bieten wir ein Beispiel der Verwendung von der FRAP-Technik Aktin Polymerisation der Lamellipodial Netzwerke bestimmen. Wir bieten auch Anleitungen und Tipps für photoaktivierungen Experimente durchzuführen, die zur cytosolischen Mobilität von Monomeren Aktin und Raten von Aktin Einbindung in Lamellipodia herangezogen werden können. Diese Techniken sind natürlich nicht nur um tracking-Aktin-Zytoskelett-Komponenten, sondern auf potenziell erforderlich moderate Anpassung bzw. Optimierung begrenzt, kann weit angewendet werden um andere Zelltypen oder verschiedene Proteinen, Strukturen, zu untersuchen und Parameter.
Protokoll
(1) Deckglas waschen und Sterilisation
- Tauchen 15 mm (Durchmesser) Deckgläser (Nr. 1) in einen 500 mL-Flasche mit einer Mischung aus 40 mL 37 % HCl und 60 mL 100 % EtOH (nicht mehr als 100 Deckgläsern pro 100 mL Waschlösung).
Hinweis: selbst wenn frisch gekauft, Deckgläsern stringent gereinigt werden vor der Aussaat Zellen auf ihrer Oberfläche. Und zwar deshalb, weil sie dünne Schichten Fett, die makroskopisch nicht sichtbar enthalten, sondern kann effizient mit Adhäsion und entsprechende Verbreitung von lebenden Zellen stören. Solche Filme mit Lösungen mit Säure oder base effizient beseitigt werden können (vgl. Fischer Et Al. ( 13), verwenden wir routinemäßig die oben beschriebene Säure/Alkohol-Gemisch. - Schütteln Sie die Flasche mit der Deckgläser für 30 min auf eine Drehung Shaker. Wählen Sie eine Geschwindigkeit, die erlaubt die Deckgläser frei verwirbelt werden, aber langsam genug, um häufige brechen zu vermeiden. Filtern Sie die Lösung um Glasscherben Stücke zu entfernen, wenn die Wiederverwendung.
- Die Deckgläser auf ein Fläschchen mit mindestens 200 mL sterilem Wasser übertragen und brüten auf eine Drehung Shaker, während das Wasser immer wieder zu ersetzen, bis der saure Geruch verschwunden ist. Für vollständige Beseitigung der HCL-EtOH Spuren werden mehrere Wäschen über mehrere Stunden empfohlen.
- Trocknen Sie einzelnen Deckgläser auf einem Blatt Filterpapier.
- Die Deckgläser am unteren Rand einer Petrischale 10 cm (Durchmesser) mit Filterpapier und Wärme-fallen trocken sterilisieren. Vermeiden Sie Autoklavieren, da Deckgläser zusammenhalten wird.
2. Behandlung von Zellen, Transfektion und Aussaat auf Deckgläsern
- Wachsen Sie B16-F1 Maus Melanomzellen nach Standardzelle Kulturbedingungen in DMEM (4,5 g/L Glukose) mit 10 % fetalen Kälberserum, 2 mM Glutamin und 1 % Penicillin-Streptomycin bei 37 ° C, 7 % CO2.
- Wachsen Sie NIH3T3 Fibroblasten-Zellen für Mikroinjektionen nach Standardzelle Kulturbedingungen (Gewebekultur Inkubator bei 37 ° C, 7 % CO2) in DMEM (4,5 g/L Glukose) mit 10 % fötalen Rinderserum, 1 mM Natrium Pyruvat, 1 X MEM nicht-essentiellen Aminosäuren , 2 mM Glutamin und 1 % Penicillin-Streptomycin.
- Wachsen Sie für Transfektionen B16-F1 Zellen zu 100 % Einmündung in eine 10 cm Teller und Passage im Verhältnis 1:5 in einer Plastikschale 3 cm (Durchmesser).
- Am selben Tag nachdem die B16-F1-Zellen für mindestens 6 h halten durften, transfizieren Sie mit 500 ng/Gericht des Photoactivatable PA-GFP-Aktin oder EGFP getaggt β-Aktin Plasmid DNA. Mischen Sie für Co Transfektionen von PA-GFP-Aktin mit mCherry-Codierung Vektoren insgesamt 1 µg Plasmid DNA pro 3 cm Schüssel.
- Transfizieren Sie B16-F1-Zellen mit dem Transfektion Reagenz (Table of Materials). Für ein 3 cm Schüssel mischen 200 µL 150 mm NaCl enthält 500 ng DNA Konstruieren mit 200 µL 150 mm NaCl mit 1 µL Transfection Reagens (d. h. DNA (µg): Reagenz (µL) Verhältnis von 1:2 verwendet wurde).
- Inkubieren Sie die Transfektion Mischung für 20 min bei Raumtemperatur (RT) und pipettieren tropfenweise auf die 3 cm-Teller, der die Zellen enthält. Vorsichtig schwenken die Schüssel zu mischen und über Nacht bei 37 ° C, 7 % CO2inkubieren.
- Bereiten Sie den Laminin-Beschichtung-Puffer mit 50 mM Tris, pH 7,4 und 150 mM NaCl.
- Für die Zellen B16-F1 Mantel 15 mm Deckgläser von 150 µL Laminin (25 µg/mL im Laminin Beschichtung Puffer) zu verbreiten und für 1 h bei RT inkubieren Beschichten Sie für die Zellen NIH3T3 die Deckgläser mit Fibronektin-Lösung (25 µg/mL in Phosphat-gepufferte Kochsalzlösung (PBS)) und 1 h bei RT inkubieren
- Laminin oder Fibronektin inkubiert Deckgläser mit PBS waschen dann Aspirieren der PBS und 2 mL von transfizierten Zellen.
- Samen der transfizierten Zellen B16-F1 (in 01:30 Verhältnis aus einer konfluierende Schüssel), am Tag nach der Transfektion auf Deckgläsern Laminin-beschichtet. Samen die NIH3T3 Fibroblasten (in 01:20 Verhältnis aus einer konfluierende Schüssel) auf Fibronektin-beschichtete Deckgläsern.
- Lassen Sie die Zellen, Laminin - oder Fibronektin-beschichtete Deckgläser über Nacht in einer Gewebekultur Inkubator bei 37 ° C vor der Mikroskopie zu verbreiten. Alternativ können Mikroskopie Experimente am selben Tag eingeleitet werden, angesichts der Tatsache, dass Zellen dürfen für mindestens 2 – 3 h zu verbreiten.
3. Montage der Mikroskopie Imaging-Kammer
- Verwenden Sie eine Wärme leitende RC-26 Aluminium imaging-Kammer für die Mikroskopie (Abbildung 1(ein). Schmieren Sie das Silikonfett rund um die Kontur der Kunststoff Versiegelung Öffnung mit einer Spritze (Abbildung 1b).
- Legen Sie das Deckglas mit den Zellen Seite nach oben auf die Kammer (Abbildung 1c).
- Legen Sie die Kunststoff Versiegelung auf der Oberseite der Abdeckung Glas, um eine sichere Abdichtung zwischen dem Deckglas und der Kammer. Beheben der Kunststoff Versiegelung (diagonal, um Deckglas Bruch zu vermeiden) durch Verschraubung der verschiebbaren Klemmen auf der Kammer, das Medium undicht (Abbildung 1-d) zu vermeiden.
- Pipette 37 ° C vorgewärmte Mikroskopie Medium in den zentralen Bereich. Verwenden Sie für mittlere in Autofluoreszenz reduziert und somit optimiert für Mikroskopie das gleiche Rezept als Kulturmedium beschrieben oben, aber mit F12-Schinken anstelle von DMEM, zusätzlich mit 20 mM HEPES für die Kultivierung der Zellen in der Abwesenheit von CO2 (Abbildung 1e).
- Legen Sie den Hitzemelder in den dafür vorgesehenen Schlitz der Kammer und verknüpfen Sie die Elektroden der Kammer mit einem TC-324B automatischer Temperaturregler Aufrechterhaltung einer konstanten Temperatur von 37 ° C (Abbildung 1-f).
- Tropfen Sie einen kleinen Immersionsöl auf das Ziel und legen Sie die Kammer darauf.
- Inkubieren Sie die Kammer mit Zellen für mindestens 10-30 min um sie zur Wiederherstellung der Temperaturabfall während der Montage und Anpassung an die Mikroskopie-Medium zu ermöglichen.
- Bevor Mikroskopie initiiert wird, ersetzen Sie das Kulturmedium im zentralen Reservoir der Kammer (rund 800 µL), unangemessene Konzentration der mittelkomponenten und Serum durch mittlere Verdunstung zu vermeiden. Verlängerte Mikroskopie-Sessions mit offenen Kammern erfordert Routine Ändern des verdampfenden Mediums.
4. Mikroinjektion Verfahren
- Beschichten der Deckgläsern, bereiten Sie die Zellen und die bildgebende Kammer montieren, wie oben beschrieben.
- Tauwetter eine Aliquote gereinigtes Protein injiziert werden (in der Regel 10 µL oder weniger) und mit den entsprechenden Mikroinjektion Puffer verdünnen.
Hinweis: Puffer Zusammensetzung kann variieren je nach Protein und Zelle Typ, aber achten Sie auf einen pH-Wert zwischen 6,95 und 8.00 Uhr nutzen und vermeiden die Verwendung von PBS, wie die meisten Zelle nicht mit PBS injiziert werden. - Bereiten Sie Rac1 Mikroinjektion Puffer mit 100 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 1 mM DTT. Die Mg2 + -Ionen sind für kleine GTPase Stabilität unerlässlich.
Hinweis: Proteinkonzentrationen normalerweise schwanken zwischen 0,1 – 1 mg/mL (max. 2 mg/mL), je nach dem Protein, Experiment und Zelle geben. - Falls zutreffend, fügen Sie fluoreszierenden Farbstoff, wie träge Dextran (0,5 µg/mL, 70 kDa hinzu) die Proteinlösung, die kann bestätigen das Vorhandensein von Nadel Flow vor Injektionen und ermöglicht die Dokumentation der erfolgreichen Injektionen nach dem Experiment.
Hinweis: Das Experiment hier richtet sich nicht an Anschluss an die Dynamik des injizierten Rac1, die nur auf direkte Fluoreszenz Kennzeichnung des Proteins möglich wäre. Kopplung von Proteinen mit Fluoreszenzfarbstoffen oder Fusion zu einem fluoreszierenden Protein ist möglich, aber vermieden werden da es birgt das Risiko einer stören die Signalisierung Funktion, insbesondere der kleinen Proteine wie der Rho-Familie GTPase Rac1 (20 kDa). - Zentrifugieren Sie die Proteinlösung bei 10.000 x g für mindestens 30 min um Protein-Aggregate zu entfernen, die zur Nadel verstopfen, wenn anwesend in der Mikroinjektion Kapillaren führen kann.
- Laden Sie eine Mikroinjektion Nadel (Mikroinjektion Kapillare) mit 1 µL des injektierungsgemisches von der Rückseite mit einem flexiblen PIPETTENSPITZE Tipp/Microloader.
- Wenn Luftblasen in der Spitze der Nadel vorhanden sind, klopfen Sie leicht die Nadel-Basis, um sie zu entfernen. Fahren Sie schnell, um zu vermeiden, Trocknen von der Spitze der Nadel, die Nadel zu Verstopfung führen können.
- Stellen Sie vorsichtig den Nadelhalter auf der Mikromanipulation Gerät. Wenn mit einem inversen Mikroskop für Phase Kontrast Bildgebung, vor dem Laden Nadel zu gewährleisten gibt es genügend Platz, um die Nadel oben oder unten bewegen, ohne Behinderung des Mikroskop-Kondensators.
- Beim Verschrauben der Microcapillary auf den Nadelhalter, Druck (20 – 50 hPa Hintergrund Druck) auf der Nadel mit einem Mikroinjektion Druckgerät vor translocating die Nadelspitze in die Zellkulturmedium.
Hinweis: Aktivieren den Druck, wenn die Nadel in das Medium ist das Medium angesaugt durch Kapillare Kraft zur Folge und damit verbieten Injektion der Lösung von Interesse wird. - Positionieren Sie die Nadel in das Sichtfeld (durch die Verwendung geringer Vergrößerung Ziele erleichtert). Hier wurde ein 40 X trocken Ziel für die Mikroinjektion Experimente verwendet.
- Positionieren Sie die Nadelspitze makroskopisch in einer vertikalen Position im Verhältnis zu der Mitte der Objektivlinse (Dies beschleunigt die Nadelspitze zu finden). Verwenden Sie das Mikroskop mit Phasenkontrast-Optik, die Nadelspitze in der Horizontalebene bezogen auf das Sichtfeld bei einer optischen Ebene gut über die Zellschicht verschieben.
Hinweis: Die Nadel erscheint zunächst als "Schatten" in das Blickfeld und die Fokusebene kann dann angepasst werden, um die Spitze zu visualisieren. Sobald die Spitze der Nadel gefunden wird, allmählich Absenken der optischen Ebene, gefolgt von der Spitze der Nadel nach unten in eine Position in der Nähe der Zellschicht. - Überprüfen Sie die Nadel fließen durch die Umstellung auf einen fluoreszierenden Kanal bei der Verwendung von fluoreszierenden Dextran und optimieren den Fluss mit Hilfe des Druckgerätes, um eine Konstante "Hintergrund" Strömung zu erhalten.
Hinweis: In diesem Artikel beschreiben wir manuelle Einspritzung, die vermittelt wird durch das brechen durch die Plasmamembran durch Berühren der Zelle Oberfläche und sanfte Bewegung der Nadelspitze während Konstante Nadel fließen. Dies ist zu unterscheiden von automatischen Injektionsgeräten, begleitet von programmierten Nadel absenken und Nadel Druckanstieg während der Injektion Ereignisse, die für die Injektion von höhere Zellzahlen, gefolgt von einer späteren Zellpopulation besser geeignet sind Analyse. Die hier beschriebene Methode ist für einzelne Zelle Analyse durch Zeitraffer Mikroskopie vor, während und nach der Mikroinjektion optimiert. - Suchen Sie eine Zelle von Interesse und senken Sie allmählich die Nadel oberhalb der Zelle.
- Wenn Sie bereit sind, microinject, senken Sie die Nadel allmählich gegen die Zelle perinukleären Region mit feinen Ritzel des Joysticks Mikromanipulator unter Beibehaltung der Zellen im Fokus.
- Für die Mikroinjektion sanft berühren der Plasmamembran der Zelle, die ausreichend sind, dringen in die Zelle, oder transiente Membran Bruch durch ein sehr sanftes Tippen auf das Mikroskop-Setup zu unterstützen.
Hinweis: Ein weißer Punkt an der Spitze der Nadel wird die Zeit des Kontaktes mit der Plasmamembran anzugeben; nach Ruptur der Membran, die Nadelspitze verschließen wird, begleitet von einem sanften Fluss der Injektionslösung in die Zelle. - Den Einspritzvorgang zu stoppen, sobald fließen in die Zelle sichtbar ist (im Idealfall innerhalb von 0,3 s) durch hochschieben der Nadelspitze in das Medium. Bei der Verwendung von fluoreszierenden Dextran können erfolgreiche Injektionen sofort durch Fluoreszenz dokumentiert werden.
- Falls gewünscht, initiieren Sie Zeitraffer Bildaufnahme vor oder nach der Mikroinjektion.
Hinweis: Lokale Anwendung von Drogen oder Inhibitoren kann an allen Schritten hier, mit Ausnahme der Mikroinjektion Ereignis schlechthinausgeführt werden. Für lokale Anwendungen die Diffusion des aktiven Moleküls kann von Fließdruck kontrolliert und dokumentiert durch Fluoreszenz und die Nadelspitze in der gewünschten Höhe positioniert werden kann. Beispiele für lokale Anwendung Experimente zu sehen, z. B. kleine und Rottner14 oder Kaverina Et al. 15 - Im Anschluss an die Mikroinjektion warten Sie, bis die Wirkung des Proteins tritt. Für verschiedene Proteine und je nach dem erwarteten Ergebnis variieren die Inkubationszeiten. Für die kleine GTPase Rac1 kann die Reaktion des Lamellipodium Bildung eingeleiteten innerhalb von 1 min. oder weniger, aber dauert ca. 10-15 min im Durchschnitt zu entfalten (Abbildung 1g, h).
- Die Lebensfähigkeit der Zellen nach der Mikroinjektion zu beurteilen.
Hinweis: Ungeeigneten oder schädliche Injektionen verursachen Zellschäden, die häufig durch unspezifische Zellenrand Widerruf oder Plasmamembran Bruch begleitet wird.- Vermeiden Sie stossen durch die oberen und unteren Plasmamembran, die für Injektionen in flachen zellulären Regionen auftreten können.
Hinweis: Injektionsvolumina sollte auf ein Minimum gehalten werden (im Idealfall < 5 % des zellulären) und werden in der Regel im Bereich von Femtoliter. Benötigten Injektionsvolumina auch durch Veränderungen in der Konzentration gesteuert werden, aber beachten Sie, dass für Proteine, Konzentrationen > 2 mg/mL kann unpraktisch wegen häufigen Nadel verstopfen werden. Allerdings hängt dies auch auf die Qualität und das Verhalten des gereinigten Proteins; z.B.Injektion von eindringmittel gekoppelt Aktin wird erschwert durch Konzentration abhängig und unvermeidbare Polymerisation in der Spitze der Nadel, und so ist es heute selten ausgeführt (siehe kleine Et Al. ( 16).
- Vermeiden Sie stossen durch die oberen und unteren Plasmamembran, die für Injektionen in flachen zellulären Regionen auftreten können.
- Vor, während oder nach der Wirkung der Mikroinjektion FRAP oder photoaktivierungen kann durchgeführt werden in der gleichen Zelle (siehe Abschnitt 5 und 6).
(5) FRAP Verfahren
- Transfizieren Zelltyp des Interesses (hier B16-F1-Zellen) mit Plasmid DNA Kodierung eine fluoreszent-tagged Proteins des Interesses (hier eine EGFP-markierte Version des β-Aktin verwendet wurde). Samen der Zellen auf Laminin-beschichtete Deckgläsern (Schritt 2.10).
- Montieren Sie die bildgebende Kammer (Abschnitt 3).
- Verwenden Sie die folgenden Einstellungen für Lamellipodial Region Immunofluoreszenz: 65 mW Laserleistung (variabel je nach experimentellen Setup und Laser-Quelle); 10 Pixel Laser Strahldurchmesser; 1 ms bleichen wohnen Zeit/Pixel; 500 ms GFP Belichtungszeit; 1.500 ms Zeitintervall. Experimentelle Ergebnisse in diesem Papier wurden mit einer 100 X 1.4NA apochromatische Zielsetzung durchgeführt.
- Führen Sie Laser Kalibrierung um Genauigkeit in den Dimensionen der Photobleached Region zu gewährleisten. Stellen Sie vor der Kalibrierung das Sichtfeld an einem fehlt jede Zellen/Fluoreszenz-Signal und beobachten Sie das Bild auf dem Display zu.
- Wählen Sie die Objektive Vergrößerung durch Anklicken des jeweiligen Vergrößerung-Buttons und reduzieren die Laserleistung (3 bis 5 mW) in der "Panel | Intensität"-Menü. Um manuelle Kalibrierung auf Visiview Software (v2.1.4) zu starten, wählen Sie die "Configure | FRAP"Menü und klicken Sie auf den" Calibrate | Stellen Sie Handbuch"Menü. Stellen Sie sicher, dass der Laser als scharfen Punkt unterschieden werden kann. Wenn nicht, entweder neu auszurichten Sie oder passen Sie die Laser-Hardware.
- Führen Sie die Kalibrierung durch die manuell Führung des Lasers auf vorher festgelegten Software XY-Koordinaten. Dies weist der Software wie den Laser auf einen frei definierbaren Bereich für die aktuelle Vergrößerung gezielt.
- Vor Auslösung des Lasers, wechseln Sie zu der GFP und initiieren Sie Bild/Zeitraffer Erwerb zu.
- Manuell zeichnen Sie die Region um Photobleached auf dem Kanal von GFP werden während der Anzeige im Displays.
- Starten Sie Immunofluoreszenz durch eine manuelle Auslösung der 405 nm Laser, mindestens 3 – 4 Frames nach Einleitung der Bildaufnahme. Erwerb von Frames vor Immunofluoreszenz ist erforderlich für die Normalisierung des Bildes bei der späteren Datenanalyse.
(6) Photoaktivierungen Verfahren
Hinweis: Software, Mikroskop-Setup und Einstellungen, mit Ausnahme der Laserleistung ähneln denen für FRAP. In photoaktivierungen, ein wichtiger Unterschied im Vergleich zu FRAP, ist das ein 405nm-Laser macht deutlich niedriger als das beschäftigt für Immunofluoreszenz verwendet werden muss, um PA-GFP ohne gleichzeitig Immunofluoreszenz aktivieren, es.
- Co transfizieren Zelltyp des Interesses (B16-F1 hier Zellen; siehe Punkt 2.5) mit Plasmid DNA Kodierung, PA-GFP-Aktin und ein anderes fluoreszent-markierten Protein (z. B. mCherry oder mCherry-Lifeact).
Hinweis: In den meisten Fällen mCherry-positiven Zellen auch positiv für die PA-GFP-Aktin-Vektor, letzteres normalerweise nicht auf den GFP-Kanal vor der photoaktivierungen gesehen werden. Um die Chance zu erhöhen, dass die mCherry-positiven Zellen auch positiv für PA-GFP, verwenden Sie ein Verhältnis 1:2 Transfektion der mCherry:PA-GFP-Aktin. Nach diesem Protokoll mehr als 90 % der Zellen mit dem Ausdruck mCherry erfolgreichen Aktivierung des PA-GFP-Aktin angezeigt. - B16-F1 Samenzellen auf Laminin-beschichtete Deckgläsern (Schritt 2.10).
- Montieren Sie die bildgebende Kammer (Abschnitt 3).
- Vor der Einleitung die photoaktivierungen Experimente, bei Bedarf führen Sie die Laser-Kalibrierung für das ausgewählte Ziel (Schritt 5,4-5,6).
- Die GFP/488 nm Bildaufnahme auf 500 ms Belichtung und 1.500 ms Zeitintervall (abhängig von den Versuchsplan) einstellen
-
Passen Sie die Softwareeinstellungen für den Erwerb der Zweikanal- oder Dreikanal-Zeitraffer-Filme durch Kennzeichnung des Platzes "Wellenlänge-Serie" und wählen die gewünschte Anzahl der Kanäle in der "Acquire | Wellenlänge"-Menü. Es wird empfohlen, dass die Zeit Zeitraffer Filme mit Phasenkontrast und GFP Kanäle erworben werden.
- Optional auch gehören des mCherry-Kanals; Zellen mit zu viel Licht aussetzen könnte jedoch lichtbedingten induzieren. Dies könnte mit Sauerstofffänger wie Oxyrase17, vermieden werden, obwohl Zelle Kammer Abdichtung eine wirksame Behandlung erfordert.
- Die transfizierten Zellen auf dem mCherry-Kanal zu finden.
- Initiieren Sie vor Auslösung des Lasers Bild/Zeitraffer Erwerb zu und manuell zeichnen Sie die Region um photoaktiviert auf dem Kanal von Phase Kontrast zu sein, während der Anzeige im Displays.
- Photoaktivierungen durch eine manuelle Auslösung der 405 nm Laser zu initiieren (Intensität fest zwischen 5 – 15 mW aus der "Panel | "Intensität" Menü), mindestens 3 – 4 Frames nach Bild Erwerb Einleitung.
(7) Datenanalyse und Präsentation der Ergebnisse der FRAP
Hinweis: Die vorgestellte Methode dient zur Untersuchung des Umsatzes eines Proteins an Standorten der dynamischen Aktin Versammlung, in diesem Fall VASP, die Mitarbeiter mit Adhäsion Websites und die Spitzen der überstehenden Lamellipodia ansammeln. Wir analysieren ihren Umsatz an der Spitze der Lamellipodium, aber die gleichen Prinzipien der Analyse können für die Untersuchung des Umsatzes der VASP oder jedes andere Protein und andere subzelluläre Kompartimente angewendet werden.
- Öffnen Sie die Zeitraffer-Filme von Visiview auf die Metamorph-Software abgeleitet. In diesem Artikel wurde Metamorph v7.8.10 verwendet.
- Leiten Sie Intensitätswerte für Photobleached Regionen manuell umreißen Regionen auf Metamorph ab. Eine Form an der Spitze der Lamellipodium, die deckt das gesamte oder einen Teil des Gebiets Photobleached, und seine Position auf unmittelbar aufeinander folgenden Einstellungen manuell anpassen, bei Bedarf (d.h., wenn der Rand hervorstehende ist), in bestellen zum Nachverfolgen von Änderungen in Lamellipodial Intensitäten der Auslosung die jeweilige Komponente während Tipp Verschiebung.
- Zur Korrektur von Hintergrund und Immunofluoreszenz Erwerb analysieren Sie Regionen innerhalb und außerhalb der Zelle. Sehen Sie Abbildung 2eine für repräsentativen Regionen der gemessenen Intensitäten.
- Während ein ROI ausgewählt ist, extrahieren Sie die Intensitätswerte auf Metamorph mithilfe des Menüs "Maßnahme | Region-Messungen". Stellen Sie sicher, dass "Verstrichene Zeit" und "Mittlere Intensität" Optionen im Menü "Konfigurieren" ausgewählt werden. Klicken Sie auf "Öffnen" und wählen Sie "Dynamic Data Exchange". Klicken Sie auf "OK", um eine Excel-Tabelle zu öffnen und klicken Sie auf die "Open Log"-Taste erneut, um die Metamorph-Werte in Excel einfügen.
Hinweis: Diese Werte werden verwendet, um die Fluoreszenz-Erholung-Kurven zu erzeugen. - Gelten Sie für die Erzeugung von Fluoreszenz Erholung Kurven an der Spitze der Lamellipodium des Photobleached Regionen (normiert auf die Intensität der Region vor Immunofluoreszenz) die folgende Gleichung:
 Gleichung 1
Gleichung 1
wo: FRAPTn ist die Photobleached Region Intensität für jeden Frame von Interesse nach Immunofluoreszenz; AusTn ist eine Region Intensität genommen außerhalb der Zelle (Hintergrund) für jeden Frame von Interesse Immunofluoreszenz folgen; InsTn ist zwei durchschnittlich innerhalb Region Intensitäten für jeden Frame von Interesse nach Immunofluoreszenz (verwendet für Erwerb Immunofluoreszenz im Laufe der Zeit normalisieren); FRAPt-1 ist die Photobleached Region Intensität vor Immunofluoreszenz; T-1 ist eine Region Intensität außerhalb der Zelle (im Hintergrund) vor Immunofluoreszenz genommen; und Inst-1 ist zwei innen Region Intensitäten für jeden Frame von Interesse vor Immunofluoreszenz gemittelt. - Verwenden Sie für jeden Zeitrahmen von Interesse Gleichung 1 eine Fluoreszenz-Erholung-Kurve mit allen Zeitrahmen untersucht werden. Die Länge der Zeit richtet sich strikt auf das Protein untersucht. Wenn unbekannt, führen Sie Vorversuchen die Fluktuationsrate des Proteins zu erwerben.
- Für die Berechnung der halbe Zeit der Erholung, fügen Sie die Werte der Fluoreszenz-Erholung-Kurve mit der entsprechenden Zeit (in Sekunden) in einem Sigma Plot (v. 12), und führen Sie eine Kurvenanpassung mit der "Dynamic Fit Assistent | Exponentiellen Anstieg auf Maximum"-Tool. Wählen Sie Mono-exponentielle (Einzelzimmer, 3 Parameter) oder Bi-exponentielle (Double, 4 Parameter) Funktionen, je nachdem die beste Kurvenanpassung.
- Verwenden Sie die folgende Formel für Mono-exponential-Funktion:
 Gleichung 2
Gleichung 2
- Verwenden Sie die folgende Formel für Bi-exponential-Funktion:
 Gleichung 3
Gleichung 3
- Fügen Sie Parameter "b" und "d" in Excel zu berechnen, die halbe Zeit der Erholung Sigma Plot (Gleichung 2 oder Gleichung 3) abgeleitet. Es gelten Sie die folgenden Gleichungen:
 Gleichung 4
Gleichung 4
oder Gleichung 5
Gleichung 5
- Wenn die Mono-Exponentialfunktion ergibt sich eine genaue Kurve passen, gelten Sie nur Gleichung 4.
- Bei Mono-exponential-Funktion eine gute Kurve passen nicht dazu führt, die Bi-exponentielle Formel Anwenden von Gleichung 4 und 5 der Gleichungzu lösen. Betrachten die resultierenden Zweieinhalbfachen der Erholung als Vertreter von zwei verschiedenen Proteinfraktionen: eine schnell und langsam den Austausch von Sekundenbruchteilen bzw..
8. Festsetzung des Lamellipodial Actin Polymerisierung von FRAP
- Zur Bestimmung der Lamellipodial-Aktin-Polymerisation-Rate, transfizieren B16-F1 Zellen mit EGFP getaggt β-Aktin, und Photobleach der Lamellipodial Region (Schritt 5.9) mit 1,5 s Zeitintervall und 500 ms GFP Exposition.
- Öffnen Sie in Metamorph, Zeitraffer-Filme von Visiview erworben und Kalibrieren der Pixel/µm-Verhältnis entsprechend der Zielsetzung von verwendet die "Maßnahme | Entfernungen zu kalibrieren"-Tool.
- Spielen Sie Zeitraffer-Film ab und auf dem Rahmen zu stoppen Sie, wenn die Lamellipodial Fluoreszenz Erholung, nach hinten in Richtung der Lamelle als Linie mündet die Lamelle erreicht hat und keine weiteren rückwärtigen Ablauf verfolgt werden kann.
- Messen Sie den Abstand in µm zwischen der Spitze der Lamellipodium und der Rückseite des wiederhergestellten Fluoreszenz. Dieser Abstand entspricht der Summe der retrograde Strömung und Vorwölbung Entfernungen.
- Alternativ, um den Vorsprung von retrograde Strömung zu trennen, markieren Sie die Lamellipodial Spitze einer Linie übereinander vor der Immunofluoreszenz. Nutzung der Linie als Referenz Punkt in nachfolgenden Frames verweisen auf die ursprüngliche Position der Lamellipodium Spitze zum Zeitpunkt der Immunofluoreszenz; der Bezugspunkt kann verwendet werden, zur Messung der Vorwölbung Abstand und retrograde Strömung.
- Notieren Sie die Zeit (in Sekunden) für die Fluoreszenz-Wiederherstellung nach Immunofluoreszenz auftreten erforderlich. Die Zeit kann von der Framerate manuell berechnet oder visualisiert durch Metamorph durch die "Maßnahme | Messungen der Region"-Tool.
- Leiten Sie die Aktin-Polymerisation-Rate mit der folgenden Gleichung (Gleichungsparameter basierend auf Metamorph Messungen von Schritten 8,4 und 8,6 ab):
 Gleichung 6
Gleichung 6
wo Aktin-Polymerisation-Rate ist in µm/min, retrograde Strömung Abstand ist in µm Lamellipodial Vorsprung Abstand ist in µm und Zeit wird in Sekunden angegeben.
9. Analyse von Protein-Diffusion und Mobilität auf Photoaktivierungen
Hinweis: Die hier vorgestellte Methode beschreibt die Analyse der Aktin-Monomer-Mobilität durch den Einsatz von photoaktivierungen von Aktin verschmolzen zu PA-GFP, wie durch Visualisierung und Quantifizierung von Proteinen Diffusion durch die Zellflüssigkeit veranschaulicht.
- Für die Messung der Diffusion von Photoactivatable Aktin Weg von einer cytosolischen Region sowie die Anhäufung innerhalb einer Lamellipodial Region, Metamorph verwenden, um die Intensität im Laufe der Zeit in folgenden Regionen zu bestimmen (dargestellt in Abbildung 3ein ): ein cytosolischen photoaktiviert Region (PA); ein Lamellipodial-Region, in welcher, die photoaktiviert Proteine voraussichtlich reichern sich im Laufe der Zeit (Lam); eine Region außerhalb der Zelle zur Normalisierung der Hintergrundfluoreszenz (raus).
- Bei der Bestimmung der Mobilität von Aktin innerhalb der Zellflüssigkeit Messen Sie verschiedene cytosolische Regionen (siehe Abbildung 3ein, Regionen R1-R5). Beachten Sie, dass Immunofluoreszenz-Erwerb in ähnlicher Weise, FRAP, Zunahme der focal- und schließlich Zelle breit Fluoreszenz bei Aktivierung nicht bestimmt werden kann.
- Übertragen Sie die Intensitätswerte für alle Regionen von Metamorph in eine Excel-Tabelle wie in Schritt 7.4 beschrieben.
- Für die Prüfung der Rate der Verschiebung der Photoactivatable Aktin Weg von der cytosolischen Region von photoaktivierungen oder seine Rate der Gründung innerhalb einer Lamellipodial Region (beide vertreten als prozentuale Intensität der cytosolischen photoaktiviert Region zum Zeitpunkt 0) , Fluoreszenz-Kurven aus den Daten im Schritt 9.3 zu generieren. Es gelten Sie die folgenden Gleichungen:
 Gleichung 7
Gleichung 7 Gleichung 8
Gleichung 8
wo: PATn ist die Intensität einer cytosolischen photoaktiviert Region für jeden Frame von Interesse nach photoaktivierungen; LAMTn ist die Intensität einer Lamellipodial Region für jeden Frame von Interesse nach photoaktivierungen; AUSTn ist die Intensität einer Region außerhalb der Zelle (Hintergrund) für jeden Frame von Interesse photoaktivierungen folgen; PA-t-1 ist die Intensität einer cytosolischen photoaktiviert Region vor photoaktivierungen; LAM-t-1 ist die Intensität einer Lamellipodial Region vor photoaktivierungen; T-1 ist die Intensität einer Region außerhalb der Zelle (im Hintergrund) vor photoaktivierungen; PA-T0 ist die Intensität einer cytosolischen photoaktiviert Region zum Zeitpunkt 0 (d. h. ersten Frame nach photoaktivierungen); undT0 ist die Intensität einer Region außerhalb der Zelle (Hintergrund) zum Zeitpunkt 0 (d. h. ersten Frame nach photoaktivierungen). - Optional zur besseren Visualisierung der Daten, Normalisieren Sie die Intensität zu 0 Kurven durch Subtraktion der Intensität des ersten Frames nach photoaktivierungen von jeder nachfolgenden Frame.
Hinweis: Die folgende Analysemethode (Schritte 9,6 – 9,8) kann auch Berechnung der cytosolischen Streuung der photoaktiviert Aktin innerhalb der Zellflüssigkeit. - Messen Sie die Intensitäten für cytosolischen Regionen, die nacheinander nach distal aus der Aktivierung Region positioniert sind.
- Um die Intensitäten dieser Regionen zum Zeitpunkt 0 Prozent Intensität der photoaktiviert Region darstellen, gelten Sie Gleichung 8, wo Lamellipodial Intensitäten mit Intensitäten für jedes cytosolischen ROI ersetzt werden. Die Größe und Anzahl der Regionen variieren je nach Zelle Größe und Ausbreitung Entfernung gemessen werden.
- Um einen messbaren Wert für die Rate des photoaktiviert Proteins infiltrieren jeder cytosolischen Region abzuleiten, fügen Sie die Zeit und die Werte der Kurve der Fluoreszenz Intensität erhöhen für jede Region in Sigma Plot (ähnlich der FRAP-Analyse in Abschnitt 7 ein), verwenden Sie Gleichung 2 und 4 Gleichung abzuleiten die Halbzeit der Fluoreszenzintensität ein Plateau zu erreichen. Vergleichen Sie die t1/2 Werte zwischen verschiedenen Versuchsgruppen.
Ergebnisse
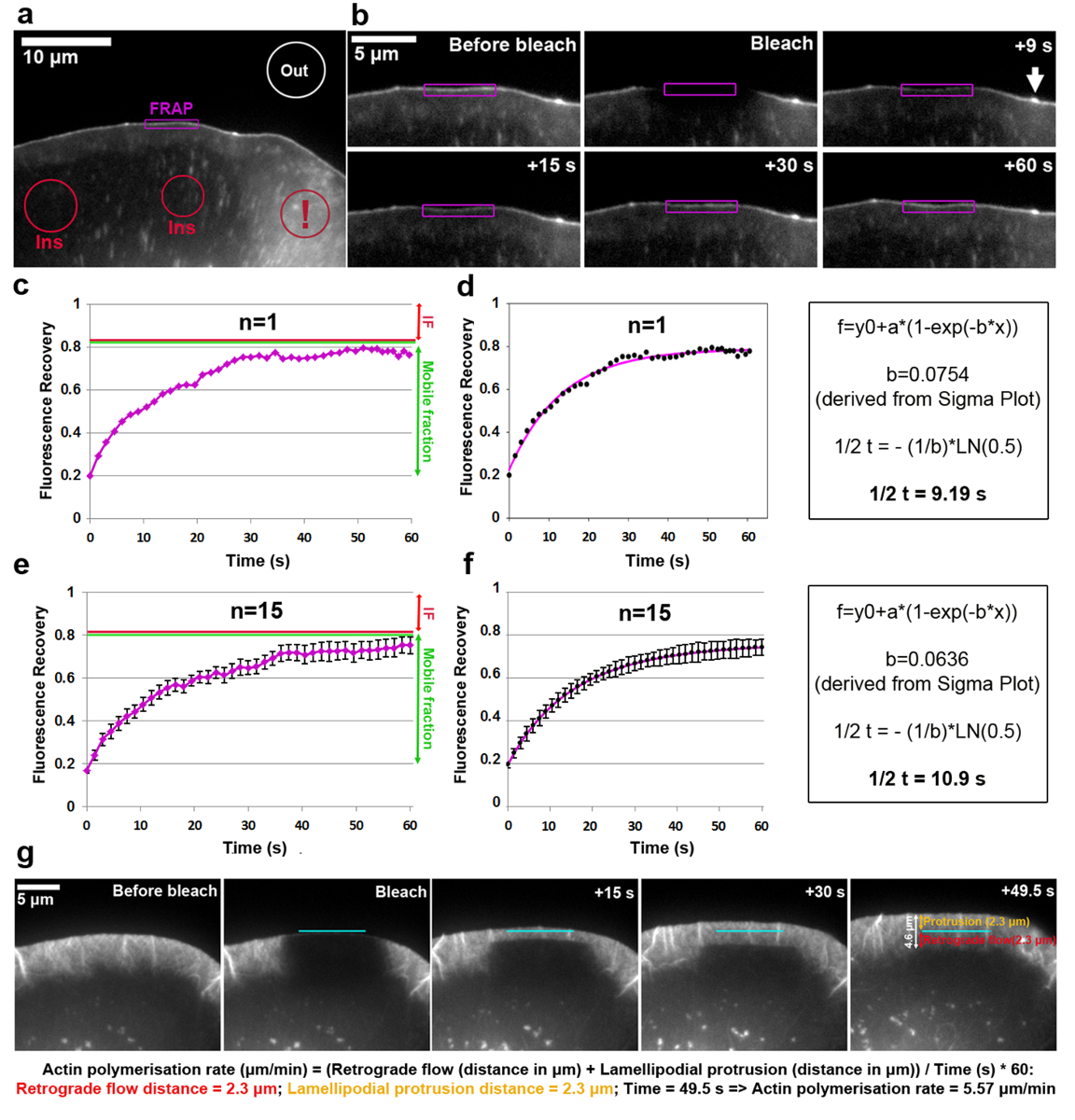
Abbildung 1 g h zeigen Phase kontrastreiche Bilder einer NIH3T3 Fibroblasten-Zelle vor und 10 min nach Mikroinjektion von Rac1, die ein kleines Rho-Familie GTPase induzieren Lamellipodia Bildung durch seine Interaktion mit der Welle Komplex fähig ist. Die Zelle wird zuerst vor der Mikroinjektion (Abbildung 1g), visualisiert, um seine Lebensfähigkeit und Morphologie, z. B. Mangel an Lamellipodia zu bestätigen. In 10 min nach Mikroinjektion hat die Zelle deutlich seine Morphologie verändert, dürfte von dieser Behandlung, und zeigt eine erfolgreiche Injektion (Abbildung 1h).
Für Einfachheit und Klarheit bieten wir als nächstes Exemplarische Ergebnisse für FRAP und photoaktivierungen Analyse in Zellen, die nicht zusätzlich mikroinjiziert worden.
Analyse des Umsatzes der VASP EGFP markiert, an der Spitze Lamellipodium zeigt Abbildung 2a-f. Beachten Sie, dass VASP richtet sich darüber hinaus, im Entstehen begriffenen und fokale Adhäsionen, klein und länglich Punkte in die Zelle innen18,19. Die Fluoreszenzintensität einer Lamellipodial Region mit einer klaren VASP Ansammlung an der Spitze war gebleicht und für jeden Zeitrahmen durch Anschluss an die Kontur des ROI vor, während und nach dem Bleichen wie die Lamellipodium nach vorne ragt gemessen. Als gebleichte EGFP-VASP Proteine durch nicht gebleicht Moleküle an diesen Standorten recycelt werden, wird allmähliche Erholung der Fluoreszenz (Abbildung 2b) beobachtet. Die FRAP Erholung Kurve erhalten auf diese Weise und normiert auf die Pre-Bleichmittel Intensität (ausgedrückt als 1) ist in Abbildung 2cersichtlich. Immunofluoreszenz Effizienz kann variieren und etwa 20 % des Wertes vor dem Bleichen in diesem Beispiel, wie aus der Value-at-t0 ermittelt wurde (das erste Bild nach Immunofluoreszenz). Die Zunahme der Fluoreszenz erreicht ein Plateau im Beispiel bei ca. 80 % der Fluoreszenz vor dem Bleichen. In einer statischen Struktur während der zeitliche Verlauf des Experiments, wie z. B. eine fokale Adhäsion nach Wiederherstellung der immobile Bruch (IF, roter Pfeil in Abbildung 2 definiert ist der Unterschied zwischen Pre-Bleichmittel Intensität und die Plateau-Fluoreszenz erreicht c, e), während die Anzahl der Fluoreszenz zwischen dem Zeitpunkt der Bleichen und vollständige Genesung wiederhergestellt wird als mobile Bruch (grünen Doppelpfeil in Abbildung 2C, e) definiert. Beachten Sie, dass in einer dynamisch verändernden Struktur wie die Lamellipodium Tipp hier analysiert, das Ausmaß der IF könnte nicht nur unbeweglich Moleküle, sondern auch eine Reduzierung des Vorsprungs Geschwindigkeit ableiten bekanntlich EGFP-VASP Intensität abhängig Parameter-18. Um die halbe Zeit der Erholung zu berechnen, entstand eine Fit Kurve auf Sigma Plot (Abbildung 2d). In diesem Fall der Wert des Parameters "b" extrahiert von der Lösung der Gleichung 2 ist gleich auf 0.0754, angewendet, bei der logarithmischen Funktion (Gleichung 4) Ergebnisse in einem geschätzten Hälfte-Zeit der Erholung der 9.19 s (Abbildung 2d , ganz rechts Panel), die relativ schnell in dieser bestimmten Zelle im Vergleich zum Durchschnitt ist vorher5veröffentlicht. Beachten ist, dass die Hälfte Wiederherstellungszeiten von Zelle zu Zelle innerhalb der gleichen Bevölkerung manchmal erheblich variieren können. Für repräsentative Ergebnisse zu erhalten, empfehlen wir daher, Bestimmung dieses Parameters als Mittelwert aus mindestens 15-20 Zellen. Zur Veranschaulichung der Grad der Abweichung, arithmetische Mittel der EGFP-VASP Erholung im Durchschnitt aus 15 Zellen für jeden Zeitpunkt waren erzeugt (Abb. 2e) und durchschnittliche Kurve passt erstellt und angezeigt auf analoge Weise (Abbildung 2 ( f).
Die Polymerisation bei der Lamellipodial-Aktin-Netzwerk umfasst die Summe vorwärts Netzwerk Vorsprung und retrograde Strömung. FRAP kann zur Messung der Aktin-Polymerisation-Rate von transfecting Zellen (in diesem Fall B16-F1) mit EGFP getaggt β-Aktin und Immunofluoreszenz einer hervorstehenden Lamellipodial Region (Abbildung 2g) angewendet werden. Für die Analyse der Lamellipodial Aktin Netzwerk Polymerisation bemisst sich die Fluoreszenz-Erholung beim Bleichen von EGFP getaggt β-Aktin im Laufe der Zeit. Als die Polymerisation von Actin Monomeren fortschreitet an den Stacheldraht Enden der Lamellipodial actinfilamente (die alle in Richtung der vorderen20hinweisen), ist das Netzwerk ständig umgesiedelt nach hinten und nach vorn, voran die Preise von denen problemlos durch polarisierte Erholung der Fluoreszenz auf Immunofluoreszenz erhalten. Fluoreszenz-Erholung von der Lamellipodium ist abgeschlossen, sobald die gebleichte Zone erreicht hat, die Übergangszone zwischen der hintere Teil der Lamellipodium und der Lamelle, charakterisiert durch eine geringere Dichte mehr horizontal angeordnet Filament-Bundles sehr viel langsamer als die Beobachtungen in den Lamellipodium umdrehen. Wie in Abbildung 2gdargestellt, kann Fluoreszenz Erholung visualisiert werden, als eine Linie horizontal auf den Rand und fließt nach hinten in Richtung der Lamelle, die ermöglicht die Messung der Entfernungen von Vorsprung und retrograde Strömung (einzeln dargestellt im rechten Bereich von Abbildung 2g als orange und rote Doppelpfeile, beziehungsweise).
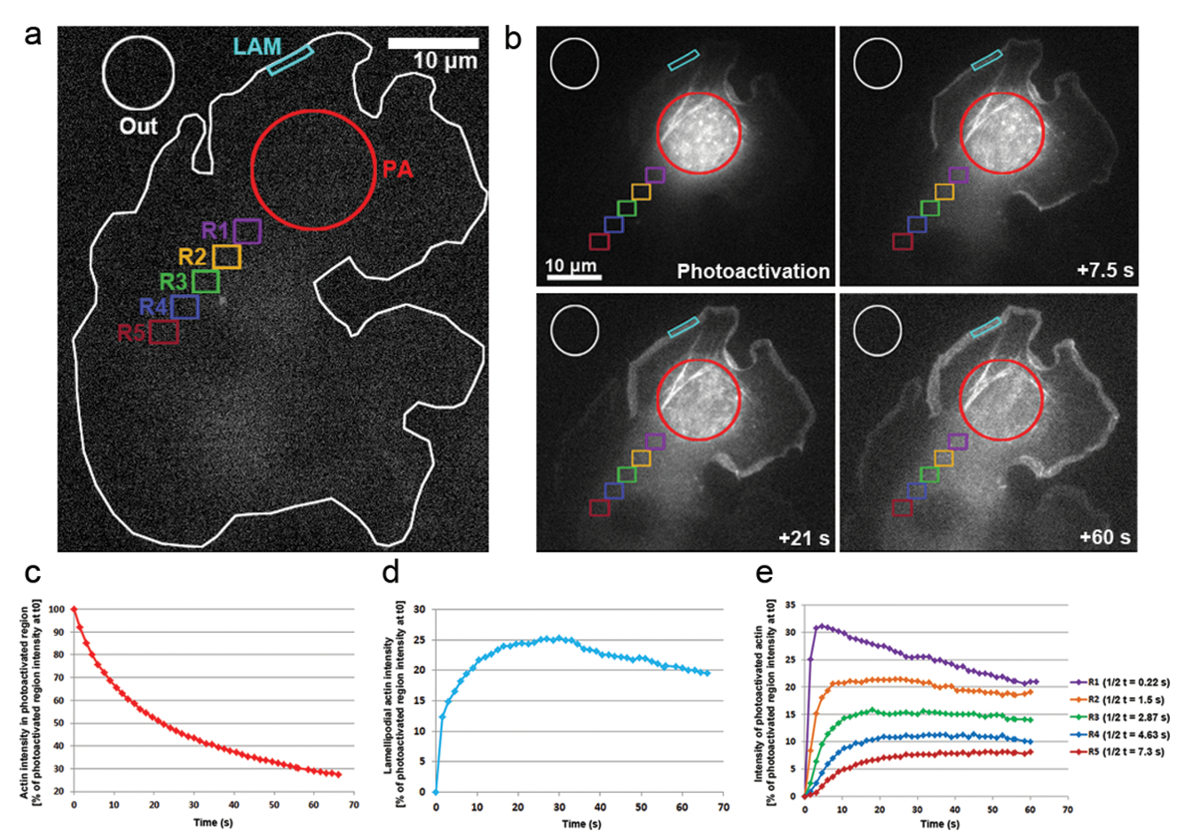
Wir haben auch photoaktivierungen in B16-F1 Zellen mit PA-GFP-Aktin, die Mobilität der Actin-Monomere innerhalb der Zellflüssigkeit und die bei ihrer Aufnahme innerhalb von hervorstehenden Lamellipodia verfolgen transfiziert angewandt. Wie in Abbildung 3a, bdargestellt, eine cytosolische Region war photoaktiviert durch die Einwirkung von 405 nm Laser, während Bilder auf die GFP erworben wurden Kanal jeder 1,5 s für die Verteilung der GFP-Tag, photoaktiviert Aktin zu visualisieren. Photoactivated GLP-Aktin ersichtlich aus der cytosolischen Region in Abbildung 3bdiffundieren. Die Rate der Fluoreszenz Intensität Rückgang der cytosolischen photoaktiviert-Region wird als prozentualer Anteil der Anfangsintensität T0 dargestellt (zuerst Rahmen nach photoaktivierungen; Abbildung 3 ( c). Photoaktiviert Aktin integriert auch an den Spitzen der Lamellipodia, wo neue Actin-Monomere, um den wachsenden Stacheldraht enden hinzugefügt werden der actinfilamente während Vorsprung verlängern. Um die Rate der Lamellipodial Einbindung abschätzen zu können, haben wir im Laufe der Zeit einer zweidimensionalen Kontur/Region von etwa 5 µm Breite und 1 µm in Höhe der Fluoreszenzintensität gemessen; die Region war ständig neu positioniert an der Spitze der Lamellipodium als es ragte. Aktin-Aufnahme wurde als Prozentsatz der Fluoreszenzintensität der cytosolischen Region T0 photoaktiviert vertreten (Abbildung 3-d). Mit fortschreitender Dehnung der actinfilamente flossen neue Actin-Monomere Lamellipodial vorne. Ein Bruchteil dieser Actin-Monomere wurde der cytosolischen Pool stochastisch abgeleitet wo Monomere photoaktiviert waren. Dadurch wird die rasche Zunahme der Fluoreszenz in Lamellipodia in den ersten 20 s nach photoaktivierungen. Als neue Monomere zu Lamellipodial vorne, zuvor eingebaute Actin Monomeren Strömung mit Fäden in Richtung der Lamelle durch retrograde Strömung hinzugefügt werden. Im Laufe der Zeit der ROI ist komplett gefüllt mit fluoreszierenden Monomeren und ein Plateau in Fluoreszenz ist erreicht (Abbildung 3-d). Ein allmählicher Rückgang der Fluoreszenz ist dann beobachtet, wenn Diffusion von photoaktiviert Monomere in der Zelle nach werden nicht photoaktiviert-Aktin-Monomere zunehmend Lamellipodial vorne neu hinzugefügt. Diese Abnahme der Fluoreszenz finden ein neues Plateau erreicht wird, sobald ein Gleichgewicht in der gesamten Zelle zwischen photoaktiviert und nicht photoaktiviert Monomere (Daten nicht gezeigt) erreicht ist.
Die Mobilität der Actin-Monomere in der Zellflüssigkeit wurde durch die Messung der Fluoreszenz-Intensitäten in Regionen gleicher Größe positioniert nach distal aus der photoaktiviert Region (wie auf Abbildung 3eine farbcodierte Regionen mit der Bezeichnung abgeleitet R1-R5). Wie in Abbildung 3edargestellt, Fluoreszenzintensität in jeder dieser Regionen sinkt schrittweise Weg von der cytosolically photoaktiviert Region, als der Anteil der photoaktiviert-Aktin-Monomere wird zunehmend verdünnt mit nicht aktivierte (d. h. nicht-fluoreszierende) Monomeren. Darüber hinaus ist der Höhepunkt der Fluoreszenz später erreicht: je weiter entfernt die gemessenen Region liegt aus der photoaktiviert Region, je länger die Zeit, die erforderlich für Actin-Monomere in diesen Regionen verbreiten. Ein repräsentativer Wert für den Grad der Aktin-Monomer-Infiltration in jeder Region kann abgeleitet werden, durch Quantifizierung der Hälfte der Zeit die Fluoreszenz-Plateau zu erreichen. Je weiter entfernt der Region, desto länger dauert es für die photoaktiviert-Aktin hinein diffundieren und somit mehr Zeit wird benötigt für das fluoreszierende Plateau erreicht werden, letztlich zu einem höheren t1/2 Wert (Abbildung 3e).

Abbildung 1 : Imaging-Kammer Montage- und Mikroinjektion Verfahren. (ein) Imaging-Kammer-Komponenten. (b) Silikon Fett ist sorgfältig um die Öffnung der Kunststoff Versiegelung verschmiert. (c) das Deckglas befindet sich mit der Zelle-Seite nach oben in die Mitte der bildgebenden Kammer öffnen. (d) eine sichere Abdichtung ist durch die Positionierung der Kunststoff Versiegelung auf dem Deckglas und durch Anziehen der Spannpratzen etabliert. (e) Mikroskopie Medium ist in den Schlitz der Kammer pipettiert. (f) der bildgebenden Kammer befindet sich auf dem Mikroskoptisch, Wärmemelder und Elektroden sind mit einer Heizeinheit auf 37 ° C voreingestellt verknüpft und Zellen dürfen für mindestens 30 min anzupassen, bevor Mikroskopie ausgelöst wird. In diesem Beispiel Mikroskoptisch ist ausgestattet mit einem Mikromanipulator für die Durchführung von Mikroinjektionen und die Mikroinjektion Nadel wird in das Medium der Zelle Deckschicht in der bildgebenden Kammer eingetaucht. (g) An NIH3T3 Fibroblasten-Zelle wird durch Phasenkontrast-Mikroskopie vor Mikroinjektion visualisiert. Das Rote Kreuz in das Fach perinukleäre gibt den Speicherort der die zukünftigen Mikroinjektion, das entspricht einer hohen zytoplasmatischen Region aufgrund der Nähe zu den sperrigen Kern. (h) 10 min nach Mikroinjektion mit Rac1, reagiert die Zelle durch prominente Bildung von Lamellipodia rund um die gesamte Zelle Peripherie (gekennzeichnet durch grüne Pfeile). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: FRAP ermöglicht die Bestimmung von Protein-Umsatz oder Lamellipodial-Aktin-Polymerisation Tarife. (einem) Vertreter Beispiel B16-F1-Zelle mit dem Ausdruck EGFP-VASP vor Immunofluoreszenz einer Lamellipodial Region, wie angegeben. Verschiedenfarbige Konturen/Formen sind beschriftet, um anzugeben, welche Regionen für Fluoreszenz-Intensität-Messungen im Laufe der Zeit galten. Hinweis Die rote Kontur mit einem Ausrufezeichen versehen, die eine cytosolische Region befindet sich in einer Gegend mit mehreren Vesikel und Zelle Oberfläche Rüschen Etiketten gekennzeichnet. Dynamische Bereiche wie diese sollte für die Auswahl von Regionen der Fluoreszenz Referenz vermieden werden, da sie von starken kurzfristigen Schwankungen der Fluoreszenz, was möglicherweise zu ungenauen Ergebnissen gekennzeichnet sind. (b) Lamellipodial Region der EGFP-VASP mit dem Ausdruck ihrer Zelle vor und nach Immunofluoreszenz. Die Erholung der Fluoreszenzsignal nach Immunofluoreszenz innerhalb der Region in lila markiert ist über einen Zeitraum visualisiert. Pfeil zeigt die Spitze des einen Microspike angereichert für VASP wahrscheinlich aufgrund der hohen Dichte der actinfilamente polymerisierenden dort19. (c) ein Beispiel für ein FRAP Erholung Kurve als abgeleitet Quantifizierung der fluoreszenten Intensität der Photobleached Lamellipodium (violette Kontur) in b rot und grüne Linien auf der rechten Seite zeigen bzw. unbeweglich und mobile Brüche. (d) A passt der FRAP Erholung Kurve in c (links) und ein Beispiel für die Berechnungsmethode verwendet, um die Hälfte Erholungszeit (Rechte Abbildung) abzuleiten. (e) ein Beispiel für eine Erholung der FRAP-Kurve abgeleitet von durchschnittlich die Fluoreszenz Erholung Kurven 15 Zellen mit SEM-Bars, den Grad der Variabilität innerhalb der Probe-Population. (f) eine Kurve Passform abgeleitet von durchschnittlich FRAP Erholung Kurve Fits 15 Zellen (links) und ein Beispiel für die Berechnungsmethode verwendet, um die Hälfte Erholungszeit (Rechte Abbildung) abzuleiten. (g) Zeitraffer Platten der überstehenden Lamellipodium einer B16-F1-Zelle mit dem Ausdruck EGFP getaggt β-Aktin, vor und nach dem Bleichen von einer Lamellipodial Region wie angegeben, durch Fluoreszenz-Erholung in der Lamellipodium im Laufe der Zeit folgten. Auf dem rechten Panel Messwerte Protrusion und retrograde Strecken stehen zur Verfügung (in Orange und rot, beziehungsweise). Berechnungen unter der Bild-Tafeln zeigen, wie die Summe der Vorwölbung und retrograde Entfernungen werden verwendet, um die Polymerisation Rate von Lamellipodial Aktin Netzwerk ableiten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: Photoaktivierungen PA-GFP-Aktin für Monomer tracking in die Zelle. (ein) A repräsentatives Beispiel einer B16-F1-Zelle mit dem Ausdruck PA-GFP-Aktin vor photoaktivierungen in einer cytosolischen Region auslösen, wie durch den roten Kreis (PA) angegeben. Verschiedenfarbige Konturen sind beschriftet, um anzugeben, welche Regionen für Fluoreszenz-Intensität-Messungen im Laufe der Zeit galten. (b) eine Darstellung der zeitlichen Verteilung der PA-GFP-Aktin Folgendes photoaktivierungen. Beachten Sie die schrittweise Verringerung der Fluoreszenz in der photoaktiviert, cytosolischen Region (roter Kreis), wie die photoaktiviert-Aktin davon diffundiert. Aufgrund ihrer Verbreitung auf der Vorder- und Montage in das Netzwerk sind Aktin-Monomere photoaktiviert allmählich in Lamellipodia (Cyan Region) und in der Zellflüssigkeit (verschiedene farblich gekennzeichneten Regionen) auf Distanz und Zeit-abhängige Weise verbessert. (c) Vertreter, zeitliche Abnahme der Fluoreszenz in der photoaktiviert cytosolischen Region (rote Kontur in b). (d) zeitliche Änderungen in Fluoreszenzintensität in der Lamellipodial Region (Cyan Kontur in b). (e) Kurven repräsentativ für die zeitlichen Veränderungen in der Fluoreszenzintensität der cytosolischen Regionen (farbcodiert in b) aufgrund der Positionierung in Variablen Abständen aus dem Bereich der photoaktivierungen. Beachten Sie wie zweieinhalbmal erreichen die Fluoreszenz plateau (angegeben in der Legende auf der rechten Seite) erhöhen mit dem Abstand der gegebenen Region auf dem Gebiet der photoaktivierungen, wahrscheinlich korrelieren mit der zunehmenden Zeiten für Diffusion von Actin-Monomere in der jeweiligen Region. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Diskussion
Hier diskutieren wir wichtige Schritte in die Techniken beschrieben in diesem Artikel, und wie sie für den Einsatz in verschiedenen experimentellen Bedingungen optimiert werden können.
Mikroinjektion ist eine Methode, die angewendet werden kann, um in den Zellen die sofortige Effekte von der Einführung exogene Proteine, Inhibitoren oder Drogen zu überwachen. Es ist besonders vorteilhaft für die Bestimmung der Funktionen der Proteine in schwierig zu transfizieren Zelltypen oder in Situationen, in denen langfristige Ausdruck nicht erwünscht ist. Anzumerken ist, dass die extrazelluläre Matrix Überleben von bestimmten Zelltypen abhängig, die sie auf ausgesät werden. Die Endothelzellen, epithelialen oder Fibroblasten-Like Zelltypen, wie auch kleinen Fisch keratozyten (siehe Dang Et al. 21 und Anderson und Cross22) können erfolgreich injiziert werden. Es gibt jedoch Ausnahmen, wie z. B. B16-F1 Zellen ausgesät auf Laminin, die bilden ein hervorragendes Modellsystem von Zellwanderung, aber sind nicht kompatibel mit Einspritzung auf diese Art von Substrat aus unbekannten Gründen. Für NIH3T3 Fibroblasten-Zellen führen wir regelmäßig Injektionen auf Fibronektin Substrat und zusätzliche Photomanipulation Techniken wie z. B. FRAP (sogar mit photoaktivierungen; für B16-F1 Zellen hier gezeigten) gleich gut in diese Fibroblasten (siehe durchgeführt werden können z. B. Köstler Et al. 3). es muss berücksichtigt werden, dass verschiedene Proteine nach ihrer funktionellen Eigenschaften und das Experiment Ziele unterschiedlich viel Zeit, Veränderungen, variierend von Sekunden bis Stunden dauern können. Ein Vorteil des Verfahrens ist, dass die Dosis/Konzentration von exogenen Agent genauer auf einzelne Zelle als z.B.gesteuert werden kann, wenn Plasmid Transfektion verwenden. Darüber hinaus ist die fluoreszierende Markierung eines Proteins keine Notwendigkeit, um seine Präsenz in der Zelle, garantieren die Flexibilität erhöhen können, wenn eine gleichzeitige Multi-Channel-Visualisierung von anderen Proteinen fluoreszent markiert erforderlich ist. Mikroinjektion ist besonders hilfreich für die Analyse von sofortiger Auswirkungen von bestimmten Proteinen oder Protein-Mischungen auf dynamische Veränderungen der Zellmorphologie oder das Zellskelett (z. B. Dang Et Al. 21 : ein Beispiel für sofortige Auswirkungen auf Migration von der Arp2/3 komplexe Inhibitor Arpin). Ein Nachteil der Technik ist die Invasivität, die Zellschäden verursachen oder Zellmorphologie beeinflussen kann. Daher ist ein wichtiger Aspekt bei der Durchführung von Mikroinjektionen die Zellviabilität Überwachung. Die hier vorgestellte Methode stützt sich auf manuelle Manipulation. Unter Bedingungen getestet, um kompatibel mit erfolgreichen Injektionen, wie z. B. Fibroblasten auf Fibronektin Substrat wachsen ermöglicht hier beschriebene manuellen Injektion-Protokoll eine in der Nähe von 100 % Erfolgsquote; Dies ist unerlässlich, wenn dieser Ansatz kombiniert mit anspruchsvoll und zeitaufwändig Follow-up-Experimente, einschließlich video-Mikroskopie oder FRAP, wie zuvor3veröffentlicht. Dies schließt nicht aus, dass gelegentlich, einzelne Zellen könnte leiden unter einer Mikroinjektion-Veranstaltung, die durch abrupte Kontrast zwischen dem Kern und Zytoplasma, gefolgt von Zelle Rand einfahren sicher erkannt werden kann. Solche seltenen experimentellen Fällen sind ausgeschlossen und somit nicht für weitere Analysen berücksichtigt.
Jedoch ein halbautomatische Ansatz ist auch allgemein verwendet, zum Beispiel mit rapid (< 300 ms) Computer-gesteuerte Nadel absenken deckungsgleich mit Einspritzung Druckanstieg, so dass die Nadel nur über jede Zelle vor der jeweiligen positioniert werden Injektion. Die Erfolgsquote der halbautomatische Injektionen ist definitionsgemäß geringer als das manuelle Methode, die oben beschrieben, einfach weil es für Geschwindigkeit optimiert ist, gefolgt von Analyse mehrerer Zellen, die erfolgreich diese Behandlung überlebt; damit verlassen es nicht auf erfolgreiche Injektion einer einzelnen Zelle. Daher sind im Gegensatz zu einzelnen Zellanalyse, halbautomatische Injektionen besser geeignet für die Analyse der Injektion Effekte aus mehreren hundert Zellen, z. B. durch Videomikroskopie bei kleiner Vergrößerung oder Zelle Fixierung und Färbung. Unabhängig von der detaillierten Ansatz beschäftigt Mikroinjektion stellt keinen Endpunkt-Assay dar, sondern ist kombinierbar mit einer Vielzahl von Techniken, einschließlich FRAP oder photoaktivierungen3.
Bei der Festlegung die Protein-Fluktuationsrate von FRAP, die Intensität des Lasers muss optimiert werden, je nach Mikroskop-Setup und Bildgebung (Vergrößerung, Ziele, etc., sowie der Zelltyp, Struktur und fluoreszierende Protein für Immunofluoreszenz). Beachten Sie, dass bei optimalen Laserleistung, effiziente bleichen mit der geringst möglichen lichtbedingten kombiniert wird zur Vermeidung von Schrumpfung oder komplette Retraktion der Struktur unter Analyse (z. B. Lamellipodia oder Filopodien) oder gar Schäden auf zellulärer Ebene. Im Idealfall sollte mindestens 70 – 80 % der Bleiche Effizienz erreicht werden, obwohl komplett bleichen durch extrem schnelle Umsatz des Proteins, behindert werden kann in dem Fall, etwas mehr als 50 % auch akzeptabel sein könnte. Optimale bleichen macht für eine vorgegebene Struktur und Fluoreszenzfarbstoff experimentell getestet werden sollte, ausgehend von einer geringen Laserleistung seine allmähliche Zunahme gefolgt. Natürlich, jeder Fluoreszenzfarbstoff kann per definitionem gebleicht werden mit Laserlicht in der Nähe von ihren Höhepunkt der Erregung (488 nm für häufig verwendete grüne Farbstoffe wie FITC oder EGFP). Jedoch mit kürzeren Wellenlängen, wie z. B. in der Nähe von UV-Laser, Laser liefern höhere Mächte und können daher auch für effiziente Bleichen von häufig verwendeten Farbstoffe eingesetzt werden. Wir beschäftigen regelmäßig 405 nm Diodenlaser (120 mW) zum Bleichen von EGFP und rot fluoreszierende Farbstoffe (z. B. mCherry), wenn auch mit etwas geringerer Effizienz bei der letzteren (Daten nicht gezeigt). Da die 405 nm-Diode auch für photoaktivierungen der PA-GLP (siehe unten) verwendet werden kann, verleiht es dieses System mit maximaler Flexibilität.
Die B16-F1 Zellstrukturen und fluoreszierende Proteine Photobleached hier wurden 405 nm-Laserleistungen zwischen 65 – 100 mW beantragt. Bei der Analyse einer Photobleached Region ist es wichtig zu prüfen, ob die vorgegebene Struktur in seine ursprüngliche Form über die Analyse Zeitraum erhalten bleibt. Zum Beispiel bei der Analyse der Umsatz von Proteinen an Lamellipodia Spitzen darauf achten ob die Krümmung der Lamellipodia im Laufe der Zeit erheblich verändert wird als Veränderungen in der Krümmung zu ungenauen Ergebnissen führen können, wenn die Region/Kontur analysiert nicht umfassen Sie vollständig die Gesamtheit der Struktur in den einzelnen gemessenen Frames. Darüber hinaus ist darauf hinzuweisen, dass Pakete in Lamellipodia, wie Microspikes, eingebettet in Fluoreszenzintensität Abweichungen verursachen könnten. Wie in Abbildung 2b (weißer Pfeil in 9 s Zeitrahmen) dargestellt, eine Microspike-artige Struktur befindet sich neben der gemessenen Photobleached Region, sondern bleibt während der gesamten Dauer der Messung außerhalb und somit führt nicht zu einer Ungenauigkeit. Für die Analyse von Protein-Umsatz sind wichtige Überlegungen bei der Auswahl der Position und Größe der Regionen analysiert ihre Fluoreszenz im Laufe der Zeit nicht erheblich durch Veränderungen der Zellmorphologie oder Faktoren anders als hart beeinflusst werden sollte, zu vermeiden Erwerb Immunofluoreszenz. Zum Beispiel sollten Strukturen erhebliche quantitative Beitrag der analysierten Struktur nicht aus der gemessenen Region während der Analyse bewegen; Darüber hinaus sollten unabhängige, fluoreszierende Entitäten wie vesikuläre Strukturen, die das Protein anziehen nicht das Feld von Interesse während der Analyse eingeben. Für die Festsetzung des Lamellipodial-Aktin-Polymerisation, sollte darauf geachtet werden, dass keine einziehbaren oder arruffano (d. h. nach oben klappbar) Lamellipodia analysiert werden, da dies die Genauigkeit der Ergebnisse stark beeinflusst. Darüber hinaus Retraktion der Lamellipodial Regionen vorkommen als schnelle rückwärtige Translokation, möglicherweise führend zu Überschätzung der Sätze der Lamellipodial-Aktin-Polymerisation. Eine weitere Überlegung ist die Entfernung der intrazellulären Normalisierung Regionen (genommen als Referenzpunkte für die Korrektur der Erwerb Immunofluoreszenz) von der tatsächlichen Position des Immunofluoreszenz, groß genug, um direkt zu vermeiden sollten beeinflussen Sie, indem Sie den Photobleached-Bereich.
Beim Einrichten der optimaler Bedingungen für photoaktivierungen PA-GFP-Tags Konstrukte sollte darauf geachtet werden, um zu vermeiden, sofort bleichen während der photoaktivierungen. In unserer Arbeit, die besten Ergebnisse wurden mit Laserleistungen 5 - 10 mal niedriger als normal beschäftigte zum Bleichen von EGFP. Für die Bildaufnahme von photoaktiviert Molekülen sollten Belichtungszeit und Zeitintervall zwischen den Frames unter Berücksichtigung der Größe der Regionen und Strukturen photoaktiviert werden optimiert und analysiert, sowie die mögliche Mobilität von photoaktiviert Proteinen subzellulärer Standorte. Für alle Arten von Fluoreszenz-Bildgebung ist Wartung der Zellviabilität entscheidend für den Erhalt der physiologisch relevante Ergebnisse.
Im Prinzip, grün-rot Ladungszustand der fluoreszierende Proteine wie mEos oder Dronpa Varianten12 stellt eine ebenso leistungsfähige Methode der folgenden Dynamik und Umsatz der subzellulären Strukturen wie die Lamellipodium (siehe z.B. Burnette Et al. 23). der Vorteil der letzteren Methode im Gegensatz zu PA-GFP wäre die Möglichkeit, Proteindynamik vor und nach der Konvertierung mit zwei unterschiedlichen Farben, ohne die Notwendigkeit, einen zusätzlichen roten fluoreszierenden Proteins Co auszudrücken zu folgen. Jedoch war in unseren Vorversuchen das Ausmaß der Veränderung von Kontrast und Intensität der Fluoreszenzsignal erreicht bei der photoaktivierungen der PA-GLP größer im Vergleich zu Photoconverted Sonden, vielleicht wegen der überlegenen spektralen Eigenschaften von Grün und rot fluoreszierende Sonden (Daten nicht gezeigt). Auf jeden Fall sind detaillierte Studien über Actin Filament Umsatz in Zellenrand Vorsprünge wie Lamellipodia oder Vaccinia Virus-induzierte Aktin Zahl bisher nur erschienen mit PA-GFP Derivate5,6,24.
Wenn man bedenkt welche Zelle Region um nach photoaktivierungen zu analysieren, mehrere Faktoren sollten berücksichtigt werden, die werden diskutiert, über das konkrete Beispiel gezeigte (Einbau von Actin-Monomere am Rande Zelle nach der Aktivierung in der Zellflüssigkeit), aber kann sicherlich zu den verschiedenen Analog wissenschaftlichen Problemen extrapoliert werden. Erstens bei der Messung der Rate der Lamellipodial Aufnahme von cytosolically photoaktiviert Proteine, zum Beispiel in verschiedenen experimentellen Bedingungen (wie in Dimchev Et Al. gezeigt ( 6), Größen der cytosolischen Regionen und ihre Abstände, Lamellipodial Kanten sollten zwischen experimentellen Gruppen vergleichbar. Es ist auch wichtig zu berücksichtigen, dass wenn Lichtaktivierung cytosolischen Regionen, die Zelle Dicke größer in Positionen näher in den Zellkern. Aktivierung dickere zelluläre Regionen kann in höheren Mengen von aktivierten Proteine führen, angesichts der Tatsache, dass die Verteilung des Proteins aktiviert werden homogen in das Zytosol verteilt wird. Zu guter Letzt können Ausdruck Niveaus des Proteins aktiviert werden in einzelnen Zellen durchaus sehr variabel sein. Aufgrund all dieser Überlegungen der Variabilität unbedingt die Einbeziehung cytosolically aktivierte Proteine an anderer Stelle in der Zelle im Verhältnis zu der gesamten Fluoreszenz erhalten nach der Aktivierung in den einzelnen Regionen vergleichen.
Wir haben beschrieben, wie Mikroinjektion kann als ein Werkzeug für die Untersuchung der Auswirkungen von Proteinen auf Zellmorphologie verwendet werden und haben dies durch den Nachweis der potenten Induktions von Lamellipodial Strukturen in NIH3T3 fibroblastenzellen mikroinjiziert mit veranschaulicht die kleine GTPase Rac1. Bisher haben wir diese Technik, um mit Arp2/3 Funktion in den Zellen mit der WCA C-terminale Domäne der Narbe/WAVE3mikroinjiziert stören eingesetzt. Verschiedene Parameter in mikroinjiziert Zellen können durch andere Tests, wie z. B. FRAP oder photoaktivierungen analysiert werden. Wir haben beschrieben, wie FRAP und photoaktivierungen für die Untersuchung der subzellulären Dynamik und Mobilität von Actin-Monomere eingesetzt werden können. FRAP betätigt wurde von unserer Gruppe vorher5 zu untersuchen, den Umsatz von Proteinen, Lamellipodia, wie z. B. VASP, Abi, Cortactin, Cofilin, Lokalisierung und capping Protein oder für die Aufklärung des Umsatzes der Komponenten in fokalen Adhäsionen in der Gegenwart und Abwesenheit von Rac4-Signalisierung. Darüber hinaus Messraten Aktin-Polymerisation kann durch Immunofluoreszenz EGFP getaggt β-Aktin5erreicht werden, aber alternative Methoden existieren. Tracking fluoreszierende Inhomogenitäten wie live Cell Imaging-kompatiblen Sonden zellulären actinfilamente, z.B. Lifeact25, Kennzeichnung kann auch Erwerbstätige6,26. Der Vorteil hierbei ist, dass die Überexpression des β-Aktin vermieden werden kann, die ist in der Lage Zelle Rand Vorsprung und Migration zu erhöhen und somit potenziell mischt sich mit den spezifischen Assay oder experimentelle Frage (siehe z. B. Kage Et Al. 26; Peckham Et al. ( 27). allerdings ein deutlichen Nachteil der Lifeact Sonde stellt seine rasche ein-/ausschalten Kinetik der Bindung an actinfilamente, so dass Bleichen von Actin Filament Strukturen gekennzeichnet durch Lifeact in Zellen nur über die Sonde Umsatz informiert aber nicht der Umsatz die actinfilamente, es an die25 bindet. Die Verfolgung von Fluoreszenz Inhomogenitäten beschäftigt bereits6,26 bietet einen praktischen Kompromiss, ähnlich zu der weit verbreiteten Verfolgung von Fluoreszenz Flecken integriert filamentösen Zellskelett Strukturen (siehe z. B. Lachs und Waterman28), aber möglicherweise nicht so geradlinig zu verwenden und so präzise wie FRAP EGFP-markierten F-Aktin-Strukturen. Photoaktivierungen wurde von uns für die Schätzung der Preise von Monomeren Aktin Einbindung in hervorstehenden Lamellipodia, sowie seine Mobilität in das Zytosol, im Zusammenhang mit experimentell dran cytosolischen F-Aktin Ebenen6angewendet. Die Technik ist nützlich, wenn Prüfung Mobilität und Verteilung von Proteinen aus relativ großen Flächen, wie cytosolischen Regionen abgeleitet. Allerdings prüft die Verteilung von Proteinen abgeleitet relativ kleine photoaktiviert Strukturen; z. B. Wachstum Zapfen können aufgrund der geringen Anzahl von fluoreszierende Moleküle aktiviert, schwache Signale und damit auch Mangel an Sensibilität schwierig sein. Mögliche alternative Techniken zur photoaktivierungen oder Ladungszustand der Fluoreszenz (siehe oben) können Inverse FRAP, gehören, die auf Immunofluoreszenz die gesamte Zelle außer den ROI setzt, gefolgt durch die Verfolgung der Mobilität von fluoreszierenden Molekülen Weg von dieser Region. Die Technik erfordert keine überexprimierenden Photoactivatable Versionen von Proteinen, aber wird immer Belastung durch eine ungewöhnlich hohe Dosis der Laserleistung, was möglicherweise zu unerwünschten Nebenwirkungen wie lichtbedingten beinhalten.
Klar, kann nicht photoaktivierungen und FRAP unterscheiden, ob Proteine als Monomeren, dimeren oder auch kleine Oligomere in Bewegung sind und ob sie in Kombination mit weiteren Bindungspartner bewegen. Informationen dieser Art erhalten Sie stattdessen von Fluoreszenz Korrelation Spektroskopie Techniken29 oder, alternativ, FLIM-FRET-30. Dennoch bilden FRAP und photoaktivierungen einfache Ansätze, um lokale und globale Proteindynamik in Zellen, unabhängig davon, das Protein des Interesses, subzelluläre Lage oder Zelltyp studierte direkt zu beurteilen.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Wir sind dankbar für finanzielle Unterstützung (Grant Nr. RO2414/5-1 bis KR), der Deutschen Forschungsgemeinschaft (DFG).
Materialien
| Name | Company | Catalog Number | Comments |
| B16-F1 mouse skin melanoma cells | American Type Culture Collection, Manassas, VA | CRL-6323 | |
| NIH-3T3 cells | American Type Culture Collection, Manassas, VA | CRL-1658 | |
| DMEM 4.5g/L glucose | Life Technologies, Thermno Fisher Scientific, Germany | 41965-039 | |
| Ham’s F-12 medium | Sigma-Aldrich | N8641 | |
| Fetal calf serum (FCS) | PAA Laboratories, Linz, Austria | A15-102 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich, Germany | F7524 | Lot054M3396 |
| MEM Non essential amino acids | Gibco, ThermoFisher Scientific, Germany | 11140035 | |
| L-Glumatine 200mM (100x) | Life Technolgies | 25030-024 | |
| Pen-Strep 5000 U/mL | Life technologies | 15070063 | |
| Sodium Pyruvate (100 mM) | Gibco, ThermoFisher Scientific, Germany | 11360-039 | |
| Laminin | Sigma-Aldrich | L-2020 | |
| Laminin coating buffer | Self-made: 50mM Tris ph7.4, 150mM NaCl | ||
| Fibronectin from human plasma | Roche Diagnostics, Mannheim, Germany | 11 051 407 001 | |
| Jetpei | Polyplus Transfection, Illkirch, France | 101-10N | |
| JetPei buffer | Polyplus Transfection, Illkirch, France | 702-50 | 150mM NaCl |
| PA-GFP-actin plasmid DNA | described in Koestler et al.2008 | ||
| pEGFP-actin plasmid DNA | Clontech, Mountain View, CA, USA | ||
| Rac1 protein for microinjection | Purified as GST-tagged version, and cleaved from GST prior to injection | ||
| Microinjection buffer | Self-made: 100mM NaCl, 50mM Tris-HCl ph7.5, 5mM MgCl2, 1mM DTT | ||
| Dextran, Texas Red, 70,000 MW, Lysine Fixable | Molecular Probes, Thermno Fisher Scientific, Germany | D1864 | |
| Microscope circular cover glasses 15mm, No.1 | Karl Hecht, Aisstent, Sondheim, Germany | 1001/15 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf, Hamburg, Germany | 5242 956 003 | |
| Eppendorf Femtotip Microinjection Capillary Tips | Eppendorf, Hamburg, Germany | 930000035 | |
| Silicone Grease | ACC Silicones, Bridgewater, England | SGM494 | |
| Aluminium Open Diamond Bath Imaging Chamber | Warner instruments | RC-26 | |
| Automatic temperature controller | Warner Instruments | TC-324B | |
| Microscope: Axio Observer | Carl Zeiss, Jena, Germany | ||
| CoolSnap-HQ2 camera | Photometrics, Tucson, AZ | ||
| Lambda DG4 light source | Sutter Instrucment, Novato, CA | ||
| Laser source | Visitron Systems | ||
| Eppendorf FemtoJet microinjector | Eppendorf, Hamburg, Germany | With built-in compressor for pressure supply | |
| Nikon Narishige Micromanipulator system | Nikon Instruments, Japan | ||
| Visiview software v2.1.4 | Visitron Systems, Puchheim, Germany | ||
| Metamorph software v7.8.10 | Molecular Devices, Sunnyvale, CA | ||
| Sigma Plot v.12 | Systat Software Inc. |
Referenzen
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 38 (10), 2887-2921 (2009).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Koestler, S. A., et al. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol Biol Cell. 24 (18), 2861-2875 (2013).
- Steffen, A., et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 126, Pt 20 4572-4588 (2013).
- Lai, F. P., et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27 (7), 982-992 (2008).
- Dimchev, G., et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol Biol Cell. 28 (10), 1311-1325 (2017).
- Koppel, D. E., Axelrod, D., Schlessinger, J., Elson, E. L., Webb, W. W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys J. 16 (11), 1315-1329 (1976).
- Patterson, G. H., Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297 (5588), 1873-1877 (2002).
- McKinney, S. A., Murphy, C. S., Hazelwood, K. L., Davidson, M. W., Looger, L. L. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 6 (2), 131-133 (2009).
- Gurskaya, N. G., et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 24 (4), 461-465 (2006).
- Lippincott-Schwartz, J., Patterson, G. H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 19 (11), 555-565 (2009).
- Kremers, G. J., Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J Vis Exp. (40), (2010).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 4988(2008).
- Small, J. V., Rottner, K. Actin-based Motility. Carlier, M. F. , Springer. Dordrecht. (2010).
- Kaverina, I., et al. Enforced polarisation and locomotion of fibroblasts lacking microtubules. Curr Biol. 10 (12), 739-742 (2000).
- Small, J., Rottner, K., Hahne, P., Anderson, K. I. Visualising the actin cytoskeleton. Microsc Res Tech. 47 (1), 3-17 (1999).
- Mikhailov, A. V., Gundersen, G. G. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskeleton. 32 (3), 173-186 (1995).
- Rottner, K., Behrendt, B., Small, J. V., Wehland, J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1 (5), 321-322 (1999).
- Svitkina, T. M., et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 160 (3), 409-421 (2003).
- Small, J. V., Isenberg, G., Celis, J. E. Polarity of actin at the leading edge of cultured cells. Nature. 272 (5654), 638-639 (1978).
- Dang, I., et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 503 (7475), 281-284 (2013).
- Anderson, K. I., Cross, R. Contact dynamics during keratocyte motility. Curr Biol. 10 (5), 253-260 (2000).
- Burnette, D. T., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 13 (4), 371-381 (2011).
- Humphries, A. C., et al. Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell Host Microbe. 12 (3), 346-359 (2012).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 5 (7), 605-607 (2008).
- Kage, F., et al. FMNL formins boost lamellipodial force generation. Nat Commun. 8, 14832(2017).
- Peckham, M., Miller, G., Wells, C., Zicha, D., Dunn, G. A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 114, Pt 7 1367-1377 (2001).
- Salmon, E. D., Waterman, C. M. How we discovered fluorescent speckle microscopy. Mol Biol Cell. 22 (21), 3940-3942 (2011).
- Machan, R., Wohland, T. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 588 (19), 3571-3584 (2014).
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J Microsc. 247 (2), 119-136 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten