Method Article
Técnicas de micromanipulação, permitindo a análise da dinâmica morfogenética e rotatividade dos reguladores do citoesqueleto
Neste Artigo
Resumo
Descrevemos como micro e photomanipulation técnicas tais como o FRAP e photoactivation permitem a determinação de parâmetros de motilidade e a spatiotemporal dinâmica de proteínas dentro de migração de células. Leituras experimentais incluem subcellular dinâmica e volume de negócios de reguladores da motilidade ou o citoesqueleto de actina subjacente.
Resumo
Examinando a spatiotemporal dinâmica de proteínas pode revelar sua importância funcional em vários contextos. Neste artigo, é discutida como fluorescente recuperação após fotobranqueamento (FRAP) e técnicas de fotoativação pode ser usada para estudar a dinâmica spatiotemporal de proteínas em locais subcellular. Também mostramos como estas técnicas permitem a determinação direta de diversos parâmetros ligados à actina do citoesqueleto regulamento e célula da motilidade. Além disso, a microinjeção de células adicionalmente é descrita como um tratamento alternativo (potencialmente anteriores ou complementando as técnicas acima mencionadas photomanipulation) gatilho efeitos instantânea de proteínas translocadas na célula morfologia e função. Micromanipulação como injeção de proteína ou aplicação local de membrana plasmática-permeável drogas ou inibidores do citoesqueleto pode servir como uma ferramenta poderosa para gravar as consequências imediatas de um determinado tratamento sobre o comportamento de célula na célula única e subcellular nível. Isto é exemplificado aqui por indução imediata de protrusão de borda de célula lamellipodial através da injeção de proteína recombinante Rac1, estabelecido um quarto de século atrás. Além disso, nós fornecemos um protocolo para determinar o volume de negócios da proteína verde fluorescente melhorada (EGFP)-VASP, uma polimerase de filamento de actina proeminentemente acumulando no lamellipodial dicas de células B16-F1, empregando o FRAP e incluindo dados associados análise e encaixe de curva. Apresentamos também orientações para estimar as taxas de polimerização de rede lamellipodial actina, como exemplificado pelas células expressando com tag EGFP β-actina. Finalmente, são dadas instruções para saber como investigar as taxas de mobilidade de monômero de actina no citoplasma celular, seguido por incorporação de actina em sites do conjunto de incandescência rápida, tais como as pontas dos salientes lamellipodia, usando o photoactivation aproxima-se. Nenhum destes protocolos é restrito aos componentes ou reguladores do citoesqueleto de actina, mas pode ser facilmente estendida para explorar de forma análoga a spatiotemporal dinâmica e função das proteínas em vários diferentes estruturas subcelulares ou funcional contextos.
Introdução
Monitoramento da dinâmica spatiotemporal de proteínas e outras moléculas em células vivas, tornou-se uma ferramenta essencial em muitos campos da biologia celular e molecular. Advanced fluorescência técnicas de microscopia, incluindo a transferência de energia de ressonância de fluorescência (FRET) e vida de FRET-fluorescência de imagem (FLIM-FRET), ou FRAP, perda de fluorescência na fotobranqueamento (FLIP) e fotoativação, bem como muitos outros permitem que para o temporal e espacial, monitoramento de interações da proteína-proteína, mudanças conformacionais, bem como determinar a cinética de difusão e localização de proteínas diferentes na célula1,2. FRAP e photoactivation técnicas, em particular, são amplamente aplicáveis para examinar os reguladores da migração actina citoesqueleto e celular. Estas técnicas podem ser aplicadas isoladamente ou em combinação com técnicas de micromanipulação adicionais como microinjeção3e envolvem a expressão de proteínas fluorescente-labeled. Eles permitem a estimativa da cinética de associação de proteína rica em actina estruturas envolvidos na migração celular, tais como filopodia ou lamellipodia, o volume de negócios de proteínas em aderências focais4, ou ramificada actina redes5. Elas também permitem a determinação das taxas de polimerização de actina lamellipodial, a avaliação da dispersão de actina monomérica dentro do citosol, a taxa de translocação de monômero subcellular actina para polimerização de filamentos de actina em salientes lamellipodia6e outros parâmetros.
FRAP é um método para visualizar e quantificar a mobilidade das proteínas dentro de uma célula viva, desenvolvido originalmente na década de 1970 por Axelrod7. Uma região de interesse (ROI) dentro de uma célula, preenchida com proteínas fluorescente-labeled, transitoriamente é exposta a um laser de alta intensidade, suficiente para causar branqueamento das moléculas de fluoróforo presentes nesta região, durante um determinado período curto de tempo. A crus, fluorescente etiquetado proteínas localizadas fora o ROI durante o clareamento, serão difusa e se infiltrar na região branqueada, dependendo de sua dinâmica spatiotemporal, causando o deslocamento de moléculas de foto ao longo do tempo. A taxa de recuperação de fluorescência em regiões branqueadas é dependente de vários factores, incluindo o tamanho e a taxa de difusão de uma determinada molécula, e claro, sua taxa de rotatividade dentro o putativo associado estrutura branqueada. Assim, proteínas solúveis vai mediar a recuperação de fluorescência dentro o ROI branqueada rapidamente através da difusão, enquanto as proteínas firmemente associado com estruturas, como aderências focais, terá mais vezes de volume de negócios, como será sua recuperação de fluorescência dependem tanto na difusão da fração solúvel da cinética de dissociação-associação e proteína da fração estrutura associada. Recuperação de fluorescência é geralmente adquirida e quantificada até atinge o nível inicial de pre-lixívia intensidade de fluorescência. No entanto, isso não ocorre se uma parte da intensidade da fluorescência inicial pertence a chamada fração imóvel, que é incapaz de ser reabastecida por difusão ou está reabastecendo em taxas muito lentas em comparação com a maioria das moléculas compreendendo o mobile fração. Para determinar a taxa de rotatividade de proteína, são geradas curvas FRAP, representando a extensão da recuperação de fluorescência ao longo do tempo. Destas curvas de recuperação, meio-tempos médios de recuperação de proteína podem ser calculadas. Através da criação de ajustes de curva da média FRAP dados e análises matemáticas, portanto, também é possível deduzir-se a taxa de rotatividade média da fração móvel constitui um composto de uma população homogênea das moléculas, ou se é composto por dois ou mais subpopulações de moléculas revirando no diferencial de taxas. Além de estimar as taxas de rotatividade de proteína por abordagens quantitativas, a recuperação das regiões de foto no lamellipodia de rastreamento também pode permitir que para quantificação exata dos parâmetros de motilidade lamellipodial como fluxo retrógrado, protrusão, e taxas de polimerização de actina. Assim, o FRAP constitui uma ferramenta versátil para ser aplicado para avaliar vários parâmetros dentro de estruturas de células vivas.
Photoactivation é um método usado para controlar a difusão e a mobilidade das proteínas ou moléculas provenientes de um local designado de celular. A técnica emprega, por exemplo, uma variante do selvagem-tipo proteína verde fluorescente (GFP), inicialmente desenvolvida por Patterson e Lippincott-Schwartz8, que é uma mutação de uma forma que permite a sua fluorescência ser altamente aumentada após a exposição ao luz ultravioleta (UV) (cerca de 400 nm; aqui, 405 nm). Conforme descrito por Patterson et al., cromóforos GFP selvagem-tipo existirem como uma população mista de fenóis neutros e fenolatos aniônicos, que produzem um pico de absorbância principais em aproximadamente 397 nm e um menor em 475 nm, respectivamente. Após irradiação da proteína com luz UV, a população sofre photoconversion, mudando para a forma aniónica. Quando animado por 488 nm, a proteína photoconverted/fotoativados exibe um 3 vezes aumento na fluorescência, insuficiente na prática para distinguir entre ativado e não está ativado GFP devido a fluorescência intrínseca de fundo elevado. No entanto, uma diminuição na intensidade de fundo foi conseguida através da introdução de uma único aminoácido mutação na sequência de GFP (substituição de histidina na posição 203). O mutante resultante do T203H, também conhecido como photoactivatable-GFP (PA-GFP) é caracterizado por uma redução significativa na absorção do pico menor, que após irradiação com luz UV é aumentada quase 100-fold quando posteriormente excitadas pela luz de nm 488. Portanto, a superexpressão de proteínas PA-GFP-etiquetado é uma abordagem amplamente utilizada, que permite a determinação da difusão e a mobilidade das moléculas dentro das células. Temos anteriormente aplicado PA-GFP-etiquetado actina para determinar a taxa de dispersão de monômeros de actina longe de regiões citosólica, permitindo não só a exploração de sua mobilidade dentro do citosol, mas também a sua taxa de incorporação para o salientes lamellipodial actina rede6. Literatura mais recente também descreve o romance, foto-conversível proteínas que podem, em princípio, ser usadas de forma análoga, mas abrigando a vantagem potencial para ser visível já antes da foto-conversão. Exemplos para este grupo de proteínas fluorescentes incluem Dendra2 e mEos29,10,11,12.
Neste artigo, vamos explicar a metodologia de células microinjecting com proteínas. Ainda mais, explicamos como esta técnica pode ser combinada com FRAP, por fotobranqueamento proteínas envolvidas na regulação de citoesqueleto de actina e motilidade, e como o FRAP curvas e intervalo de recuperação de frações móveis podem ser derivadas. Além disso, nós fornecemos um exemplo de como a FRAP técnica pode ser usada para determinar taxas de polimerização de actina de redes lamellipodial. Nós também fornecemos as instruções e dicas sobre como executar o photoactivation experiências, que podem ser usadas para determinar a mobilidade citosólica da actina monomérica e taxas de incorporação de actina em lamellipodia. Estas técnicas, claro, não são apenas limitado ao acompanhamento de componentes do citoesqueleto de actina, mas potencialmente necessária adaptação moderada ou otimização, pode ser amplamente aplicada a outros tipos de célula, ou para investigar diferentes proteínas, estruturas, e parâmetros.
Protocolo
1. lamela lavagem e esterilização
- Mergulhe a 15 mm (diâmetro) capa óculos (no. 1) em um balão de 500 mL, contendo uma mistura de 40 mL 37% HCl e 60 mL 100% EtOH (lamelas não mais de 100 por 100 mL de solução de lavagem).
Nota: mesmo que recém comprados, lamelas devem ser rigorosamente limpos antes da semeadura de células em suas superfícies. Isto é porque eles podem conter filmes finos de gordura, que são macroscopicamente invisível, mas com eficiência podem interferir com a aderência e espalhamento adequado de células vivas. Considerando que tais filmes podem ser eficientemente removidos com soluções contendo ácido ou base (ver Fischer et al 13), usamos rotineiramente a mistura de álcool/ácido descrita acima. - Agite o frasco contendo os capa óculos durante 30 min num agitador de rotação. Escolha uma velocidade que permite que os vidros de tampa ser rodado livremente, mas lenta o suficiente para evitar ruptura frequente. Filtra a solução para remover pedaços de vidro quebrado se reutilizando.
- Transferir os óculos de capa para um frasco contendo pelo menos 200 mL de água estéril e incubar um agitador de rotação, enquanto repetidamente, substituindo a água até que o cheiro ácido desapareceu. Várias lavagens ao longo de várias horas são recomendadas para a completa eliminação de HCL-EtOH vestígios.
- Óculos de cobertura individuais em uma folha de papel de filtro a seco.
- Lugar, os óculos de cobertura no fundo de um prato de Petri de 10 cm (diâmetro) cobertos com papel de filtro e calor seco-esterilizar. Evite autoclave causará capa óculos para ficar juntos.
2. tratamento de células e Transfection semeadura em lamelas
- Desenvolvem-se células de melanoma B16-F1 do mouse de acordo com as condições de cultura celular padrão em DMEM (glicose de 4,5 g/L) contendo soro fetal bezerro de 10%, glutamina 2 mM e 1% penicilina-estreptomicina a 37 ° C, 7% de CO2.
- Desenvolvem-se células de fibroblastos NIH3T3 para microinjeções de acordo com as condições de cultura de célula padrão (cultura de tecidos incubadora a 37 ° C, 7% CO2) em DMEM (glicose de 4,5 g/L) contendo 10% fetal de soro bovino, piruvato de sódio 1 mM, 1 x MEM não aminoácidos essenciais , glutamina 2 mM e 1% penicilina-estreptomicina.
- Para transfections, desenvolvem-se células B16-F1 a confluência de 100% em um prato de 10 cm e passagem na proporção de 1:5 em um prato de plástico de 3 cm (diâmetro).
- No mesmo dia, após as células B16-F1 foram autorizadas a aderir pelo menos 6 h, transfect com 500 ng/prato de photoactivatable PA-GFP-actina ou β-actina EGFP-Tag do plasmídeo DNA. Para co transfections de PA-GFP-actina com vetores de mCherry-codificação, misture um total de 1 µ g de plasmídeo por prato de 3cm.
- Transfect as células B16-F1 com o reagente de transfeccao (Tabela de materiais). Por um prato de 3cm, misturar 200 µ l de 150 mM de NaCl contendo 500 ng de DNA construir com 200 µ l de NaCl contendo 1 µ l de reagente de Transfeccao de 150 mM (ou seja, DNA (µ g): utilizou-se relação de reagente (µ l) de 1:2).
- Incube a mistura de Transfeccao para 20 min em temperatura ambiente (RT) e pipeta drop-wise para o prato de 3 cm, que contém as células. Agite suavemente o prato para misturar e incubar durante uma noite a 37 ° C, 7% de CO2.
- Prepare o buffer de revestimento de laminina contendo 50 mM Tris, pH 7,4 e 150 mM de NaCl.
- Para as células B16-F1, casaco 15mm capa óculos por espalhar 150 µ l de laminina (25 µ g/mL no buffer de revestimento laminina) e incube por 1h no RT Para as células NIH3T3, revestir os vidros de cobertura com solução de fibronectina (25 µ g/mL de tampão fosfato salino (PBS)) e incube por 1h no RT
- Lavar vidros de tampa laminina ou fibronectina-incubados com PBS, em seguida, Aspire a PBS e adicionar 2 mL de células transfectadas.
- Semente de células transfectadas B16-F1 (em 01:30 relação de um prato confluente), no dia após a transfeccao, em lamelas laminina-revestido. Os fibroblastos NIH3T3 de sementes (em 01:20 relação de um prato confluente) em lamelas de fibronectina-revestido.
- Permitir que as células a se espalhar na laminina - ou fibronectina-revestido capa óculos durante a noite em uma incubadora de cultura de tecidos, a 37 ° C antes da microscopia. Alternativamente, experimentos de microscopia podem ser iniciados no mesmo dia, dado que as células são permitidas para espalhar pelo menos 2-3 h.
3. montagem da câmara de imagem de microscopia
- Use um calor condutivo RC-26 de alumínio câmara para microscopia (Figura 1a) de imagem. Mancha de graxa de silicone ao redor do contorno da abertura do selador plástico utilizando uma seringa (Figura 1b).
- Coloque o vidro com o lado células para cima na câmara (Figura 1c).
- Coloque o selador de plástico por cima do vidro de tampa para fazer uma vedação segura entre a lamínula e a câmara. Corrigi o selador plástico (na diagonal para evitar a ruptura da lamela) apertando os grampos de deslizamento para a câmara para evitar o meio vazando (Figura 1d).
- Meio de microscopia pré-aquecido pipeta 37 ° C para a área central. Por meio de autofluorescência de reduzido e, portanto, otimizado para microscopia, usar a mesma receita como meio de cultura descrito acima, mas com F12-presunto em vez de DMEM, adicionalmente, contendo 20 mM HEPES para cultivo de células, na ausência de CO2 (Figura 1e).
- Insira o detector de calor no slot designado da câmara e vincular os eletrodos da câmara para um controlador de temperatura automático de TC-324. oB manter uma temperatura constante de 37 ° C (Figura 1f).
- Coloque uma pequena gota de óleo de imersão sobre o objectivo e a câmara no topo.
- Incube a câmara com células pelo menos 10 a 30 min permitir-lhes para recuperar a queda de temperatura durante a montagem e se adaptar ao meio de microscopia.
- Antes de microscopia é iniciada, substitua o meio de cultura no reservatório central da câmara (aproximadamente 800 µ l) para evitar a concentração inadequada de componentes médias e soro devido a evaporação média. Sessões de microscopia prolongada com câmaras abertas exigirá mudança de rotina do meio de evaporação.
4. microinjection procedimento
- Revestir as lamelas, preparar as células e montar a câmara de imagem conforme descrito acima.
- Descongelar uma alíquota de proteína purificada para ser injetado (tipicamente 10 µ l ou menos) e dilui-lo com o buffer de microinjeção apropriado.
Nota: Buffer composição pode variar de acordo com o tipo de proteína e células, mas tome cuidado para usar um pH entre 6,95 e 8,00 e evitar o uso de PBS, como a maioria das células não gostam de ser injetado com PBS. - Para o microinjection Rac1, prepare tampão contendo 100 mM de NaCl, pH de Tris-HCl 50 mM 7.5, 5 mM MgCl2, 1 milímetro DTT. Os íons de Mg2 + são essenciais para a estabilidade de GTPase pequena.
Nota: As concentrações de proteína normalmente variam entre 0,1 a 1 mg/mL (máximo 2 mg/mL), dependendo da proteína, o tipo de experimento e célula tipo. - Se for o caso, adicione corante fluorescente, como o dextran inerte (0,5 µ g/mL, 70 kDa) à solução da proteína, que pode confirmar a presença de fluxo da agulha antes de injeções e permite a documentação de injeções bem sucedidas após o experimento.
Nota: O experimento aqui não visa a seguir a dinâmica do Rac1 injetado, que só seria possível mediante rotulagem de fluorescência direta da proteína. Acoplamento de proteínas com corantes fluorescentes ou fusão com uma proteína fluorescente é possível, mas evitar aqui como que abriga o risco de interferir com a função de sinalização, em especial de pequenas proteínas como o Rho-família GTPase Rac1 (20 kDa). - Centrifugue a solução da proteína a 10.000 x g, durante pelo menos 30 min para remover agregados de proteínas que podem levar ao entupimento se estiver presente nos capilares da microinjeção de agulha.
- Carrega uma agulha de microinjeção (microinjeção capilar) com 1 µ l de mistura de injeção do lado traseiro usando uma ponta de ponta/microloader de pipeta flexível.
- Se as bolhas de ar estão presentes na ponta da agulha, bata levemente a base da agulha para removê-los. Prossiga rapidamente para evitar a secagem da ponta da agulha, que pode causar entupimento da agulha.
- Ajuste cuidadosamente o suporte da agulha no dispositivo de micromanipulação. Se usando um microscópio invertido para a imagem latente de contraste de fase, antes de carregar a agulha, certifique-se de não há espaço suficiente para mover a agulha de cima e para baixo sem obstruir o condensador do microscópio.
- Após lixar a conta para o suporte da agulha, aplica pressão (20 – 50 hPa fundo) na agulha usando um dispositivo de pressão microinjeção antes de que se a ponta da agulha no meio de cultura celular.
Nota: Ativando a pressão quando a agulha estiver no meio de resultará no meio sendo sugado pela força capilar e, portanto, proibir a injecção da solução de interesse. - Posicione a agulha no campo de visão (facilitado usando objectivos de baixa ampliação). Um objectivo de seco 40 X para experimentos de microinjeção foi usado aqui.
- Posicione a ponta da agulha macroscopicamente na posição vertical em relação ao meio da lente objetiva (isso irá acelerar a encontrar a ponta da agulha). Use o microscópio com óptica de contraste de fase para mover a ponta da agulha no plano horizontal em relação ao campo de visão, em um plano ótico bem acima da camada de células.
Nota: A agulha aparecerá inicialmente como uma sombra no campo de visão e do plano de foco, então, pode ser ajustado para visualizar a ponta. Uma vez que encontra-se a ponta da agulha, diminuir gradualmente o plano ótico seguido pela ponta da agulha para baixo uma posição perto da camada de células. - Verificar o fluxo de agulha por mudar para um canal fluorescente quando utilizar dextran fluorescente e sintonizar o fluxo usando o dispositivo de pressão para obter um fluxo constante de "fundo".
Nota: Neste artigo, descrevemos injeção manual, que é mediada por romper a membrana plasmática através de tocar o celular de superfície e suave movimento da ponta da agulha durante o fluxo constante de agulha. Isto deve ser distinguido de dispositivos automáticos de injeção acompanhados por aumento de pressão reduzindo e agulha agulha programados durante eventos de injeção, que são mais apropriados para a injeção de maior número de células seguido por uma população de células mais tarde análise. O método descrito aqui é otimizado para análise de célula única pela microscopia de lapso de tempo, antes, durante e depois da microinjeção. - Encontrar uma célula de interesse e diminuir gradualmente a agulha acima dela.
- Quando estiver pronto para microinjeção, abaixe a agulha gradualmente em direção a região perinuclear de célula usando o pinhão bem do joystick micromanipulador, mantendo as células em foco.
- Para microinjeção, toque suavemente a membrana plasmática da célula, que pode ser suficiente para penetrar na célula, ou ruptura de membrana transiente de auxílios por um toque muito suave na configuração do microscópio.
Nota: Um ponto branco na ponta da agulha indicará o tempo de contato com a membrana plasmática; após ruptura de membrana, vai selar a ponta da agulha, acompanhado por um fluxo suave de solução de injeção dentro da célula. - Parar o processo de injeção, assim que o fluxo para a célula é visível (idealmente dentro de 0.3 s) movendo-se até a ponta da agulha para o meio. Ao usar fluorescente dextrano, injeções de sucesso podem ser documentadas imediatamente por fluorescência.
- Se desejar, inicie a aquisição de imagem de lapso de tempo, antes ou depois da microinjeção.
Nota: A aplicação Local de drogas ou inibidores pode ser executada em todos os passos aqui, exceto a microinjeção evento por si. Para aplicativos locais, a difusão da molécula ativa pode ser controlada pela pressão do fluxo e documentada por fluorescência, e a ponta da agulha pode ser posicionada na altura desejada. Para exemplos de experiências de aplicação local, consulte , por exemplo, pequenas Rottner e14 ou Kaverina et al . 15 - Seguindo a microinjeção, esperar até que o efeito da proteína ocorre. Para proteínas diferentes e, dependendo do resultado esperado, tempos de incubação podem variar. Para a pequena GTPase Rac1, a resposta de formação de lamellipodium pode ser iniciada dentro de 1 min ou menos, mas leva cerca de 10-15 minutos em média para se desenvolver plenamente (Figura 1g, h).
- Julga a viabilidade das células após a microinjeção.
Nota: Injeções inadequadas ou prejudiciais podem causar danos celulares, que é frequentemente acompanhado de retração borda celular específico ou ruptura da membrana plasmática.- Evitam tocar na ferida através da parte superior e inferior membrana plasmática, que pode ocorrer para injeções em regiões planas celulares.
Nota: Os volumes de injeção devem ser mantidos a um mínimo (idealmente < 5% do volume celular) e geralmente será na faixa de femtoliter. Necessários volumes de injeção também podem ser controlados por alterações na concentração, mas note que para proteínas, concentrações > 2 mg/mL pode tornar-se impraticável devido ao entupimento da agulha frequente. No entanto, isso também depende da qualidade e comportamento da proteína purificada; por exemplo, injeção de actina fluorescente-acoplado é complicada pela polimerização de concentração-dependente e inevitável na ponta da agulha, e então é raramente executada hoje (ver pequenas et al 16).
- Evitam tocar na ferida através da parte superior e inferior membrana plasmática, que pode ocorrer para injeções em regiões planas celulares.
- Antes, durante ou após o efeito da microinjeção, FRAP ou photoactivation pode ser executada na mesma célula (ver seção 5 e 6).
5. procedimento do FRAP
- Transfect o tipo de célula de interesse (aqui células B16-F1) com plasmídeo codificação de uma proteína fluorescente-etiquetadas de interesse (aqui, foi usada uma versão EGFP-Tag do β-actina). Sementes das células para laminina-revestido lamelas (passo 2.10).
- Monte a câmara de imagem (secção 3).
- Use as seguintes configurações para lamellipodial região fotobranqueamento: 65 mW de potência do laser (variável de acordo com a fonte de instalação e laser experimental); diâmetro de feixe de laser pixel 10; 1 ms lixívia Habitai tempo/pixel; tempo de exposição GFP 500 ms; intervalo de tempo de 1.500 ms. Resultados experimentais neste trabalho foram realizados com um objetivo apocromático 100 X 1.4NA.
- Realize a calibração do laser para garantir a precisão nas dimensões da região de foto. Antes da calibração, mover o campo de visão para uma área sem qualquer sinal de células/fluorescência e observar a imagem no visor.
- Selecione a ampliação objetiva clicando no botão respectivo ampliação e reduzir a potência do laser (3-5 mW) no "painel | Menu de intensidade". Para iniciar a calibração manual em Visiview software (v2.1.4), selecione o "Configure | FRAP"menu e clique sobre o" calibrar | Menu de ajuste Manual". Certifique-se de que o laser pode ser distinguido como um ponto afiado. Se não, ou mudar o foco ou ajustar o hardware do laser.
- Execute a calibração orientando manualmente o laser para pre-determinado software X-Y-coordenadas. Isso instrui o software como alvejar especificamente o laser para uma região definida pelo usuário para a ampliação atual.
- Antes de acionar o laser, mude para o canal GFP e iniciar aquisição de lapso de tempo/imagem.
- Desenhe manualmente a região para ser foto no canal de GFP, enquanto visualiza o ecrã.
- Inicie o fotobranqueamento por um gatilho manual do laser 405 nm, pelo menos 3-4 frames após a iniciação de aquisição de imagem. Adquirir quadros antes fotobranqueamento é necessária para a normalização da imagem em posterior análise de dados.
6. Photoactivation procedimento
Nota: Software, instalação de microscópio e configurações, exceto para a potência do laser, são semelhantes de FRAP. Na fotoativação, uma diferença importante em relação ao FRAP, é um poder de 405nm-laser significativamente inferior que empregavam para fotobranqueamento deve ser usado, para ativar o PA-GFP sem simultaneamente fotobranqueamento isso.
- Co transfect o tipo de célula de interesse (B16-F1 células aqui; consulte a etapa 2.5) com Plasmídeo codificação PA-GFP-actina e outra proteína fluorescente-etiquetadas (por exemplo, mCherry ou mCherry-Lifeact).
Nota: Na maioria dos casos, as células mCherry-positivo também será positivas para o vetor PA-GFP-actina, este último não é normalmente visto no canal antes da fotoativação de GFP. Para aumentar a chance de que as células mCherry-positivos também são positivas para PA-GFP, use uma proporção de Transfeccao de 1:2 de mCherry:PA-GFP-actina. Seguir este protocolo, mais de 90% das células expressando mCherry exibido ativação bem-sucedida de PA-GFP-actina. - As sementes das células B16-F1 para laminina-revestido lamelas (passo 2.10).
- Monte a câmara de imagem (secção 3).
- Antes de iniciar os experimentos de fotoativação, se necessário, realizar a calibração do laser para o objetivo selecionado (passo 5.4 – 5.6).
- Defina a aquisição de imagens do GFP/488 nm ao intervalo de tempo exposição e 1.500 ms 500 ms (dependendo do projeto experimental).
-
Ajustar as configurações de software para a aquisição de canal duplo ou triplo-canal filmes de lapso de tempo marcando a Praça "Série de comprimento de onda" e seleccionando o número desejado de canais no "Acquire | Menu de comprimento de onda". É recomendável que os filmes de lapso de tempo são adquiridos com contraste de fase e canais GFP.
- Opcionalmente, também incluem o canal mCherry; no entanto, expondo as células com demasiada luz pode induzir Fotodano. Isto poderia ser evitado com os catadores de oxigênio como Oxyrase17, apesar de tratamento eficaz requer célula câmara de vedação.
- Encontre as células transfectadas no canal mCherry.
- Antes de acionar o laser, iniciar aquisição de lapso de tempo/imagem e desenhar manualmente a região para ser fotoativados no canal de contraste de fase, enquanto visualiza o ecrã.
- Iniciar a fotoativação por um gatilho manual do laser 405 nm (intensidade definida entre 5 a 15 mW do "painel | Menu de intensidade"), quadros de pelo menos 3-4 após a iniciação de aquisição de imagem.
7. dados análise e apresentação dos resultados da FRAP
Nota: O método apresentado é usado para investigar o volume de negócios de uma proteína acumular-se nos locais de assembly dinâmico actina, nesse caso, VASP, que associa com sites de adesão e as dicas de salientes lamellipodia. Estamos analisando seu volume de negócios na ponta de lamellipodium, mas os mesmos princípios de análise podem ser aplicados para investigar o volume de negócios da VASP ou qualquer outro tipo de proteína e outros compartimentos subcellular.
- Abra os lapso de tempo filmes derivados de Visiview no software da metamorfose. Neste artigo, utilizou-se a metamorfose v7.8.10.
- Derive os valores de intensidade para regiões foto delineando manualmente as respectivas regiões na metamorfose. Desenhar uma forma na ponta do lamellipodium que cobre a totalidade ou parte da área de foto e manualmente ajusta a sua posição nos quadros subsequentes se necessário (ou seja, se a borda é salientes), em ordem para controlar as alterações nas intensidades de lamellipodial de o respectivo componente durante o deslocamento de ponta.
- Para a correção de fundo e fotobranqueamento aquisição, analise regiões dentro e fora da célula. Consulte a Figura 2uma para regiões representativas das intensidades medidas.
- Enquanto um ROI é seleccionado, extrair seus valores de intensidade na metamorfose usando o menu "medida | Medições da região". Certifique-se de "Tempo decorrido" e "Intensidade média" opções são selecionadas no menu "Configurar". Clique em "Abrir log" e selecione "Dynamic Data Exchange". Clique em "Okey" para abrir uma planilha do Excel e clique no botão "Abrir log" novamente para colar os valores de metamorfose em Excel.
Nota: Estes valores são usados para gerar as curvas de recuperação de fluorescência. - Para a geração de curvas de recuperação de fluorescência na ponta lamellipodium das regiões de foto (normalizada para a intensidade da região antes de fotobranqueamento), aplica a seguinte equação:
 Equação 1
Equação 1
Onde: FRAPTn é a intensidade de região de foto para cada quadro de interesse seguindo o fotobranqueamento; Fora aTn é uma intensidade de região tomada fora da célula (fundo) para cada quadro de interesse seguindo fotobranqueamento; InsTn é dois em média dentro região intensidades para cada quadro de interesse seguindo o fotobranqueamento (usado para normalizar para aquisição fotobranqueamento ao longo do tempo); FRAPT-1 é a intensidade de região foto antes fotobranqueamento; T-1 é uma intensidade de região tomada fora da célula (fundo), antes de fotobranqueamento; e dois em média dentro região intensidades para cada quadro de interesse antes de fotobranqueamento InsT-1 . - Para cada período de tempo de interesse, use a equação 1 para obter uma curva de recuperação de fluorescência contendo todos os prazos para ser investigado. O comprimento de tempo é estritamente dependente da proteína sob investigação. Quando desconhecido, realiza experimentos preliminares para adquirir a taxa de rotatividade da proteína.
- Para calcular o intervalo de recuperação, colar os valores da curva de recuperação de fluorescência com o correspondente tempo (em segundos) em uma trama de Sigma (v.12) e executar uma curva de ajuste usando o "Dynamic Fit assistente | Ferramenta de crescimento exponencial ao máximo". Selecione mono-exponencial (single, 3 parâmetros) ou bi-exponencial (duplo, 4 parâmetros) funções, dependendo da curva de melhor ajuste.
- Use a seguinte fórmula para a função mono-exponencial:
 Equação 2
Equação 2
- Use a seguinte fórmula para função bi-exponencial:
 Equação 3
Equação 3
- Colar parâmetros "b" e "d", derivado de Sigma Plot (equação 2 ou 3 equação) em Excel para calcular o intervalo de recuperação. Aplicam-se as seguintes equações:
 Equação 4
Equação 4
ou Equação 5
Equação 5
- Quando a função mono-exponencial resulta em uma curva exata, aplicam-se somente a equação 4.
- Quando a função mono-exponencial não resulta em uma boa curva de ajuste, aplica a fórmula bi-exponencial, resolvendo tanto a equação 4 e a equação 5. Considere os metade-dois vezes resultantes de recuperação como a representação de duas frações de proteína diferentes: uma rápida e uma fração lentamente troca, respectivamente.
8. determinar a taxa de polimerização de actina Lamellipodial por FRAP
- Para determinar a taxa de polimerização de actina lamellipodial, transfect células B16-F1 com EGFP-tag β-actina, e photobleach da região de lamellipodial (etapa 5.9) usando 1,5 intervalo s e exposição GFP de 500 ms.
- Em metamorfose, abra os lapso de tempo filmes adquiridos de Visiview e calibrar a proporção de pixel/µm de acordo com o objetivo de usado pela "medida | Ferramenta de calibrar distâncias".
- Reproduzir o filme lapso de tempo e parar no frame quando a recuperação de fluorescência lamellipodial, que flui para trás em direção a lamela como uma linha, chegou-se a lamela e não mais para a retaguarda fluxo pode ser rastreado.
- Medir a distância em µm entre a ponta da lamellipodium e parte de trás da fluorescência recuperada. Esta distância corresponde à soma do fluxo retrógrado e distâncias de protrusão.
- Como alternativa, para separar a protrusão de fluxo retrógrado, marca a ponta de lamellipodial com um quadro de uma linha antes do fotobranqueamento. Uso a linha como referência apontar em quadros posteriores para se referir a posição original da ponta do lamellipodium no momento da fotobranqueamento; o ponto de referência pode ser usado para medir a distância de protrusão e fluxo retrógrado.
- Observe o tempo (em segundos) necessário para a recuperação de fluorescência após fotobranqueamento ocorra. O tempo pode ser calculado manualmente a partir da taxa de quadros ou visualizado pela metamorfose através da "medida | Ferramenta de medição da região".
- Derive a taxa de polimerização de actina, usando a seguinte equação (com alguns parâmetros da equação baseados em medições de metamorfose de passos 8.4 e 8.6):
 Equação 6
Equação 6
onde taxa de polimerização de actina é em µm/min, distância do fluxo retrógrado é em µm, lamellipodial protrusão distância é em µm e tempo é em segundos.
9. análise da difusão de proteína e mobilidade em cima Photoactivation
Nota: O método aqui apresentado descreve a análise da mobilidade de monômero de actina empregando photoactivation de actina fundida ao PA-GFP, como ilustrado pela visualização e quantificação de difusão de proteína através do citosol.
- Para medição da difusão da actina photoactivatable longe de uma região citosólica, bem como o acúmulo dentro de uma região de lamellipodial, use metamorfose para determinar a intensidade ao longo do tempo nas seguintes regiões (ilustrado na Figura 3,um ): uma região citosólica fotoativados (PA); uma região de lamellipodial, no qual fotoativados proteínas espera-se que se acumulam ao longo do tempo (Lam); uma região fora o celular usado para normalização da fluorescência do fundo (para fora).
- Ao determinar a mobilidade de actina dentro do citosol, medir distintas regiões citosólica (veja a Figura 3um, regiões R1-R5). Note-se que a aquisição de fotobranqueamento não pode ser determinada de forma semelhante ao FRAP, devido a um aumento na fluorescência focal - e eventualmente toda a célula após a ativação.
- Transferi os valores de intensidade para todas as regiões da metamorfose em uma planilha do Excel, conforme descrito na etapa 7,4.
- Para analisar a taxa de deslocamento de photoactivatable actina longe da região citosólica de fotoativação ou sua taxa de incorporação dentro de uma região de lamellipodial (ambos representados como intensidade percentual da região citosólica fotoativados no tempo 0) , gerar curvas de fluorescência dos dados no passo 9.3. Aplicam-se as seguintes equações:
 Equação 7
Equação 7 Equação 8
Equação 8
Onde: PATn é a intensidade de uma região citosólica fotoativados para cada quadro de interesse seguindo o photoactivation; LAMTn é a intensidade de uma região de lamellipodial para cada quadro de interesse seguindo o photoactivation; FORATn é a intensidade de uma região tomada fora da célula (fundo) para cada quadro de interesse seguindo o photoactivation; PAT-1 é a intensidade de uma região citosólica fotoativados antes photoactivation; LAMT-1 é a intensidade de uma região lamellipodial antes photoactivation; T-1 é a intensidade de uma região tomada fora da célula (fundo), antes de fotoativação; PAT0 é a intensidade de uma região citosólica fotoativados no tempo 0 (ou seja, primeiro quadro após fotoativação); eT0 é a intensidade de uma região tomada fora da célula (fundo) no tempo 0 (ou seja, primeiro quadro após fotoativação). - Opcionalmente, para melhor visualização dos dados, normalize as curvas de intensidade para 0 subtraindo-se a intensidade do primeiro quadro após a fotoativação de cada quadro subsequente.
Nota: O seguinte método de análise (etapas 9,6-9,8) também permite calcular a dispersão citosólica da actina fotoativados dentro do citosol. - Medir as intensidades para regiões citosólica, que consecutivamente são posicionadas distalmente da região de ativação.
- Para representar as intensidades destas regiões como intensidade percentual da região fotoativados no tempo 0, aplica a equação 8, onde lamellipodial as intensidades são substituídas com intensidades para cada ROI citosólica. O tamanho e o número das regiões podem variar dependendo da distância de tamanho e dispersão de célula a ser medido.
- Para derivar um valor quantificável para a taxa de proteína fotoativados infiltrando cada região citosólica, Cole trama do Sigma (semelhante à análise FRAP na secção 7) o tempo e os valores da curva de aumento de intensidade de fluorescência para cada região, use Equação 2 e equação 4 para derivar o intervalo da intensidade de fluorescência, atingindo um platô. Compare os valores de1/2 t entre diferentes grupos experimentais.
Resultados
Figura 1 g h mostrar imagens de contraste de fase de uma célula de fibroblasto NIH3T3 prévia e 10 min pós-microinjeção de Rac1, que é uma pequena GTPase de Rho-família capaz de induzir a formação de lamellipodia por meio de sua interação com a onda complexa. A célula é visualizada primeiro antes da microinjeção (Figura 1g), para confirmar sua viabilidade e morfologia, por exemplo, a falta de lamellipodia. Em 10 min após microinjeção, a célula claramente mudou sua morfologia, o que é esperada deste tratamento e indica uma sucesso injeção (Figura 1h).
Pela simplicidade e clareza, em seguida fornecemos resultados exemplares para análise FRAP e photoactivation nas células, que não tenham sido injetadas adicionalmente.
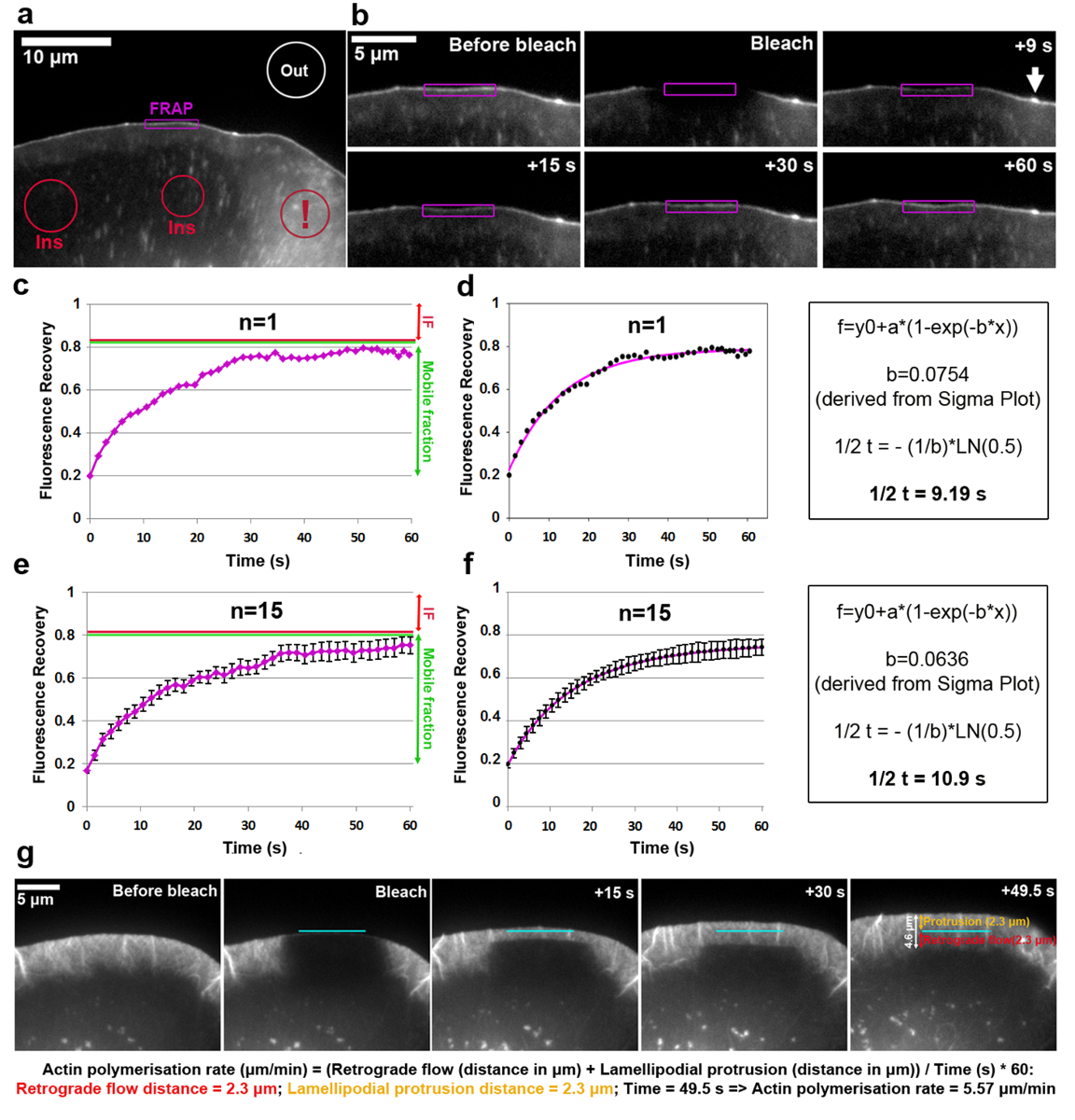
Análise do volume de negócios da VASP EGFP marcados na ponta de lamellipodium é mostrado na Figura 2a f. Observe que a VASP além destinos para nascentes e focais aderências, alongadas e pequenos pontos na cela interior18,19. A intensidade de fluorescência de uma região lamellipodial com uma acumulação de VASP clara na ponta foi descorada e medida para cada período de tempo, antes, durante e após o clareamento, como o lamellipodium se projeta para a frente, seguindo o contorno do ROI. Branqueada EGFP-VASP proteínas estão sendo recicladas por moléculas não-branqueada nesses locais, uma recuperação gradual da fluorescência é observar (Figura 2b). A curva de recuperação FRAP obtidos desta forma e normalizado para a intensidade de pré-lixívia (expressa em 1) pode ser vista na Figura 2c. Eficiência de fotobranqueamento pode variar e foi cerca de 20% do valor antes de branqueamento neste exemplo, como determinado a partir do valor no t0 (o primeiro quadro após fotobranqueamento). O aumento da fluorescência atinge um platô no exemplo mostrado em aproximadamente 80% da fluorescência antes de branqueamento. Em uma estrutura estática durante o tempo da experiência, como uma adesão focal, a diferença entre a intensidade de pré-lixívia e a fluorescência do planalto chegou após recuperação é definida como a fração de imóvel (se, seta vermelha na Figura 2 c, e), Considerando que a quantidade de fluorescência recuperado entre o tempo de recuperação completa e branqueamento é definida como a fração móvel (verde seta dupla em Figura 2c, i). Observe que em uma estrutura dinamicamente mudança como a ponta de lamellipodium analisada aqui, na medida do que pode não só representam moléculas imóveis, mas também derivar de uma redução da velocidade de protrusão, como intensidade EGFP-VASP é conhecida por depender disto parâmetro18. Para calcular o intervalo de recuperação, uma ajuste de curva foi criada na trama Sigma (Figura 2d). Neste caso, o valor do parâmetro "b" extraído de resolver a equação 2 é igual a 0.0754, que, quando aplicado à função logarítmica (equação 4) resulta em um intervalo estimado de recuperação de 9.19 s (Figura 2d , painel de extrema-direita), que é relativamente rápido nessa célula específica em comparação com a média publicada anteriormente5. Deve-se notar que meio-tempos de recuperação podem às vezes variar significativamente de uma célula para outra dentro da mesma população. Portanto, para a obtenção de resultados representativos, recomendamos determinar este parâmetro como uma média pelo menos 15-20 células. Para ilustrar o grau de variação, média aritmética da VASP-EGFP recuperação em média de 15 células para cada ponto de tempo foram gerados (Figura 2e), e curva média se encaixa criados e exibidos de forma análoga (Figura 2 f).
A taxa de polimerização da rede lamellipodial actina compreende a soma do fluxo retrógrado e rede para a frente. FRAP pode ser aplicado para medir a taxa de polimerização de actina pelas células transfecting (no caso B16-F1) com EGFP-tag β-actina e fotobranqueamento uma região saliente de lamellipodial (Figura 2g). Para análise de polimerização de rede lamellipodial actina, a recuperação de fluorescência após branqueamento de EGFP-tag β-actina é avaliada ao longo do tempo. Como a polimerização de progride de monômeros de actina nas extremidades farpadas de lamellipodial filamentos de actina (que todos apontam para a frente20), a rede é constantemente translocados para a retaguarda e progredindo para a frente, as taxas do que podem ser facilmente obtidas através da recuperação de fluorescência após fotobranqueamento polarizada. Recuperação de fluorescência da lamellipodium está completa quando a zona branqueada atingiu a zona de transição entre a parte posterior da lamellipodium e a lamela, que é caracterizada por uma menor densidade de mais pacotes de filamento dispostas horizontalmente virando-se muito mais lentamente do que o que se observa no lamellipodium. Conforme ilustrado na Figura 2g, recuperação de fluorescência pode ser visualizada como uma linha horizontal para a borda e retornando em direção a lamela, que permite medir as distâncias de protrusão e fluxo retrógrado (representado individualmente no painel de extrema-direito da Figura 2g como flechas bicéfala laranja e vermelhas, respectivamente).
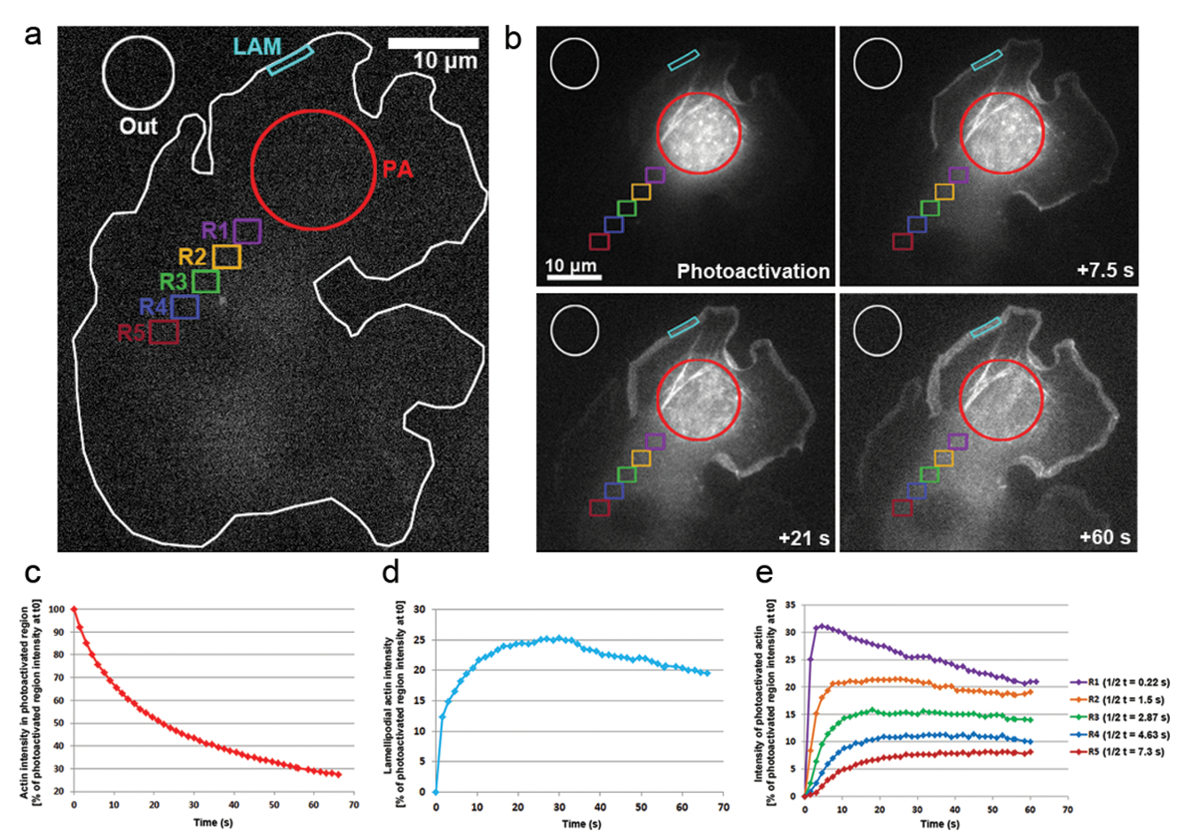
Temos também aplicado photoactivation em B16-F1 células transfectadas com PA-GFP-actina para controlar a mobilidade dos monômeros de actina dentro do citosol e a taxa de sua incorporação dentro salientes lamellipodia. Conforme ilustrado na Figura 3, b, uma região citosólica foi fotoativados por exposição ao laser 405 nm, enquanto imagens foram adquiridas no GFP canal cada 1,5 s para visualizar a distribuição de GFP-etiquetado, fotoativados actina. GFP-actina de Photoactivated pode ser visto difusão fora da região citosólica em Figura 3b. A taxa de diminuição de intensidade de fluorescência na região citosólica fotoativados é representada como a porcentagem da intensidade inicial no t0 (primeiro frame após fotoativação; Figura 3 c). fotoativados actina também integra-se nas pontas de lamellipodia, onde novos monômeros de actina são adicionados aos fins farpados crescentes da alongando filamentos de actina durante a protrusão. Para estimar a taxa de incorporação de lamellipodial, Nós medimos a intensidade da fluorescência ao longo do tempo de um contorno bidimensional/região de aproximadamente 5 µm de largura e 1 µm de altura; a região era constantemente re-posicionada na ponta da lamellipodium como que se projetava. Incorporação de actina foi representada como a porcentagem da intensidade de fluorescência da região citosólica fotoativados no t0 (Figura 3d). Como alongamento de filamentos de actina progrediu, monômeros de actina novos foram incorporados na frente lamellipodial. Uma fração destes monômeros de actina estocàstica foi derivada da piscina citosólica onde monômeros foram fotoativados. Isso resulta no rápido aumento da fluorescência em lamellipodia no primeiro 20 s após a fotoativação. Como monômeros novos estão sendo adicionados para a frente de lamellipodial, fluxo de monômeros de actina anteriormente incorporado com filamentos em direção a lamela por fluxo retrógrado. Ao longo do tempo, o ROI é completamente preenchido com monômeros fluorescentes e um platô em fluorescência é atingido (Figura 3d). Uma diminuição gradual da fluorescência é então observada quando, após a difusão dos monômeros fotoativados por toda a célula, não-fotoativados monômeros de actina estão cada vez mais sendo re-adicionados para a frente de lamellipodial. Esta diminuição na fluorescência irá encontrar um novo patamar, que será atingido assim um equilíbrio em toda a célula entre monômeros fotoativo e fotoativados não é alcançado (dados não mostrados).
A mobilidade dos monômeros de actina por todo o citosol derivou-se por medir intensidades de fluorescência em regiões de igual tamanho posicionado distalmente da região fotoativados (exemplificada na Figura 3um por regiões de cor codificada rotulado R1-R5). Conforme ilustrado na Figura 3e, a intensidade de fluorescência em cada uma destas regiões está diminuindo gradualmente longe do cytosolically fotoativados região, como a fração de monômeros de actina fotoativados torna-se cada vez mais diluída com Não está ativado monômeros (ou seja, não-fluorescente). Além disso, o pico de fluorescência é alcançado mais tarde: quanto mais distante a região medida está localizada da região fotoativados, quanto mais o tempo necessário para monômeros de actina que se espalham nestas regiões. Um valor representativo para o grau de infiltração de monômero de actina em cada região pode ser derivado por quantificar a metade do tempo de alcançar o planalto de fluorescência. Quanto mais distante a região, mais tempo demora para a actina fotoativados para se espalham, e assim, mais tempo é necessário para o planalto fluorescente ser alcançado, finalmente levando para um valor de1/2 t superior (Figura 3e).

Figura 1 : Procedimento de montagem e microinjeção de câmara de imagem. (um) componentes de câmara de imagem. (b) Silicone graxa é manchada cuidadosamente ao redor da abertura de um aferidor de plástico. (c) a lamela é posicionado com a celular-virados para cima no centro da câmara de imagem de abertura. selo (d) um seguro é estabelecido pelo posicionamento do aferidor de plástico em cima da lamela e apertando os grampos do lado. (e), microscopia médio é pipetado na ranhura da câmara. (f), a câmara de imagem é posicionada no palco microscópio, eletrodos e detector de calor estão ligados a uma unidade de aquecimento, pré-ajuste a 37 ° C e as células são permitidas para adaptar-se pelo menos 30 min antes de microscopia é iniciada. Neste exemplo, o estágio de microscópio também é equipado com um micromanipulador para a realização de microinjeções, e a agulha de microinjeção é mergulhada no meio de cobrir a camada de células na câmara de imagens. (g), An NIH3T3 celular de fibroblasto é visualizado antes microinjeção por microscopia de contraste de fase. A cruz vermelha no compartimento perinuclear indica a localização do microinjection futura, o que corresponde a uma região citoplasmática alta devido a proximidade com o núcleo volumoso. (h) 10 min após microinjeção com Rac1, a célula reage por proeminente formação de lamellipodia em torno da periferia de toda a célula (indicada pelas setas verdes). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: FRAP permite a determinação das taxas de polimerização de actina proteína rotatividade ou lamellipodial. (um) representante exemplo de célula B16-F1 expressando EGFP-VASP antes fotobranqueamento de uma região de lamellipodial, conforme indicado. Diferentemente coloridas contornos/formas são etiquetadas para indicar quais regiões foram consideradas para medições de intensidade de fluorescência ao longo do tempo. Nota o contorno vermelho marcado com um ponto de exclamação, que rotula uma região citosólica posicionada em uma área que contém múltiplas vesículas e babados de superfície celular. Áreas dinâmicas como este devem ser evitadas para selecionar regiões de referência de fluorescência, como eles são caracterizados por fortes flutuações de curto prazo da fluorescência, potencialmente causando resultados imprecisos. (b) Lamellipodial região da célula expressando EGFP-VASP antes e depois de fotobranqueamento. A recuperação do sinal fluorescente depois fotobranqueamento dentro da região marcado em roxo é visualizada ao longo do tempo. A seta indica a ponta de um microspike, enriquecido para VASP, provavelmente devido à alta densidade de filamentos de actina Demetron lá19. (c) um exemplo de um FRAP curva de recuperação como derivado de quantificar a intensidade fluorescente de lamellipodium a foto (contorno roxo) no b. vermelho e linhas verdes à direita indicam, respectivamente, fracções de imóveis e móveis. (d), um ajuste da curva de recuperação o FRAP em c (painel esquerdo) e um exemplo do método de cálculo utilizado para obter o intervalo de recuperação (painel direito). (e), um exemplo de uma recuperação FRAP curva derivada de uma média das curvas de recuperação de fluorescência de 15 células, com barras de SEM, indicando o grau de variabilidade dentro da população de amostra. (f), uma curva de ajuste derivado em média os ajustes de curva de recuperação FRAP 15 células (painel esquerdo) e um exemplo do método de cálculo utilizado para obter o intervalo de recuperação (painel direito). (g) Time-Lapse painéis de salientes lamellipodium de uma célula B16-F1 expressando com tag EGFP β-actina, antes e depois de branqueamento de uma região lamellipodial conforme indicado, seguida pela recuperação de fluorescência no lamellipodium ao longo do tempo. No painel do lado direito, os valores medidos para protrusão e retrógradas distâncias são fornecidas (em laranja e vermelho, respectivamente). Cálculos sob os painéis de imagem revelam como a soma de protuberância e retrógradas distâncias são usados para derivar a taxa de polimerização da rede lamellipodial de actina. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Photoactivation de PA-GFP-actina de monômero de rastreamento em toda a célula. (um) A exemplo representativo de uma célula B16-F1 expressando PA-GFP-actina antes de acionar o photoactivation numa região citosólica, conforme indicado pelo círculo vermelho (PA). Diferentemente coloridos contornos são etiquetados para indicar quais regiões foram consideradas para medições de intensidade de fluorescência ao longo do tempo. (b), uma ilustração da distribuição temporal dos seguintes PA-GFP-actina photoactivation. Note que a redução gradual da fluorescência no fotoativados, região citosólica (círculo vermelho), como a actina fotoativados difunde-se longe dela. Devido a sua difusão para a frente e montagem da rede, fotoativados monômeros de actina são gradualmente reforçados em lamellipodia (região de ciano) e por todo o citosol (regiões com códigos de cores diferentes), em uma forma de distância e tempo-dependente. (c) declínio temporal, representante da fluorescência na região citosólica fotoativados (contorno vermelho em b). (d) Temporal alterações na intensidade de fluorescência na região de lamellipodial (ciano contorno em b). (e) curvas representativas das mudanças temporais na intensidade da fluorescência das regiões citosólica (Color-coded em b), devido ao posicionamento em distâncias variáveis da área de fotoativação. Observe como meio-tempos de alcançar a fluorescência Planalto (indicado na legenda à direita) aumenta com a distância de dada região para a área de fotoativação, provável correlacionando com o aumento vezes necessários para difusão dos monômeros de actina para o respectiva região. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Aqui vamos discutir passos críticos nas técnicas descritas neste artigo, e como eles podem ser otimizados para aplicação em diferentes condições experimentais.
Microinjeção é um método que pode ser aplicado para monitorar nas células, os efeitos imediatos de introduzir proteínas exógenas, inibidores ou drogas. Pode ser particularmente vantajoso para determinar as funções das proteínas em difícil para transfect tipos de células ou em situações em que a expressão a longo prazo não é desejada. Deve-se notar que a sobrevivência de certos tipos de célula varia de acordo com a matriz extracelular, que são semeados no. Mais endoteliais, epiteliais ou fibroblastos, como tipos de células, mesmo pequenas, como keratocytes de peixe (ver Dang et al 21 e Anderson e Cruz22) podem ser injetados com sucesso. No entanto, existem exceções, como as células B16-F1 semeadas na laminina, que constituem um excelente modelo sistema de migração celular, mas que são incompatíveis com injeção neste tipo de substrato por motivo desconhecido. Para as células de fibroblastos NIH3T3, executamos rotineiramente injeções em substrato de fibronectina e técnicas photomanipulation adicionais tais como FRAP (mesmo com photoactivation; exibido por células B16-F1 aqui) podem ser igualmente bem executada nesses fibroblastos (ver por exemplo, Köstler et al 3). ele também deve ser considerado que as proteínas diferentes, de acordo com suas propriedades funcionais e os objetivos do experimento, podem levar a diferentes quantidades de tempo para causar alterações, variando de segundos a horas. Uma vantagem da técnica é que a dose/concentração do agente exógeno podem ser controlada com mais precisão no nível da célula única do que por exemplo, quando usando a transfeccao Plasmideo. Além disso, marcação fluorescente de uma proteína não é uma necessidade para garantir a sua presença na célula, o que pode aumentar a flexibilidade se visualização simultânea de multi-canal de outras proteínas fluorescente-etiquetadas é necessária. Microinjeção pode ser particularmente útil para analisar os efeitos instantâneos de proteínas específicas ou misturas de proteínas em mudanças dinâmicas de morfologia celular ou citoesqueleto (por exemplo, Dang et al 21 para obter um exemplo de efeitos instantâneos na migração por Arp2/3 complexo inibidor Arpin). Uma desvantagem da técnica é sua capacidade de invasão, que pode causar danos celulares ou influenciar a morfologia celular. Portanto, uma consideração importante ao realizar microinjeções está monitorando a viabilidade celular. O método apresentado aqui baseia-se na manipulação manual. Em condições testadas para serem compatíveis com injeções bem sucedidas, como fibroblastos, crescendo em substrato de fibronectina, o protocolo de injeção manual descrito aqui permite uma taxa de sucesso de 100% perto; Isto é essencial ao combinar esta abordagem com experimentos de acompanhamento sofisticados e demorados incluindo vídeo microscopia ou FRAP, como publicado anteriormente3. Isto não exclui que, ocasionalmente, células individuais podem sofrer de um evento de microinjeção, que pode com segurança ser reconhecido por mudanças abruptas de contraste do núcleo e do citoplasma, seguido de retração de borda de célula. Tais casos raros experimentais são excluídos e, portanto, não são considerados para mais análises.
No entanto, uma abordagem de meio automático também é comumente usada, por exemplo, empregando rápida (< 300 ms) agulha de máquina controlada reduzindo coincidente com aumento de pressão de injeção, para que a agulha só deve ser colocado acima de cada célula antes da respectiva injeção. A taxa de sucesso de injeções de metade-automático é por definição mais baixa do que a abordagem manual descrita acima, simplesmente porque ele é otimizado para velocidade, seguido de análise de várias células que sobreviveram com sucesso este tratamento; assim, ele não depende de injeção bem sucedida de uma célula individual. Portanto, em oposição à análise da única célula, injeções de metade-automático são mais adequadas para analisar os efeitos da injeção de células várias centenas, por exemplo, pelo vídeo microscopia na ampliação baixa ou mediante fixação da pilha e coloração. Independentemente da abordagem detalhada empregada, microinjeção não constitui um ensaio de ponto de extremidade, mas pode ser combinada com uma variedade de técnicas, incluindo FRAP ou fotoativação3.
Ao determinar a taxa de rotatividade de proteína por FRAP, a intensidade do laser deve ser otimizada, dependendo das condições de instalação e de imagem do microscópio (ampliação, objectivos, etc., bem como o tipo de célula, estrutura e proteína fluorescente para fotobranqueamento). Note que a potência ideal do laser, clareamento eficiente é combinado com o Fotodano menos possível, para evitar o encolhimento ou completar a retração da estrutura sob análise (por exemplo, lamellipodia ou filopodia) ou até mesmo danos a nível celular. Idealmente, pelo menos 70 a 80% de eficiência de clareamento deve ser alcançado, embora branqueamento completo pode ser prejudicado pelo volume de negócios extremamente rápida da proteína, em que caso, qualquer coisa acima de 50% também pode ser aceitável. Óptimo poder clareador para uma determinada estrutura e tintura fluorescente deve ser testado experimentalmente, a partir de um poder do laser de baixa seguido por seu aumento gradual. Claro, qualquer tintura fluorescente pode por definição ser branqueada com laser luz perto de seu pico de excitação (488 nm para corantes verdes usados com frequência como FITC ou EGFP). No entanto, lasers com comprimentos de onda mais curtos, tais como lasers de perto-UV, entregam poderes superiores e, portanto, também podem ser usados para clareamento eficiente de corantes comumente usados. Nós empregamos rotineiramente um 405 nm laser diode (120 mW) para branqueamento de EGFP e vermelhos corantes fluorescentes (como mCherry), embora com uma eficiência ligeiramente inferior no caso dos último (dados não mostrados). Como a 405 nm-diodo também pode ser usado para fotoativação de PA-GFP (veja abaixo), ele dota este sistema com a máxima flexibilidade.
Para as estruturas de célula B16-F1 e proteínas fluorescentes foto aqui, 405 poderes nm-laser entre 65 e 100 mW foram aplicados. Ao analisar uma região da foto, é importante considerar se a determinada estrutura é preservada em sua forma original com a análise da período de tempo. Por exemplo, ao analisar o volume de negócios de proteínas no lamellipodia dicas, cuidados devem ser tomados se a curvatura do lamellipodia é significativamente alterada ao longo do tempo, como alterações na curvatura podem levar a resultados imprecisos se a região/contorno analisado não totalmente abrange a totalidade da estrutura em cada quadro medido. Além disso, note-se que pacotes incorporados em lamellipodia, como microspikes, podem causar desvios na intensidade de fluorescência. Conforme ilustrado na Figura 2b (seta branca no prazo de 9 s), uma estrutura de microspike situa-se próximo à região de foto medido, mas permanece fora dele durante toda a duração da medição e, portanto, não causa qualquer imprecisão. Para a análise do volume de negócios de proteína, considerações importantes quando selecionando a localização e tamanho de analisados regiões são que sua fluorescência ao longo do tempo não deve ser significativamente influenciada por alterações na morfologia celular ou fatores além de duro para evitar aquisição de fotobranqueamento. Por exemplo, estruturas, proporcionando significativa contribuição quantitativa para a estrutura analisada não devem mover fora da região medida durante a análise; Além disso, entidades independentes, fluorescentes, tais como estruturas vesiculares que atraem a proteína não devem entrar no campo de interesse durante a análise. Para determinar a taxa de polimerização de actina lamellipodial, tenha cuidado que não retracção ou ruffling (ou seja, para cima de dobramento) lamellipodia são analisados, como isso influenciará fortemente a precisão dos resultados. Além disso, retração das regiões lamellipodial pode aparecer como rápida translocação para a retaguarda, potencialmente levando à superestimação dos índices de polimerização de actina lamellipodial. Uma consideração adicional é a distância das regiões intracelulares normalização (tomado como posições de referência para a correção de aquisição fotobranqueamento) da posição real de fotobranqueamento, que deve ser grande o suficiente para evitar direto influenciar-se pela área de foto.
Quando a criação de condições óptimas para fotoativação de PA-GFP-etiquetado construções, deve ter cuidado para evitar o branqueamento instantâneo durante a fotoativação. Em nosso trabalho, os melhores resultados foram obtidos com os poderes do laser 5 - 10 vezes menor do que o normalmente empregado para o branqueamento da EGFP. Para aquisição de imagens de moléculas fotoativo, tempo de exposição e intervalo de tempo entre os quadros devem ser otimizados, considerando o tamanho das regiões e estruturas para ser fotoativados e analisados, bem como a mobilidade potencial de fotoativo proteínas para outros locais subcellular. Quanto a todos os tipos de imagens de fluorescência, a manutenção da viabilidade celular é crucial para a obtenção de resultados relevantes fisiologicamente.
Em princípio, verde e vermelho photoconversion de proteínas fluorescentes como mEos ou Dronpa variantes12 constitui um método igualmente poderoso da seguinte dinâmica e volume de negócios de estruturas subcelulares, tais como o lamellipodium (veja por exemplo, Et al . Burnette 23). a vantagem do último método em vez de PA-GFP seria a possibilidade de acompanhar a dinâmica da proteína antes e depois da conversão, com duas cores distintas, sem a necessidade de co expressar uma proteína fluorescente vermelha adicional. No entanto, em nossos experimentos preliminares, a extensão da mudança de contraste e intensidade do sinal fluorescente alcançado após fotoativação de PA-GFP foram maiores em comparação com sondas photoconverted, talvez devido às características espectrais superiores de verde contra vermelho sondas fluorescentes (dados não mostrados). Em qualquer caso, estudos detalhados sobre a rotatividade de filamento de actina em saliências de borda de célula como lamellipodia ou caudas de actina induzida pelo vírus Vaccinia até agora só foram publicados usando PA-GFP derivados5,6,24.
Ao considerar que região da célula para analisar após fotoativação, vários fatores devem ter em conta, que são discutidos usando o exemplo específico mostrado aqui (incorporação de monômeros de actina na borda de célula após a ativação no citosol), mas Certamente pode ser extrapolada para vários problemas científicos análogos. Primeiro, quando medir a taxa de incorporação de lamellipodial de cytosolically fotoativados proteínas, por exemplo, em distintas condições experimentais (como mostrado em Dimchev et al 6), tamanhos de regiões citosólica e suas distâncias a lamellipodial as bordas devem ser comparáveis entre os grupos experimentais. Também é importante considerar que, quando photoactivating regiões citosólica, a espessura da célula é maior em posições mais perto para o núcleo. Ativar regiões celulares mais grossas pode resultar em quantidades mais elevadas de proteínas ativadas, dado que a distribuição da proteína a ser ativado é distribuída homogênea no citosol. Por último, os níveis de expressão da proteína a ser ativado certamente podem ser altamente variáveis em células individuais. Devido a todas estas considerações de variabilidade, é crucial comparar níveis de incorporação de proteínas cytosolically registrados em outro lugar na célula em relação a fluorescência total obtida após a ativação em regiões específicas.
Descrevemos como microinjeção pode ser usado como uma ferramenta para investigar os efeitos das proteínas na morfologia celular e ter exemplificado isso demonstrando a potente indução de lamellipodial estruturas em NIH3T3 células de fibroblastos injetadas com o pequena GTPase Rac1. Anteriormente nós aplicamos esta técnica para interferir com a função de Arp2/3 em células injetadas com o domínio C-terminal WCA da cicatriz/WAVE3. Vários parâmetros em células microinjected podem ser analisados por outros ensaios, tais como o FRAP ou fotoativação. Descrevemos como FRAP e photoactivation podem ser empregados para investigar a subcellular dinâmica e mobilidade de monômeros de actina. FRAP tem sido usado por nosso grupo anteriormente5 para investigar o volume de negócios de proteínas, localizando-se a lamellipodia, tais como a VASP, cofilin, Abi, ciclina e tampando a proteína, ou para elucidar o volume de negócios de componentes em aderências focais na presença e ausência de Rac4de sinalização. Além disso, taxas de polimerização de actina de medição pode ser realizado por fotobranqueamento β-actina marcados EGFP5, mas existem métodos alternativos. Heterogeneidades fluorescentes de rastreamento, como visto por sondas de imagem compatível com célula viva rotulando os filamentos de actina celulares, tais como Lifeact,25, também pode ser empregados6,26. A vantagem aqui é que a superexpressão de β-actina pode ser evitado, que é capaz de aumentar a protrusão de borda de célula e migração e, portanto, potencialmente interfere com o ensaio específico ou pergunta experimental (ver por exemplo, Kage et al 26. o; Peckham et al 27). no entanto, uma clara desvantagem da sonda Lifeact constitui sua rápida de ligar/desligar cinética de ligação a filamentos de actina, para que o branqueamento de estruturas de filamentos de actina etiquetadas por Lifeact nas células fornece informações apenas sobre o volume de negócios de sonda, Mas não o volume de negócios dos filamentos de actina, ao qual, liga-se a25. O acompanhamento das heterogeneidades de fluorescência anteriormente empregado6,26 fornece um compromisso prático, muito semelhante do rastreamento amplamente utilizado de fluorescência speckles incorporada filamentosas do citoesqueleto estruturas (ver, por exemplo, salmão e Waterman28), mas pode não ser tão direta para usar e tão precisa quanto FRAP de estruturas com tag EGFP F-Actina. Photoactivation tem sido aplicado por nós para estimar as taxas de actina monomérico incorporação salientes lamellipodia, bem como a sua mobilidade em todo o citoplasma, no contexto da F-Actina citosólica experimentalmente atento níveis6. A técnica é útil quando examinar a mobilidade e a distribuição de proteínas provenientes de áreas relativamente grandes, tais como regiões citosólica. No entanto, examinar a distribuição de proteínas derivadas de fotoativados relativamente pequenas estruturas; por exemplo, os cones de crescimento podem ser um desafio devido ao baixo número de moléculas fluorescentes ativados, os sinais fracos e assim, falta de sensibilidade. Técnicas alternativas potenciais photoactivation ou photoconversion de fluorescência (veja acima) podem incluir inverso FRAP, que se baseia em fotobranqueamento toda a célula excepto o ROI, seguido de acompanhamento a mobilidade das moléculas fluorescentes de nesta região. A técnica não requer superexpressão versões photoactivatable de proteínas, mas sempre envolverá a exposição a uma dose invulgarmente elevada de poder do laser, causando potencialmente indesejáveis efeitos colaterais, como Fotodano.
Claramente, a fotoativação e FRAP não consegue distinguir se as proteínas estão se movendo como monômeros, dímeros ou mesmo pequenas oligómeros e se movem-se em combinação com os parceiros de ligação adicionais. Informações desse tipo podem ser obtidas em vez de fluorescência espectroscopia de correlação técnicas29 ou, alternativamente, FLIM-FRET30. Não obstante, FRAP e photoactivation constituem abordagens simples para avaliar diretamente a dinâmica local e global de proteínas nas células, independentemente da proteína de interesse, Localização subcellular ou tipo de célula estudado.
Divulgações
Os autores não têm nada para divulgar.
Agradecimentos
Agradecemos a Fundação de pesquisa o alemão (DFG) apoio financeiro (grant Nr. RO2414/5-1 para KR).
Materiais
| Name | Company | Catalog Number | Comments |
| B16-F1 mouse skin melanoma cells | American Type Culture Collection, Manassas, VA | CRL-6323 | |
| NIH-3T3 cells | American Type Culture Collection, Manassas, VA | CRL-1658 | |
| DMEM 4.5g/L glucose | Life Technologies, Thermno Fisher Scientific, Germany | 41965-039 | |
| Ham’s F-12 medium | Sigma-Aldrich | N8641 | |
| Fetal calf serum (FCS) | PAA Laboratories, Linz, Austria | A15-102 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich, Germany | F7524 | Lot054M3396 |
| MEM Non essential amino acids | Gibco, ThermoFisher Scientific, Germany | 11140035 | |
| L-Glumatine 200mM (100x) | Life Technolgies | 25030-024 | |
| Pen-Strep 5000 U/mL | Life technologies | 15070063 | |
| Sodium Pyruvate (100 mM) | Gibco, ThermoFisher Scientific, Germany | 11360-039 | |
| Laminin | Sigma-Aldrich | L-2020 | |
| Laminin coating buffer | Self-made: 50mM Tris ph7.4, 150mM NaCl | ||
| Fibronectin from human plasma | Roche Diagnostics, Mannheim, Germany | 11 051 407 001 | |
| Jetpei | Polyplus Transfection, Illkirch, France | 101-10N | |
| JetPei buffer | Polyplus Transfection, Illkirch, France | 702-50 | 150mM NaCl |
| PA-GFP-actin plasmid DNA | described in Koestler et al.2008 | ||
| pEGFP-actin plasmid DNA | Clontech, Mountain View, CA, USA | ||
| Rac1 protein for microinjection | Purified as GST-tagged version, and cleaved from GST prior to injection | ||
| Microinjection buffer | Self-made: 100mM NaCl, 50mM Tris-HCl ph7.5, 5mM MgCl2, 1mM DTT | ||
| Dextran, Texas Red, 70,000 MW, Lysine Fixable | Molecular Probes, Thermno Fisher Scientific, Germany | D1864 | |
| Microscope circular cover glasses 15mm, No.1 | Karl Hecht, Aisstent, Sondheim, Germany | 1001/15 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf, Hamburg, Germany | 5242 956 003 | |
| Eppendorf Femtotip Microinjection Capillary Tips | Eppendorf, Hamburg, Germany | 930000035 | |
| Silicone Grease | ACC Silicones, Bridgewater, England | SGM494 | |
| Aluminium Open Diamond Bath Imaging Chamber | Warner instruments | RC-26 | |
| Automatic temperature controller | Warner Instruments | TC-324B | |
| Microscope: Axio Observer | Carl Zeiss, Jena, Germany | ||
| CoolSnap-HQ2 camera | Photometrics, Tucson, AZ | ||
| Lambda DG4 light source | Sutter Instrucment, Novato, CA | ||
| Laser source | Visitron Systems | ||
| Eppendorf FemtoJet microinjector | Eppendorf, Hamburg, Germany | With built-in compressor for pressure supply | |
| Nikon Narishige Micromanipulator system | Nikon Instruments, Japan | ||
| Visiview software v2.1.4 | Visitron Systems, Puchheim, Germany | ||
| Metamorph software v7.8.10 | Molecular Devices, Sunnyvale, CA | ||
| Sigma Plot v.12 | Systat Software Inc. |
Referências
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 38 (10), 2887-2921 (2009).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Koestler, S. A., et al. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol Biol Cell. 24 (18), 2861-2875 (2013).
- Steffen, A., et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 126, 4572-4588 (2013).
- Lai, F. P., et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27 (7), 982-992 (2008).
- Dimchev, G., et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol Biol Cell. 28 (10), 1311-1325 (2017).
- Koppel, D. E., Axelrod, D., Schlessinger, J., Elson, E. L., Webb, W. W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys J. 16 (11), 1315-1329 (1976).
- Patterson, G. H., Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297 (5588), 1873-1877 (2002).
- McKinney, S. A., Murphy, C. S., Hazelwood, K. L., Davidson, M. W., Looger, L. L. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 6 (2), 131-133 (2009).
- Gurskaya, N. G., et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 24 (4), 461-465 (2006).
- Lippincott-Schwartz, J., Patterson, G. H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 19 (11), 555-565 (2009).
- Kremers, G. J., Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J Vis Exp. (40), (2010).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 4988 (2008).
- Small, J. V., Rottner, K., Carlier, M. F. . Actin-based Motility. , (2010).
- Kaverina, I., et al. Enforced polarisation and locomotion of fibroblasts lacking microtubules. Curr Biol. 10 (12), 739-742 (2000).
- Small, J., Rottner, K., Hahne, P., Anderson, K. I. Visualising the actin cytoskeleton. Microsc Res Tech. 47 (1), 3-17 (1999).
- Mikhailov, A. V., Gundersen, G. G. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskeleton. 32 (3), 173-186 (1995).
- Rottner, K., Behrendt, B., Small, J. V., Wehland, J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1 (5), 321-322 (1999).
- Svitkina, T. M., et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 160 (3), 409-421 (2003).
- Small, J. V., Isenberg, G., Celis, J. E. Polarity of actin at the leading edge of cultured cells. Nature. 272 (5654), 638-639 (1978).
- Dang, I., et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 503 (7475), 281-284 (2013).
- Anderson, K. I., Cross, R. Contact dynamics during keratocyte motility. Curr Biol. 10 (5), 253-260 (2000).
- Burnette, D. T., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 13 (4), 371-381 (2011).
- Humphries, A. C., et al. Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell Host Microbe. 12 (3), 346-359 (2012).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 5 (7), 605-607 (2008).
- Kage, F., et al. FMNL formins boost lamellipodial force generation. Nat Commun. 8, 14832 (2017).
- Peckham, M., Miller, G., Wells, C., Zicha, D., Dunn, G. A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 114, 1367-1377 (2001).
- Salmon, E. D., Waterman, C. M. How we discovered fluorescent speckle microscopy. Mol Biol Cell. 22 (21), 3940-3942 (2011).
- Machan, R., Wohland, T. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 588 (19), 3571-3584 (2014).
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J Microsc. 247 (2), 119-136 (2012).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados