
Method Article
Micromanipulation Techniques Allowing Analysis of Morphogenetic Dynamics and Turnover of Cytoskeletal Regulators
In This Article
Summary
We describe how micro- and photomanipulation techniques such as FRAP and photoactivation enable the determination of motility parameters and the spatiotemporal dynamics of proteins within migrating cells. Experimental readouts include subcellular dynamics and turnover of motility regulators or of the underlying actin cytoskeleton.
Abstract
Examining the spatiotemporal dynamics of proteins can reveal their functional importance in various contexts. In this article, it is discussed how fluorescent recovery after photobleaching (FRAP) and photoactivation techniques can be used to study the spatiotemporal dynamics of proteins in subcellular locations. We also show how these techniques enable straightforward determination of various parameters linked to actin cytoskeletal regulation and cell motility. Moreover, the microinjection of cells is additionally described as an alternative treatment (potentially preceding or complementing the aforementioned photomanipulation techniques) to trigger instantaneous effects of translocated proteins on cell morphology and function. Micromanipulation such as protein injection or local application of plasma membrane-permeable drugs or cytoskeletal inhibitors can serve as powerful tool to record immediate consequences of a given treatment on cell behavior at the single cell and subcellular level. This is exemplified here by immediate induction of lamellipodial cell edge protrusion by the injection of recombinant Rac1 protein, as established a quarter-century ago. In addition, we provide a protocol for determining the turnover of enhanced green fluorescent protein (EGFP)-VASP, an actin filament polymerase prominently accumulating at lamellipodial tips of B16-F1 cells, employing FRAP and including associated data analysis and curve fitting. We also present guidelines for estimating the rates of lamellipodial actin network polymerization, as exemplified by cells expressing EGFP-tagged β-actin. Finally, instructions are given for how to investigate the rates of actin monomer mobility within the cell cytoplasm, followed by actin incorporation at sites of rapid filament assembly, such as the tips of protruding lamellipodia, using photoactivation approaches. None of these protocols is restricted to components or regulators of the actin cytoskeleton, but can easily be extended to explore in analogous fashion the spatiotemporal dynamics and function of proteins in various different subcellular structures or functional contexts.
Introduction
Monitoring the spatiotemporal dynamics of proteins and other molecules in living cells has become an essential tool in many fields of cell and molecular biology. Advanced fluorescence microscopy techniques including fluorescence resonance energy transfer (FRET) and FRET-fluorescence lifetime imaging (FRET-FLIM), or FRAP, fluorescence loss in photobleaching (FLIP) and photoactivation as well as many others allow for the temporal and spatial tracking of protein-protein interactions, conformational changes, as well as determining the kinetics of diffusion and localization of different proteins in the cell1,2. FRAP and photoactivation techniques, in particular, are widely applicable for examining the regulators of the actin cytoskeleton and cell migration. These techniques can be applied alone or in combination with additional micromanipulation techniques such as microinjection3, and involve the expression of fluorescently-labeled proteins. They allow the estimation of the kinetics of protein association to actin-rich structures involved in cell migration, such as filopodia or lamellipodia, the turnover of proteins in focal adhesions4, or branched actin networks5. They also enable the determination of lamellipodial actin polymerization rates, the assessment of the dispersion of monomeric actin within the cytosol, the rate of subcellular actin monomer translocation to polymerizing actin filaments in protruding lamellipodia6, and other parameters.
FRAP is a method for visualizing and quantifying the mobility of proteins within a living cell, originally developed in the 1970s by Axelrod7. A region of interest (ROI) within a cell, populated with fluorescently-labeled proteins, is transiently exposed to a laser of high intensity, sufficient to cause bleaching of the fluorophore molecules present in this region during a given short period of time. The unbleached, fluorescently labeled proteins located outside the ROI during bleaching, will diffuse and infiltrate the bleached region depending on their spatiotemporal dynamics, causing the displacement of photobleached molecules over time. The rate of fluorescence recovery in bleached regions is dependent on various factors, including the size and the rate of diffusion of a given molecule, and of course its turnover rate within the putative associated bleached structure. Thus, soluble proteins will mediate the recovery of fluorescence within the bleached ROI rapidly through diffusion, while proteins tightly associated with structures, such as focal adhesions, will have longer turnover times, as their fluorescence recovery will depend both on the diffusion of the soluble fraction of the protein and dissociation-association kinetics of the structure-associated fraction. Fluorescence recovery is usually acquired and quantified until the initial level of pre-bleach intensity of fluorescence is reached. However, this does not occur if a part of the initial fluorescence intensity belongs to the so-called immobile fraction, which is unable to be replenished by diffusion or is replenishing at very slow rates as compared to the majority of molecules comprising the mobile fraction. To determine the rate of protein turnover, FRAP curves are generated, representing the extent of fluorescence recovery over time. From these recovery curves, average half-times of protein recovery can be calculated. By creating curve fits of the average FRAP data, and hence mathematical analyses, it is also possible to deduce whether the average turnover rate of the mobile fraction constitutes a composite of one homogeneous population of molecules, or whether it is composed of two or more subpopulations of molecules turning over at differential rates. In addition to estimating protein turnover rates by quantitative approaches, tracking the recovery of photobleached regions in lamellipodia can also allow for accurate quantification of lamellipodial motility parameters such as retrograde flow, protrusion, and actin polymerization rates. Thus, FRAP constitutes a versatile tool to be applied for assessing various parameters within structures of living cells.
Photoactivation is a method used to track the diffusion and mobility of proteins or molecules originating from a designated cellular location. The technique employs, for instance, a variant of wild-type green fluorescent protein (GFP), initially developed by Patterson and Lippincott-Schwartz8, which is mutated in a manner that allows its fluorescence to be highly increased upon exposure to ultraviolet (UV) light (around 400 nm; here, 405 nm). As described by Patterson et al., wild-type GFP chromophores exist as a mixed population of neutral phenols and anionic phenolates, which produce a major absorbance peak at approximately 397 nm and a minor one at 475 nm, respectively. Upon irradiation of the protein with UV light, the population undergoes photoconversion, shifting towards the anionic form. When excited by 488 nm, the photoconverted/photoactivated protein exhibits a 3-fold increase in fluorescence, insufficient in practice for distinguishing between activated and non-activated GFP due to the high intrinsic background fluorescence. However, a decrease in background intensity has been achieved by introducing a single amino acid mutation into the GFP sequence (histidine substitution at position 203). The resulting T203H mutant, also known as photoactivatable-GFP (PA-GFP) is characterized by a significant reduction in absorbance of the minor peak, which upon irradiation with UV light is increased nearly 100-fold when subsequently excited by 488 nm light. Hence, overexpression of PA-GFP-tagged proteins is a widely used approach, which allows the determination of diffusion and motility of molecules within cells. We have previously applied PA-GFP-tagged actin to determine the rate of dispersion of actin monomers away from cytosolic regions, allowing not only exploration of their mobility within the cytosol, but also their incorporation rate into the protruding lamellipodial actin network6. More recent literature also describes novel, photo-convertible proteins that can in principle be used in an analogous fashion, but harboring the potential advantage to be visible already before photo-conversion. Examples for this group of fluorescent proteins include Dendra2 and mEos29,10,11,12.
In this article, we explain the methodology of microinjecting cells with proteins. We further explain how this technique can be combined with FRAP, by photobleaching proteins involved in actin cytoskeleton regulation and motility, and how FRAP curves and half-time of recovery of mobile fractions can be derived. In addition, we provide an example of how the FRAP technique can be used to determine actin polymerization rates of lamellipodial networks. We also provide instructions and tips on how to perform photoactivation experiments, which can be used to determine cytosolic mobility of monomeric actin and rates of actin incorporation into lamellipodia. These techniques, of course, are not only limited to tracking actin cytoskeleton components, but upon potentially required moderate adaption or optimization, can be widely applied to other cell types or to investigate different proteins, structures, and parameters.
Protocol
1. Coverslip Washing and Sterilization
- Immerse 15 mm (diameter) cover glasses (no.1) in a 500 mL flask containing a mixture of 40 mL 37% HCl and 60 mL 100% EtOH (not more than 100 coverslips per 100 mL washing solution).
NOTE: Even if freshly purchased, coverslips must be stringently cleaned before seeding cells onto their surfaces. This is because they may contain thin films of grease, which are macroscopically invisible, but can efficiently interfere with adhesion and appropriate spreading of live cells. Whereas such films can be efficiently removed with solutions containing acid or base (see Fischer et al.13), we routinely use the acid/alcohol mixture described above. - Shake the flask containing the cover glasses for 30 min on a rotation shaker. Choose a speed that allows the cover glasses to be swirled freely, but slow enough to avoid frequent breaking. Filtrate the solution to remove broken glass pieces if reusing.
- Transfer the cover glasses to a flask containing at least 200 mL of sterile water and incubate on a rotation shaker, while repeatedly replacing the water until the acidic smell has vanished. Multiple washes over several hours are recommended for complete elimination of HCL-EtOH traces.
- Dry individual cover glasses on a sheet of filter paper.
- Place the cover glasses at the bottom of a 10 cm (diameter) Petri dish covered with filter paper, and heat dry-sterilize. Avoid autoclaving as this will cause cover glasses to stick together.
2. Treatment of Cells, Transfection, and Seeding onto Coverslips
- Grow B16-F1 mouse melanoma cells according to standard cell culture conditions in DMEM (4.5 g/L glucose) containing 10% fetal calf serum, 2 mM glutamine, and 1% penicillin-streptomycin at 37 °C, 7% CO2.
- Grow NIH3T3 fibroblast cells for microinjections according to standard cell culture conditions (tissue culture incubator at 37 °C, 7% CO2) in DMEM (4.5 g/L glucose) containing 10% fetal bovine serum, 1 mM sodium pyruvate, 1x MEM non-essential amino acids, 2 mM glutamine, and 1% penicillin-streptomycin.
- For transfections, grow B16-F1 cells to 100% confluence in a 10 cm dish and passage at a 1:5 ratio into a 3 cm (diameter) plastic dish.
- On the same day, after the B16-F1 cells were allowed to adhere for at least 6 h, transfect with 500 ng/dish of photoactivatable PA-GFP-actin or EGFP-tagged β-actin plasmid DNA. For co-transfections of PA-GFP-actin with mCherry-encoding vectors, mix a total of 1 µg of plasmid DNA per 3 cm dish.
- Transfect the B16-F1 cells with the transfection reagent (Table of Materials). For a 3 cm dish, mix 200 µL of 150 mM NaCl containing 500 ng of DNA construct with 200 µL of 150 mM NaCl containing 1 µL of transfection reagent (i.e., DNA (µg):reagent (µL) ratio of 1:2 was used).
- Incubate the transfection mixture for 20 min at room temperature (RT) and pipet drop-wise onto the 3 cm dish containing the cells. Gently swirl the dish to mix and incubate overnight at 37 °C, 7% CO2.
- Prepare the laminin coating buffer containing 50 mM Tris, pH 7.4 and 150 mM NaCl.
- For the B16-F1 cells, coat 15 mm cover glasses by spreading 150 µL of laminin (25 µg/mL in laminin coating buffer) and incubate for 1 h at RT. For the NIH3T3 cells, coat the cover glasses with fibronectin solution (25 µg/mL in phosphate-buffered saline (PBS)) and incubate for 1 h at RT.
- Wash laminin- or fibronectin-incubated cover glasses with PBS, then aspirate the PBS and add 2 mL of transfected cells.
- Seed the transfected B16-F1 cells (in 1:30 ratio from a confluent dish), on the day after transfection, onto laminin-coated coverslips. Seed the NIH3T3 fibroblasts (in 1:20 ratio from a confluent dish) onto fibronectin-coated coverslips.
- Allow the cells to spread on laminin- or fibronectin-coated cover glasses overnight in a tissue culture incubator at 37 °C prior to microscopy. Alternatively, microscopy experiments can be initiated on the same day, given that cells are allowed to spread for at least 2–3 h.
3. Assembly of Microscopy Imaging Chamber
- Use a heat conductive RC-26 aluminum imaging chamber for microscopy (Figure 1a). Smear the silicone grease around the contour of the plastic sealer opening using a syringe (Figure 1b).
- Place the cover glass with the cells side-up on the chamber (Figure 1c).
- Place the plastic sealer on top of the cover glass to make a secure seal between the coverslip and the chamber. Fix the plastic sealer (diagonally to avoid coverslip breakage) by screwing the sliding clamps onto the chamber to avoid the medium leaking (Figure 1d).
- Pipette 37 °C pre-heated microscopy medium into the central area. For medium reduced in autofluorescence and thus optimized for microscopy, use the same recipe as culture medium described above, but with F12-HAM instead of DMEM, additionally containing 20 mM HEPES for culturing of cells in the absence of CO2 (Figure 1e).
- Insert the heat detector into the designated slot of the chamber and link the electrodes of the chamber to a TC-324B automatic temperature controller maintaining a constant temperature of 37 °C (Figure 1f).
- Place a small drop of immersion oil onto the objective and place the chamber on top.
- Incubate the chamber with cells for at least 10–30 min to allow them to recover from the temperature drop during mounting and to adapt to the microscopy medium.
- Before microscopy is initiated, replace the culture medium in the central reservoir of the chamber (roughly 800 µL) to avoid inappropriate concentration of medium components and serum due to medium evaporation. Prolonged microscopy sessions with open chambers will require routine changing of the evaporating medium.
4. Microinjection Procedure
- Coat the coverslips, prepare the cells, and assemble the imaging chamber as described above.
- Thaw an aliquot of purified protein to be injected (typically 10 µL or less) and dilute it with the appropriate microinjection buffer.
NOTE: Buffer composition may vary according to protein and cell type, but take care to use a pH between 6.95 and 8.00 and avoid using PBS, as most cell don't like to be injected with PBS. - For Rac1 microinjection, prepare buffer containing 100 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 1 mM DTT. The Mg2+ ions are essential for small GTPase stability.
NOTE: Protein concentrations normally vary between 0.1–1 mg/mL (maximum 2 mg/mL), depending on the protein, type of experiment, and cell type. - If applicable, add fluorescent dye, such as inert dextran (0.5 µg/mL, 70 kDa) to the protein solution, which can confirm the presence of needle flow before injections and permits the documentation of successful injections after the experiment.
NOTE: The experiment here is not aimed at following the dynamics of injected Rac1, which would only be possible upon direct fluorescence labeling of the protein. Coupling of proteins with fluorescent dyes or fusion to a fluorescent protein is possible, but avoided here as it harbors the risk of interfering with the signaling function, in particular of small proteins like the Rho-family GTPase Rac1 (20 kDa). - Centrifuge the protein solution at 10,000 x g for at least 30 min to remove protein aggregates that can lead to needle clogging if present in the microinjection capillary.
- Load a microinjection needle (microinjection capillary) with 1 µL of injection mixture from the back side using a flexible pipette tip/microloader tip.
- If air bubbles are present in the needle tip, gently tap the needle base in order to remove them. Proceed rapidly to avoid drying of the needle tip, which could cause needle clogging.
- Carefully adjust the needle holder on the micromanipulation device. If using an inverted microscope for phase contrast imaging, before the needle loading, ensure there is enough space to move the needle up and down without obstructing the microscope condenser.
- Upon screwing the microcapillary onto the needle holder, apply pressure (20–50 hPa background pressure) to the needle using a microinjection pressure device before translocating the needle tip into the cell culture medium.
NOTE: Activating the pressure when the needle is in the medium will result in the medium being sucked up by capillary force, and thus prohibit injection of the solution of interest. - Position the needle in the field of view (facilitated by using low magnification objectives). A 40X dry objective for microinjection experiments was used here.
- Position the needle tip macroscopically in a vertical position relative to the middle of the objective lens (this will accelerate finding the needle tip). Use the microscope with phase-contrast optics to move the needle tip in the horizontal plane relative to the field of view, at an optical plane well-above the cell layer.
NOTE: The needle will initially appear as a shadow in the field of view and the focus plane can then be adjusted to visualize the tip. Once the tip of the needle is found, gradually lower the optical plane followed by the needle tip down to a position close to the cell layer. - Check the needle flow by switching to a fluorescent channel when using fluorescent dextran, and tune the flow using the pressure device to obtain a constant "background" flow.
NOTE: In this article, we describe manual injection, which is mediated by breaking through the plasma membrane via touching the cell surface and gentle movement of the needle tip during constant needle flow. This must be distinguished from automatic injection devices accompanied by programmed needle lowering and needle pressure increase during injection events, which are more appropriate for the injection of higher cell numbers followed by a later cell population analysis. The method described here is optimized for single cell analysis by time-lapse microscopy before, during, and after the microinjection. - Find a cell of interest and gradually lower the needle above the cell.
- When ready to microinject, lower the needle gradually towards the cell perinuclear region using the fine pinion of the micromanipulator joystick, while keeping the cells in focus.
- For microinjection, gently touch the plasma membrane of the cell, which may suffice to penetrate the cell, or aid transient membrane rupture by a very gentle tap on the microscope setup.
NOTE: A white dot at the needle tip will indicate the time of contact with the plasma membrane; following membrane rupture, the needle tip will reseal, accompanied by a gentle flow of injection solution into the cell. - Stop the injection process as soon as flow into the cell is visible (ideally within 0.3 s) by moving up the needle tip into the medium. When using fluorescent dextran, successful injections can be immediately documented by fluorescence.
- If desired, initiate time-lapse image acquisition before or after the microinjection.
NOTE: Local application of drugs or inhibitors can be executed at all steps here, except for the microinjection event per se. For local applications, the diffusion of the active molecule can be controlled by flow pressure, and documented by fluorescence, and the needle tip can be positioned at the desired height. For examples of local application experiments, see e.g., Small and Rottner14 or Kaverina et al.15 - Following the microinjection, wait until the effect of the protein occurs. For different proteins and depending on the expected outcome, incubation times may vary. For the small GTPase Rac1, the response of lamellipodium formation can be initiated within 1 min or less, but takes approximately 10-15 min on average to fully develop (Figure 1g, h).
- Judge the viability of cells following the microinjection.
NOTE: Inappropriate or harmful injections can cause cell damage, which is frequently accompanied by non-specific cell-edge retraction or plasma membrane rupture.- Avoid poking through both the top and bottom plasma membrane, which can occur for injections in flat cellular regions.
NOTE: Injection volumes should be kept to a minimum (ideally <5% of the cellular volume) and will usually be in the femtoliter range. Required injection volumes can also be controlled by changes in concentration, but note that for proteins, concentrations >2 mg/mL may become impractical due to frequent needle clogging. However, this also depends on the quality and behavior of the purified protein; e.g., injection of fluorescently-coupled actin is complicated by concentration-dependent and unavoidable polymerization in the needle tip, and so it is rarely executed today (see Small et al.16).
- Avoid poking through both the top and bottom plasma membrane, which can occur for injections in flat cellular regions.
- Before, during, or after the effect of the microinjection, FRAP or photoactivation can be performed on the same cell (see Section 5 and 6).
5. FRAP Procedure
- Transfect the cell type of interest (B16-F1 cells here) with plasmid DNA encoding a fluorescently-tagged protein of interest (here, a EGFP-tagged version of β-actin was used). Seed the cells onto laminin-coated coverslips (step 2.10).
- Assemble the imaging chamber (Section 3).
- Use the following settings for lamellipodial region photobleaching: 65 mW laser power (variable according to experimental setup and laser source); 10 pixel laser beam diameter; 1 ms bleach dwell time/pixel; 500 ms GFP exposure time; 1,500 ms time interval. Experimental results in this paper were performed with an 100X 1.4NA apochromatic objective.
- Perform laser calibration to ensure accuracy in the dimensions of the photobleached region. Prior to calibration, move the field of view to an area lacking any cells/fluorescence signal and observe the picture on the display.
- Select the objective magnification by clicking the respective magnification button and reduce the laser power (3–5 mW) in the "Panel | Intensity" menu. To initiate manual calibration on Visiview software (v2.1.4), select the "Configure | FRAP" menu and click on the "Calibrate | Adjust Manual" menu. Ensure that the laser can be distinguished as a sharp dot. If not, either refocus or adjust the laser hardware.
- Perform the calibration by manually guiding the laser to pre-determined software X-Y coordinates. This instructs the software how to specifically target the laser to a user-defined region for the current magnification.
- Before triggering the laser, switch to the GFP channel and initiate image/time lapse acquisition.
- Manually draw the region to be photobleached on the GFP channel, while viewing the display.
- Initiate photobleaching by a manual trigger of the 405 nm laser, at least 3–4 frames after initiation of image acquisition. Acquiring frames prior to photobleaching is required for normalization of the image in later data analysis.
6. Photoactivation Procedure
NOTE: Software, microscope setup, and settings, except for the laser power, are similar to those for FRAP. In photoactivation, an important difference as compared to FRAP, is that a 405nm-laser power significantly lower than that employed for photobleaching must be used, to activate PA-GFP without simultaneously photobleaching it.
- Co-transfect the cell type of interest (B16-F1 cells here; see step 2.5) with plasmid DNA encoding PA-GFP-actin and another fluorescently-labeled protein (e.g., mCherry or mCherry-Lifeact).
NOTE: In most cases, mCherry-positive cells will also be positive for the PA-GFP-actin vector, the latter not normally seen on the GFP channel prior to photoactivation. To enhance the chance that the mCherry-positive cells are also positive for PA-GFP, use a 1:2 transfection ratio of mCherry:PA-GFP-actin. Following this protocol, more than 90% of the cells expressing mCherry displayed successful activation of PA-GFP-actin. - Seed the B16-F1 cells onto laminin-coated coverslips (step 2.10).
- Assemble the imaging chamber (Section 3).
- Before initiating the photoactivation experiments, if needed, perform the laser calibration for the selected objective (step 5.4–5.6).
- Set the GFP/488 nm image acquisition to 500 ms exposure and 1,500 ms time interval (depending on the experimental design).
- Adjust the software settings for acquiring either dual-channel or triple-channel time-lapse movies by marking the "Wavelength series" square and selecting the desired number of channels in the "Acquire | Wavelength" menu. It is recommended that the time lapse movies are acquired with phase contrast and GFP channels.
- Optionally, also include the mCherry channel; however, exposing cells with too much light might induce photodamage. This could be avoided with oxygen scavengers such as Oxyrase17, although effective treatment requires cell chamber sealing.
- Find the transfected cells on the mCherry channel.
- Before triggering the laser, initiate image/time lapse acquisition and manually draw the region to be photoactivated on the phase contrast channel, while viewing the display.
- Initiate photoactivation by a manual trigger of the 405 nm laser (intensity set between 5–15 mW from the "Panel | Intensity" menu), at least 3–4 frames after image acquisition initiation.
7. Data Analysis and Presentation of FRAP Results
NOTE: The method presented is used for investigating the turnover of a protein accumulating at sites of dynamic actin assembly, in this case VASP, which associates with adhesion sites and the tips of protruding lamellipodia. We are analyzing its turnover at the lamellipodium tip, but the same principles of analysis can be applied for investigating the turnover of VASP or any other protein and other subcellular compartments.
- Open the time-lapse movies derived from Visiview on the Metamorph software. In this article, Metamorph v7.8.10 was used.
- Derive intensity values for photobleached regions by manually outlining respective regions on Metamorph. Draw a shape at the tip of the lamellipodium that covers the entire or part of the photobleached area, and manually adjust its position on subsequent frames if needed (i.e., if the edge is protruding), in order to track changes in lamellipodial intensities of the respective component during tip displacement.
- For correction of background and photobleaching acquisition, analyze regions inside and outside of the cell. See Figure 2a for representative regions of measured intensities.
- While an ROI is selected, extract its intensity values on Metamorph by using the menu "Measure | Region Measurements". Ensure "Elapsed time" and "Average Intensity" options are selected in the "Configure" menu. Click "Open log" and select "Dynamic Data Exchange". Click "OK" to open an Excel spreadsheet and click the "Open log" button again to paste the Metamorph values into Excel.
NOTE: These values are used to generate the fluorescence recovery curves. - For generating fluorescence recovery curves at the lamellipodium tip of photobleached regions (normalized to the region intensity before photobleaching), apply the following equation:
 Equation 1
Equation 1
where: FRAPTn is the photobleached region intensity for each frame of interest following photobleaching; OutTn is a region intensity taken outside of the cell (background) for each frame of interest following photobleaching; InsTn is two averaged inside region intensities for each frame of interest following photobleaching (used to normalize for acquisition photobleaching over time); FRAPT-1 is the photobleached region intensity before photobleaching; OutT-1 is a region intensity taken outside of the cell (background) before photobleaching; and InsT-1 is two averaged inside region intensities for each frame of interest before photobleaching. - For each time frame of interest, use Equation 1 to obtain a fluorescence recovery curve containing all time frames to be investigated. The length of time is strictly dependent on the protein under investigation. When unknown, perform preliminary experiments to acquire the turnover rate of the protein.
- For calculating the half-time of recovery, paste the values of the fluorescence recovery curve with the corresponding time (in seconds) into a Sigma plot (v.12), and perform a curve fit using the "Dynamic Fit Wizard | Exponential rise to maximum" tool. Select mono-exponential (single, 3 parameters) or bi-exponential (double, 4 parameters) functions, depending on the best curve fit.
- Use the following formula for mono-exponential function:
 Equation 2
Equation 2 - Use the following formula for bi-exponential function:
 Equation 3
Equation 3 - Paste parameters "b" and "d" derived from Sigma Plot (Equation 2 or Equation 3) into Excel to calculate the half-time of recovery. Apply the following equations:
 Equation 4
Equation 4
or
 Equation 5
Equation 5 - When the mono-exponential function results in an accurate curve fit, apply only Equation 4.
- When mono-exponential function does not result in a good curve fit, apply the bi-exponential formula by solving both Equation 4 and Equation 5. Consider the resulting two half-times of recovery as representing two different protein fractions: a rapidly and a slowly exchanging fraction, respectively.
8. Determining the Lamellipodial Actin Polymerization Rate by FRAP
- To determine the lamellipodial actin polymerization rate, transfect B16-F1 cells with EGFP-tagged β-actin, and photobleach the lamellipodial region (step 5.9) using 1.5 s time interval and 500 ms GFP exposure.
- In Metamorph, open the time-lapse movies acquired from Visiview and calibrate the pixel/µm ratio according to the objective used by the "Measure| Calibrate distances" tool.
- Play the time-lapse movie and stop it on the frame when the lamellipodial fluorescence recovery, which flows backwards towards the lamella as a line, has reached the lamella and no further rearward flow can be tracked.
- Measure the distance in µm between the tip of the lamellipodium and the back of recovered fluorescence. This distance corresponds to the sum of the retrograde flow and protrusion distances.
- Alternatively, to separate the protrusion from retrograde flow, mark the lamellipodial tip with a line one frame before the photobleaching. Use the line as a reference point in subsequent frames to refer to the original position of the lamellipodium tip at the time of photobleaching; the reference point can be used to measure the protrusion distance and retrograde flow.
- Note the time (in seconds) required for fluorescence recovery after photobleaching to occur. The time can be calculated manually from the frame rate or visualized by Metamorph through the "Measure | Region measurements" tool.
- Derive the actin polymerization rate by using the following equation (with some equation parameters based on Metamorph measurements from steps 8.4 and 8.6):
 Equation 6
Equation 6
where actin polymerization rate is in µm/min, retrograde flow distance is in µm, lamellipodial protrusion distance is in µm, and time is in seconds.
9. Analysis of Protein Diffusion and Mobility Upon Photoactivation
NOTE: The method presented here describes the analysis of actin monomer mobility by employing photoactivation of actin fused to PA-GFP, as illustrated by visualization and quantification of protein diffusion through the cytosol.
- For measuring the diffusion of photoactivatable actin away from a cytosolic region, as well as the accumulation within a lamellipodial region, use Metamorph to determine the intensity over time in the following regions (illustrated in Figure 3a): a cytosolic photoactivated region (PA); a lamellipodial region, in which photoactivated proteins are expected to accumulate over time (Lam); a region outside of the cell used for normalization of background fluorescence (OUT).
- When determining the mobility of actin within the cytosol, measure distinct cytosolic regions (see Figure 3a, regions R1-R5). Note that the photobleaching acquisition cannot be determined in a similar fashion to FRAP, due to an increase in focal- and eventually cell-wide fluorescence upon activation.
- Transfer the intensity values for all regions from Metamorph into an Excel spreadsheet, as described in step 7.4.
- For examining the rate of displacement of photoactivatable actin away from the cytosolic region of photoactivation or its rate of incorporation within a lamellipodial region (both represented as percent intensity of the cytosolic photoactivated region at time 0), generate fluorescence curves from the data in step 9.3. Apply the following equations:
 Equation 7
Equation 7
 Equation 8
Equation 8
where: PATn is the intensity of a cytosolic photoactivated region for each frame of interest following photoactivation; LAMTn is the intensity of a lamellipodial region for each frame of interest following photoactivation; OUTTn is the intensity of a region taken outside of the cell (background) for each frame of interest following photoactivation; PAT-1 is the intensity of a cytosolic photoactivated region before photoactivation; LAMT-1 is the intensity of a lamellipodial region before photoactivation; OUTT-1 is the intensity of a region taken outside of the cell (background) before photoactivation; PAT0 is the intensity of a cytosolic photoactivated region at time 0 (i.e., first frame after photoactivation); and OUTT0 is the intensity of a region taken outside of the cell (background) at time 0 (i.e., first frame after photoactivation). - Optionally, for better visualization of the data, normalize the intensity curves to 0 by subtracting the intensity of the first frame after photoactivation from each subsequent frame.
NOTE: The following analysis method (steps 9.6–9.8) also allows calculating the cytosolic dispersion of photoactivated actin within the cytosol. - Measure the intensities for cytosolic regions, which are consecutively positioned distally from the activation region.
- To represent the intensities of these regions as percent intensity of the photoactivated region at time 0, apply Equation 8, where lamellipodial intensities are replaced with intensities for each cytosolic ROI. The size and number of the regions may vary depending on cell size and dispersal distance to be measured.
- To derive a quantifiable value for the rate of photoactivated protein infiltrating each cytosolic region, paste the time and values of the curve of fluorescence intensity increase for each region into Sigma plot (similar to the FRAP analysis in Section 7), use Equation 2 and Equation 4 to derive the half-time of the fluorescence intensity reaching a plateau. Compare the t1/2 values between different experimental groups.
Results
Figure 1g, h show phase contrast images of an NIH3T3 fibroblast cell prior and 10 min post-microinjection of Rac1, which is a small Rho-family GTPase capable of inducing lamellipodia formation through its interaction with the WAVE complex. The cell is first visualized before the microinjection (Figure 1g), to confirm its viability and morphology, e.g., lack of lamellipodia. At 10 min post-microinjection, the cell has clearly changed its morphology, which is expected from this treatment, and indicates a successful injection (Figure 1h).
For simplicity and clarity, we next provide exemplary results for FRAP and photoactivation analysis in cells, which have not been additionally microinjected.
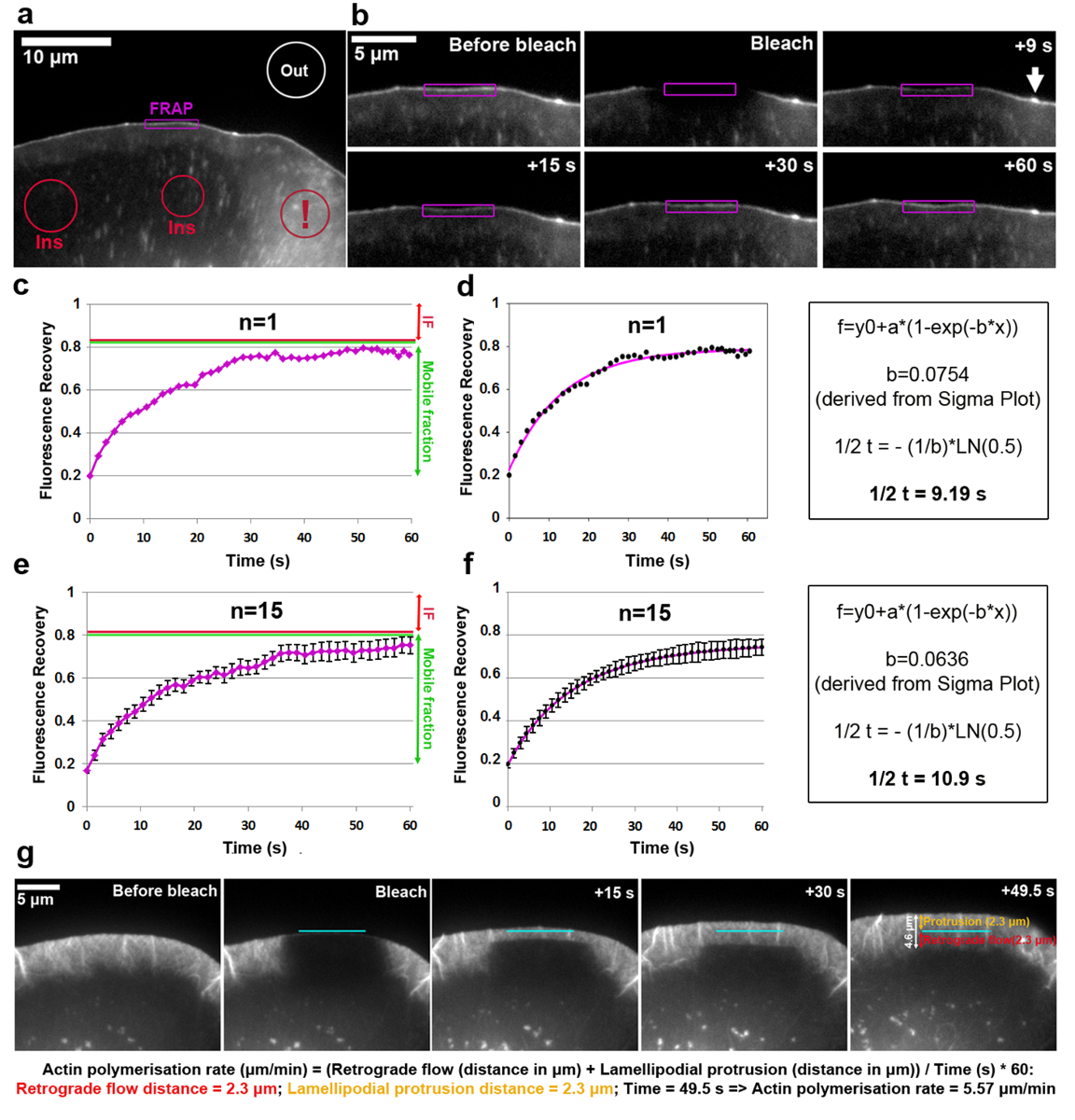
Analysis of the turnover of EGFP-tagged VASP at the lamellipodium tip is shown in Figure 2a-f. Note that VASP in addition targets to nascent and focal adhesions, small and elongated dots in the cell interior18,19. The fluorescence intensity of a lamellipodial region with a clear VASP accumulation at the tip was bleached and measured for each time frame, by following the contour of the ROI before, during, and after bleaching as the lamellipodium protrudes forwards. As bleached EGFP-VASP proteins are being recycled by non-bleached molecules at these sites, gradual recovery of fluorescence is observed (Figure 2b). The FRAP recovery curve obtained in this fashion and normalized to the pre-bleach intensity (expressed as 1) can be seen in Figure 2c. Photobleaching efficiency can vary and was approximately 20% of the value before bleaching in this example, as determined from the value at t0 (the first frame after photobleaching). The increase of fluorescence reaches a plateau in the example shown at roughly 80% of the fluorescence before bleaching. In a static structure during the time course of the experiment, such as a focal adhesion, the difference between the pre-bleach intensity and the plateau fluorescence reached after recovery is defined as the immobile fraction (IF, red arrow in Figure 2c, e), whereas the amount of fluorescence recovered between the time of bleaching and full recovery is defined as the mobile fraction (green double-headed arrow in Figure 2c, e). Note that in a dynamically changing structure such as the lamellipodium tip analyzed here, the extent of the IF might not only represent immobile molecules, but also derive from a reduction of protrusion speed, as EGFP-VASP intensity is known to depend on this parameter18. To calculate the half-time of recovery, a fit curve was created on Sigma plot (Figure 2d). In this case, the value of the "b" parameter extracted from solving Equation 2 is equal to 0.0754, which when applied to the logarithmic function (Equation 4) results in an estimated half-time of recovery of 9.19 s (Figure 2d, far right panel), which is relatively fast in this particular cell as compared to the average published previously5. It must be noted that recovery half-times may sometimes vary significantly from cell to cell within the same population. Therefore, for obtaining representative results, we recommend determining this parameter as an average from at least 15-20 cells. To illustrate the degree of variance, arithmetic means of EGFP-VASP recovery averaged from 15 cells for each time-point were generated (Figure 2e), and average curve fits created and displayed in an analogous fashion (Figure 2f).
The polymerization rate of the lamellipodial actin network comprises the sum of forward network protrusion and retrograde flow. FRAP can be applied for measuring the actin polymerization rate by transfecting cells (in this case B16-F1) with EGFP-tagged β-actin and photobleaching a protruding lamellipodial region (Figure 2g). For analysis of lamellipodial actin network polymerization, the fluorescence recovery upon bleaching of EGFP-tagged β-actin is assessed over time. As the polymerization of actin monomers progresses at the barbed ends of lamellipodial actin filaments (which all point towards the front20), the network is constantly translocated rearwards and progressing forwards, the rates of which can be easily obtained through polarized recovery of fluorescence upon photobleaching. Fluorescence recovery of the lamellipodium is complete as soon as the bleached zone has reached the transition zone between the rear part of the lamellipodium and the lamella, which is characterized by a lower density of more horizontally-arranged filament bundles turning over much more slowly than what is observed in the lamellipodium. As illustrated in Figure 2g, fluorescence recovery can be visualized as a line horizontal to the edge and flowing backwards towards the lamella, which allows measuring the distances of protrusion and retrograde flow (individually represented in the far right panel of Figure 2g as orange and red double-headed arrows, respectively).
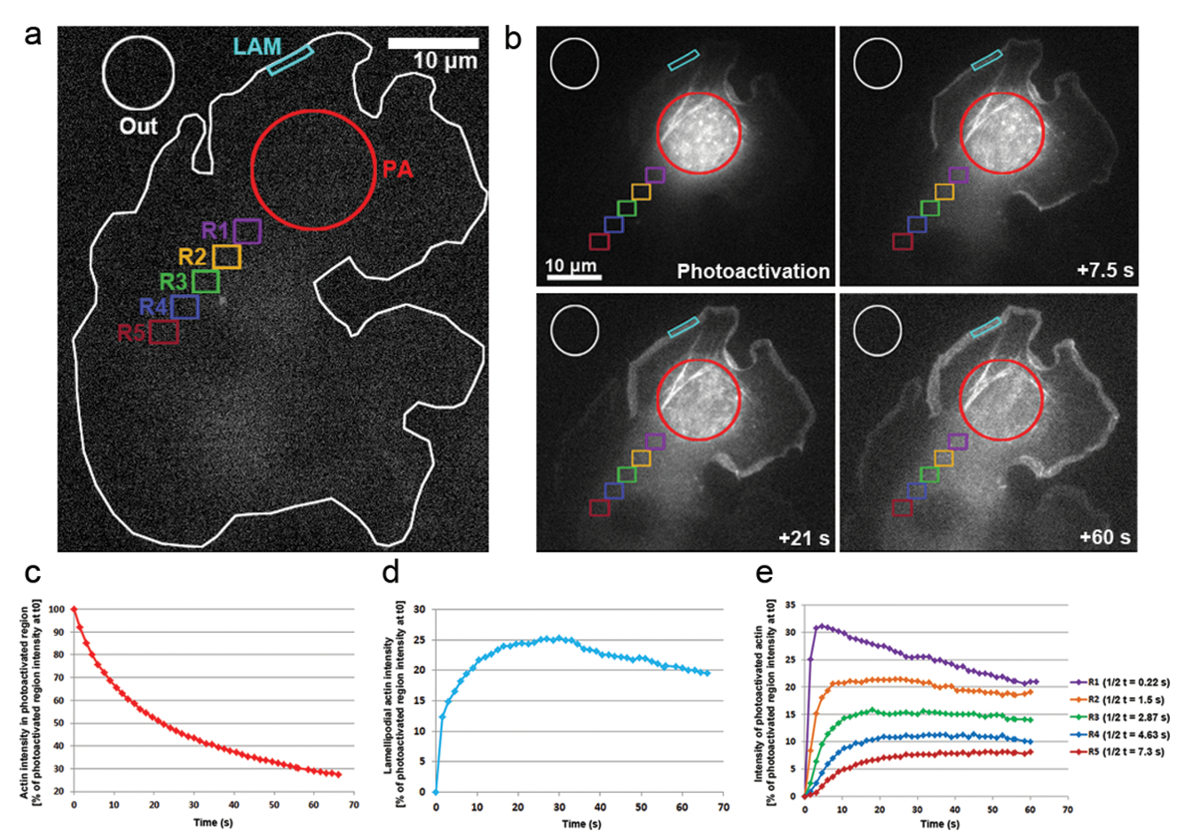
We have also applied photoactivation in B16-F1 cells transfected with PA-GFP-actin to track the mobility of actin monomers within the cytosol and the rate of their incorporation within protruding lamellipodia. As illustrated in Figure 3a, b, a cytosolic region was photoactivated by exposure to a 405 nm laser, while images were acquired on the GFP channel every 1.5 s for visualizing the distribution of GFP-tagged, photoactivated actin. Photoactivated GFP-actin can be seen diffusing out of the cytosolic region in Figure 3b. The rate of fluorescence intensity decrease in the photoactivated cytosolic region is represented as the percentage of the initial intensity at t0 (first frame after photoactivation; Figure 3c). Photoactivated actin also integrates at the tips of lamellipodia, where new actin monomers are added to the growing barbed ends of elongating actin filaments during protrusion. To estimate the rate of lamellipodial incorporation, we measured the fluorescence intensity over time of a two-dimensional contour/region of approximately 5 µm in width and 1 µm in height; the region was constantly re-positioned at the tip of the lamellipodium as it protruded. Actin incorporation was represented as the percentage of fluorescence intensity of the photoactivated cytosolic region at t0 (Figure 3d). As elongation of actin filaments progressed, new actin monomers were incorporated at the lamellipodial front. A fraction of these actin monomers was stochastically derived from the cytosolic pool where monomers were photoactivated. This results in the rapid increase of fluorescence in lamellipodia in the first 20 s after photoactivation. As new monomers are being added to the lamellipodial front, previously incorporated actin monomers flow with filaments towards the lamella by retrograde flow. Over time, the ROI is completely filled with fluorescent monomers and a plateau in fluorescence is reached (Figure 3d). A gradual drop in fluorescence is then observed when, following diffusion of photoactivated monomers throughout the cell, non-photoactivated actin monomers are increasingly being re-added to the lamellipodial front. This decrease in fluorescence will find a new plateau, which will be reached as soon as a balance in the entire cell between photoactivated and non-photoactivated monomers is reached (data not shown).
The mobility of actin monomers throughout the cytosol was derived by measuring fluorescence intensities in regions of equal size positioned distally from the photoactivated region (exemplified on Figure 3a by color coded regions labeled R1-R5). As illustrated in Figure 3e, fluorescence intensity in each of these regions is gradually decreasing away from the cytosolically photoactivated region, as the fraction of photoactivated actin monomers becomes increasingly diluted with non-activated (i.e., non-fluorescent) monomers. Furthermore, the peak of fluorescence is reached later: the more distant the measured region is located from the photoactivated region, the longer the time that is required for actin monomers to diffuse into these regions. A representative value for the degree of actin monomer infiltration into each region can be derived by quantifying the half time of reaching the fluorescence plateau. The more distant the region, the longer it takes for the photoactivated actin to diffuse into it, and thus more time is required for the fluorescent plateau to be reached, ultimately leading to a higher t1/2 value (Figure 3e).

Figure 1: Imaging chamber assembly and microinjection procedure. (a) Imaging chamber components. (b) Silicone grease is carefully smeared around the opening of a plastic sealer. (c) The coverslip is positioned with the cell-side facing up into the center of the imaging chamber opening. (d) A secure seal is established by positioning the plastic sealer on top of the coverslip and by tightening the side clamps. (e) Microscopy medium is pipetted into the chamber slot. (f) The imaging chamber is positioned on the microscope stage, heat detector and electrodes are linked to a heating unit pre-set to 37 °C, and cells are allowed to adapt for at least 30 min before microscopy is initiated. In this example, the microscope stage is also equipped with a micromanipulator for performing microinjections, and the microinjection needle is dipped into the medium covering the cell layer in the imaging chamber. (g) An NIH3T3 fibroblast cell is visualized before microinjection by phase-contrast microscopy. The red cross in the perinuclear compartment indicates the location of the future microinjection, which corresponds to a high cytoplasmic region due to the close proximity to the bulky nucleus. (h) 10 min following microinjection with Rac1, the cell reacts by prominent formation of lamellipodia around the entire cell periphery (indicated by green arrows). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: FRAP allows determining rates of protein turnover or lamellipodial actin polymerization. (a) Representative example of B16-F1 cell expressing EGFP-VASP before photobleaching of a lamellipodial region as indicated. Differently colored contours/shapes are labeled to indicate which regions were considered for fluorescence intensity measurements over time. Note the red contour marked with an exclamation mark, which labels a cytosolic region positioned in an area containing multiple vesicles and cell surface ruffles. Dynamic areas like this should be avoided for selecting regions of fluorescence reference, as they are characterized by strong short-term fluctuations of fluorescence, potentially causing inaccurate results. (b) Lamellipodial region of the EGFP-VASP expressing cell before and after photobleaching. The recovery of fluorescent signal after photobleaching within the region marked in purple is visualized over time. Arrow indicates the tip of a microspike, enriched for VASP likely due to the high density of actin filaments polymerizing there19. (c) An example of a FRAP recovery curve as derived from quantifying the fluorescent intensity of the photobleached lamellipodium (purple contour) in b. Red and green lines on the right indicate, respectively, immobile and mobile fractions. (d) A fit of the FRAP recovery curve in c (left panel) and an example of the calculation method used to derive the recovery half time (right panel). (e) An example of a FRAP recovery curve derived from averaging the fluorescence recovery curves of 15 cells, with SEM bars indicating the degree of variability within the sample population. (f) A curve fit derived from averaging the FRAP recovery curve fits of 15 cells (left panel) and an example of the calculation method used to derive the recovery half time (right panel). (g) Time-lapse panels of protruding lamellipodium of a B16-F1 cell expressing EGFP-tagged β-actin before and after bleaching of a lamellipodial region as indicated, followed by fluorescence recovery in the lamellipodium over time. On the far right panel, values measured for protrusion and retrograde distances are provided (in orange and red, respectively). Calculations under the image panels reveal how the sum of protrusion and retrograde distances are used to derive polymerization rate of the lamellipodial actin network. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Photoactivation of PA-GFP-actin for monomer tracking throughout the cell. (a) A representative example of a B16-F1 cell expressing PA-GFP-actin before triggering photoactivation in a cytosolic region as indicated by the red circle (PA). Differently colored contours are labeled to indicate which regions were considered for fluorescence intensity measurements over time. (b) An illustration of the temporal distribution of PA-GFP-actin following photoactivation. Note the gradual reduction of fluorescence in the photoactivated, cytosolic region (red circle), as the photoactivated actin diffuses away from it. Due to their diffusion to the front and assembly into the network, photoactivated actin monomers are gradually enhanced in lamellipodia (cyan region) and throughout the cytosol (different color-coded regions) in a distance- and time-dependent fashion. (c) Representative, temporal decline of fluorescence within the photoactivated cytosolic region (red contour in b). (d) Temporal changes in fluorescence intensity in the lamellipodial region (cyan contour in b). (e) Curves representative of the temporal changes in fluorescence intensity of cytosolic regions (color-coded in b) due to positioning in variable distances from the area of photoactivation. Note how half-times of reaching the fluorescence plateau (indicated in legend on the right) increase with the distance of given region to the area of photoactivation, likely correlating with the increased times needed for diffusion of actin monomers into the respective region. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Here we discuss critical steps in the techniques described in this article, and how they can be optimized for application in different experimental conditions.
Microinjection is a method that can be applied to monitor in cells the instant effects from introducing exogenous proteins, inhibitors, or drugs. It can be particularly advantageous for determining the functions of proteins in difficult to transfect cell types or in situations when long-term expression is not desired. It must be noted that survival of certain cell types varies depending on the extracellular matrix they are seeded on. Most endothelial, epithelial, or fibroblast-like cell types, even small ones like fish keratocytes (see Dang et al.21 and Anderson and Cross22) can be successfully injected. However, there are exceptions, such as B16-F1 cells seeded on laminin, which constitute an excellent model system of cell migration, but are incompatible with injection on this type of substratum for unknown reason. For NIH3T3 fibroblast cells, we routinely perform injections on fibronectin substratum, and additional photomanipulation techniques such as FRAP (even with photoactivation; shown for B16-F1 cells here) can be equally well performed in these fibroblasts (see e.g., Köstler et al.3). It must also be considered that different proteins, according to their functional properties and the experiment goals, may take different amounts of time to cause changes, varying from seconds to hours. An advantage of the technique is that the dosage/concentration of exogenous agent can be controlled more accurately at the single cell level than e.g., when using plasmid transfection. In addition, fluorescent tagging of a protein is not a necessity to guarantee its presence in the cell, which can increase flexibility if simultaneous multi-channel visualization of other fluorescently-tagged proteins is required. Microinjection can be particularly useful for analyzing instant effects of specific proteins or protein mixtures on dynamic changes of cell morphology or the cytoskeleton (e.g., Dang et al.21 for an example of instant effects on migration by the Arp2/3 complex inhibitor Arpin). A disadvantage of the technique is its invasiveness, which can cause cell damage or influence cell morphology. Therefore, an important consideration when performing microinjections is monitoring the cell viability. The method introduced here relies on manual manipulation. In conditions tested to be compatible with successful injections, such as fibroblasts growing on fibronectin substratum, the manual injection protocol described here allows a near 100% success rate; this is essential when combining this approach with sophisticated and time-consuming follow-up experiments including video microscopy or FRAP, as published previously3. This does not exclude that occasionally, individual cells might suffer from a microinjection event, which can be safely recognized by abrupt changes of contrast of both the nucleus and cytoplasm, followed by cell edge retraction. Such rare experimental cases are excluded and thus not considered for further analyses.
However, a half-automatic approach is also commonly used, for instance employing rapid (<300 ms) machine-controlled needle lowering coincident with injection pressure increase, so that the needle only has to be positioned above each cell prior to respective injection. The success rate of half-automatic injections is by definition lower than the manual approach described above, simply because it is optimized for speed, followed by analysis of multiple cells that successfully survived this treatment; thus it does not rely on successful injection of an individual cell. Therefore, as opposed to single cell analysis, half-automatic injections are better suited for analyzing injection effects of several hundred cells, e.g., by video microscopy at low magnification or upon cell fixation and staining. Irrespective of the detailed approach employed, microinjection does not constitute an end-point assay, but can be combined with a variety of techniques, including FRAP or photoactivation3.
When determining the protein turnover rate by FRAP, the intensity of the laser must be optimized, depending on the microscope setup and imaging conditions (magnification, objectives, etc., as well as the cell type, structure, and fluorescent protein for photobleaching). Note that at optimal laser power, efficient bleaching is combined with the least possible photodamage, to avoid shrinkage or complete retraction of the structure under analysis (e.g., lamellipodia or filopodia) or even damage at the cellular level. Ideally, at least 70–80% of bleaching efficiency should be achieved, although complete bleaching may be hampered by extremely rapid turnover of the protein, in which case, anything above 50% might also be acceptable. Optimal bleaching power for a given structure and fluorescent dye should be experimentally tested, starting from a low laser power followed by its gradual increase. Of course, any fluorescent dye can by definition be bleached with laser light close to its peak of excitation (488 nm for frequently used green dyes such as FITC or EGFP). However, lasers with shorter wavelengths, such as near-UV lasers, deliver higher powers and can thus also be used for efficient bleaching of commonly used dyes. We routinely employ a 405 nm diode laser (120 mW) for bleaching of both EGFP and red fluorescent dyes (such as mCherry), albeit with slightly lower efficiency in case of the latter (data not shown). As the 405 nm-diode can also be used for photoactivation of PA-GFP (see below), it endows this system with maximal flexibility.
For the B16-F1 cell structures and fluorescent proteins photobleached here, 405 nm-laser powers between 65–100 mW were applied. When analyzing a photobleached region, it is important to consider whether the given structure is preserved in its original shape over the analysis time period. For instance, when analyzing turnover of proteins at lamellipodia tips, care should be taken whether the curvature of lamellipodia is significantly altered over time, as changes in curvature might lead to inaccurate results if the region/contour analyzed does not fully encompass the entirety of the structure in each measured frame. In addition, it should be noted that bundles embedded into lamellipodia, such as microspikes, might cause deviations in fluorescence intensity. As illustrated in Figure 2b (white arrow in 9 s time frame), a microspike-like structure is situated next to the measured photobleached region, but remains outside of it throughout the duration of measurement, and thus does not cause any inaccuracy. For analysis of protein turnover, important considerations when selecting location and size of analyzed regions are that their fluorescence over time should not be significantly influenced by changes in cell morphology or factors other than hard to avoid acquisition photobleaching. For instance, structures providing significant quantitative contribution to the analyzed structure should not move out of the measured region during analysis; in addition, unrelated, fluorescent entities such as vesicular structures that attract the protein should not enter the field of interest during analysis. For determining the rate of lamellipodial actin polymerization, care should be taken that no retracting or ruffling (i.e., upwards folding) lamellipodia are analyzed, as this will strongly influence the accuracy of the results. In addition, retraction of lamellipodial regions might appear as rapid rearward translocation, potentially leading to overestimation of rates of lamellipodial actin polymerization. An additional consideration is the distance of intracellular normalization regions (taken as reference positions for the correction of acquisition photobleaching) from the actual position of photobleaching, which should be large enough to avoid direct influence by the photobleached area.
When setting up optimal conditions for photoactivation of PA-GFP-tagged constructs, care should be taken to avoid instant bleaching during photoactivation. In our work, the best results were obtained with laser powers 5-10 times lower than normally employed for bleaching of EGFP. For image acquisition of photoactivated molecules, exposure time and time interval between frames should be optimized by considering the size of regions and structures to be photoactivated and analyzed, as well as the potential mobility of photoactivated proteins to other subcellular locations. As for all types of fluorescence imaging, maintenance of cell viability is crucial for obtaining physiologically relevant results.
In principle, green-to-red photoconversion of fluorescent proteins such as mEos or Dronpa variants12 constitutes an equally powerful method of following dynamics and turnover of subcellular structures such as the lamellipodium (see e.g., Burnette et al.23). The advantage of the latter method as opposed to PA-GFP would be the possibility to follow protein dynamics before and after conversion with two distinct colors, without the need to co-express an additional red fluorescent protein. However, in our preliminary experiments, the extent of contrast change and intensity of fluorescent signal achieved upon photoactivation of PA-GFP was larger as compared to photoconverted probes, perhaps due to the superior spectral features of green versus red fluorescent probes (data not shown). In any case, detailed studies on actin filament turnover in cell-edge protrusions such as lamellipodia or Vaccinia virus-induced actin tails have so far only been published using PA-GFP derivatives5,6,24.
When considering which cell region to analyze following photoactivation, several factors should be taken into account, which are discussed using the specific example shown here (incorporation of actin monomers at the cell edge upon activation in the cytosol), but can certainly be extrapolated to various analogous scientific problems. First, when measuring the rate of lamellipodial incorporation of cytosolically photoactivated proteins, for instance, in distinct experimental conditions (as shown in Dimchev et al.6), sizes of cytosolic regions and their distances to lamellipodial edges should be comparable between experimental groups. It is also important to consider that when photoactivating cytosolic regions, the cell thickness is greater in positions closer to the nucleus. Activating thicker cellular regions might result in higher amounts of activated proteins, given that the distribution of the protein to be activated is homogenously distributed in the cytosol. Lastly, expression levels of the protein to be activated can certainly be highly variable in individual cells. Due to all these considerations of variability, it is crucial to compare incorporation levels of cytosolically activated proteins elsewhere in the cell relative to the total fluorescence obtained upon activation in the specific regions.
We have described how microinjection can be used as a tool for investigating the effects of proteins on cell morphology and have exemplified this by demonstrating the potent induction of lamellipodial structures in NIH3T3 fibroblast cells microinjected with the small GTPase Rac1. We have previously applied this technique to interfere with Arp2/3 function in cells microinjected with the C-terminal WCA domain of Scar/WAVE3. Various parameters in microinjected cells can be analyzed by other assays, such as FRAP or photoactivation. We have described how FRAP and photoactivation can be employed for investigating the subcellular dynamics and mobility of actin monomers. FRAP has been used by our group previously5 to investigate the turnover of proteins localizing to lamellipodia, such as VASP, Abi, cortactin, cofilin, and capping protein, or for elucidating the turnover of components in focal adhesions in the presence and absence of Rac signaling4. Moreover, measuring actin polymerization rates can be accomplished by photobleaching EGFP-tagged β-actin5, but alternative methods exist. Tracking fluorescent inhomogeneities as seen by live cell imaging-compatible probes labeling cellular actin filaments, such as Lifeact25, can also be employed6,26. The advantage here is that the overexpression of β-actin can be avoided, which is capable of increasing cell edge protrusion and migration, and thus potentially interferes with the specific assay or experimental question (see e.g., Kage et al.26; Peckham et al.27). However, a distinct disadvantage of the Lifeact probe constitutes its rapid on/off kinetics of binding to actin filaments, so that bleaching of actin filament structures labeled by Lifeact in cells provides information only on the probe turnover, but not the turnover of the actin filaments, to which it binds25. The tracking of fluorescence inhomogeneities employed previously6,26 does provide a practical compromise, much similar to the widely used tracking of fluorescence speckles incorporated into filamentous cytoskeletal structures (see e.g., Salmon and Waterman28), but may not be as straight forward to use and as precise as FRAP of EGFP-tagged F-actin structures. Photoactivation has been applied by us for estimating the rates of monomeric actin incorporation into protruding lamellipodia, as well as its mobility throughout the cytosol, in the context of experimentally tuned cytosolic F-actin levels6. The technique is useful when examining mobility and distribution of proteins derived from relatively large areas, such as cytosolic regions. However, examining the distribution of proteins derived from relatively small photoactivated structures; e.g., growth cones might be challenging due to the low numbers of fluorescent molecules activated, weak signals, and thus lack of sensitivity. Potential alternative techniques to photoactivation or photoconversion of fluorescence (see above) may include inverse FRAP, which relies on photobleaching the entire cell except the ROI, followed by tracking the mobility of fluorescent molecules away from this region. The technique does not require overexpressing photoactivatable versions of proteins, but will always involve exposure to an unusually high dose of laser power, potentially causing undesired side effects such as photodamage.
Clearly, photoactivation and FRAP cannot distinguish whether proteins are moving as monomers, dimers, or even small oligomers, and whether they move in combination with additional binding partners. Information of that kind can be obtained instead from fluorescence correlation spectroscopy techniques29 or, alternatively, FLIM-FRET30. Nonetheless, FRAP and photoactivation constitute straightforward approaches to directly assess local and global protein dynamics in cells, irrespective of the protein of interest, subcellular location, or cell type studied.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We are grateful to the German Research Foundation (DFG) for financial support (grant Nr. RO2414/5-1 to KR).
Materials
| Name | Company | Catalog Number | Comments |
| B16-F1 mouse skin melanoma cells | American Type Culture Collection, Manassas, VA | CRL-6323 | |
| NIH-3T3 cells | American Type Culture Collection, Manassas, VA | CRL-1658 | |
| DMEM 4.5g/L glucose | Life Technologies, Thermno Fisher Scientific, Germany | 41965-039 | |
| Ham’s F-12 medium | Sigma-Aldrich | N8641 | |
| Fetal calf serum (FCS) | PAA Laboratories, Linz, Austria | A15-102 | |
| Fetal bovine serum (FBS) | Sigma-Aldrich, Germany | F7524 | Lot054M3396 |
| MEM Non essential amino acids | Gibco, ThermoFisher Scientific, Germany | 11140035 | |
| L-Glumatine 200mM (100x) | Life Technolgies | 25030-024 | |
| Pen-Strep 5000 U/mL | Life technologies | 15070063 | |
| Sodium Pyruvate (100 mM) | Gibco, ThermoFisher Scientific, Germany | 11360-039 | |
| Laminin | Sigma-Aldrich | L-2020 | |
| Laminin coating buffer | Self-made: 50mM Tris ph7.4, 150mM NaCl | ||
| Fibronectin from human plasma | Roche Diagnostics, Mannheim, Germany | 11 051 407 001 | |
| Jetpei | Polyplus Transfection, Illkirch, France | 101-10N | |
| JetPei buffer | Polyplus Transfection, Illkirch, France | 702-50 | 150mM NaCl |
| PA-GFP-actin plasmid DNA | described in Koestler et al.2008 | ||
| pEGFP-actin plasmid DNA | Clontech, Mountain View, CA, USA | ||
| Rac1 protein for microinjection | Purified as GST-tagged version, and cleaved from GST prior to injection | ||
| Microinjection buffer | Self-made: 100mM NaCl, 50mM Tris-HCl ph7.5, 5mM MgCl2, 1mM DTT | ||
| Dextran, Texas Red, 70,000 MW, Lysine Fixable | Molecular Probes, Thermno Fisher Scientific, Germany | D1864 | |
| Microscope circular cover glasses 15mm, No.1 | Karl Hecht, Aisstent, Sondheim, Germany | 1001/15 | |
| Eppendorf Femtotips Microloader Tips | Eppendorf, Hamburg, Germany | 5242 956 003 | |
| Eppendorf Femtotip Microinjection Capillary Tips | Eppendorf, Hamburg, Germany | 930000035 | |
| Silicone Grease | ACC Silicones, Bridgewater, England | SGM494 | |
| Aluminium Open Diamond Bath Imaging Chamber | Warner instruments | RC-26 | |
| Automatic temperature controller | Warner Instruments | TC-324B | |
| Microscope: Axio Observer | Carl Zeiss, Jena, Germany | ||
| CoolSnap-HQ2 camera | Photometrics, Tucson, AZ | ||
| Lambda DG4 light source | Sutter Instrucment, Novato, CA | ||
| Laser source | Visitron Systems | ||
| Eppendorf FemtoJet microinjector | Eppendorf, Hamburg, Germany | With built-in compressor for pressure supply | |
| Nikon Narishige Micromanipulator system | Nikon Instruments, Japan | ||
| Visiview software v2.1.4 | Visitron Systems, Puchheim, Germany | ||
| Metamorph software v7.8.10 | Molecular Devices, Sunnyvale, CA | ||
| Sigma Plot v.12 | Systat Software Inc. |
References
- Day, R. N., Davidson, M. W. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 38 (10), 2887-2921 (2009).
- Ishikawa-Ankerhold, H. C., Ankerhold, R., Drummen, G. P. Advanced fluorescence microscopy techniques--FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 17 (4), 4047-4132 (2012).
- Koestler, S. A., et al. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Mol Biol Cell. 24 (18), 2861-2875 (2013).
- Steffen, A., et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 126, Pt 20 4572-4588 (2013).
- Lai, F. P., et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27 (7), 982-992 (2008).
- Dimchev, G., et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol Biol Cell. 28 (10), 1311-1325 (2017).
- Koppel, D. E., Axelrod, D., Schlessinger, J., Elson, E. L., Webb, W. W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys J. 16 (11), 1315-1329 (1976).
- Patterson, G. H., Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 297 (5588), 1873-1877 (2002).
- McKinney, S. A., Murphy, C. S., Hazelwood, K. L., Davidson, M. W., Looger, L. L. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 6 (2), 131-133 (2009).
- Gurskaya, N. G., et al. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat Biotechnol. 24 (4), 461-465 (2006).
- Lippincott-Schwartz, J., Patterson, G. H. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 19 (11), 555-565 (2009).
- Kremers, G. J., Piston, D. Photoconversion of purified fluorescent proteins and dual-probe optical highlighting in live cells. J Vis Exp. (40), (2010).
- Fischer, A. H., Jacobson, K. A., Rose, J., Zeller, R. Preparation of slides and coverslips for microscopy. CSH Protoc. 2008, 4988(2008).
- Small, J. V., Rottner, K. Actin-based Motility. Carlier, M. F. , Springer. Dordrecht. (2010).
- Kaverina, I., et al. Enforced polarisation and locomotion of fibroblasts lacking microtubules. Curr Biol. 10 (12), 739-742 (2000).
- Small, J., Rottner, K., Hahne, P., Anderson, K. I. Visualising the actin cytoskeleton. Microsc Res Tech. 47 (1), 3-17 (1999).
- Mikhailov, A. V., Gundersen, G. G. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskeleton. 32 (3), 173-186 (1995).
- Rottner, K., Behrendt, B., Small, J. V., Wehland, J. VASP dynamics during lamellipodia protrusion. Nat Cell Biol. 1 (5), 321-322 (1999).
- Svitkina, T. M., et al. Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol. 160 (3), 409-421 (2003).
- Small, J. V., Isenberg, G., Celis, J. E. Polarity of actin at the leading edge of cultured cells. Nature. 272 (5654), 638-639 (1978).
- Dang, I., et al. Inhibitory signalling to the Arp2/3 complex steers cell migration. Nature. 503 (7475), 281-284 (2013).
- Anderson, K. I., Cross, R. Contact dynamics during keratocyte motility. Curr Biol. 10 (5), 253-260 (2000).
- Burnette, D. T., et al. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 13 (4), 371-381 (2011).
- Humphries, A. C., et al. Clathrin potentiates vaccinia-induced actin polymerization to facilitate viral spread. Cell Host Microbe. 12 (3), 346-359 (2012).
- Riedl, J., et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 5 (7), 605-607 (2008).
- Kage, F., et al. FMNL formins boost lamellipodial force generation. Nat Commun. 8, 14832(2017).
- Peckham, M., Miller, G., Wells, C., Zicha, D., Dunn, G. A. Specific changes to the mechanism of cell locomotion induced by overexpression of beta-actin. J Cell Sci. 114, Pt 7 1367-1377 (2001).
- Salmon, E. D., Waterman, C. M. How we discovered fluorescent speckle microscopy. Mol Biol Cell. 22 (21), 3940-3942 (2011).
- Machan, R., Wohland, T. Recent applications of fluorescence correlation spectroscopy in live systems. FEBS Lett. 588 (19), 3571-3584 (2014).
- Becker, W. Fluorescence lifetime imaging--techniques and applications. J Microsc. 247 (2), 119-136 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved