
Method Article
Droplet Barcode-basierte Einzelzelle Transkriptom des adulten Säugetier-Geweben
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die allgemeine Prozesse und Qualitätskontrollen für die Zubereitung von gesunder Erwachsenen einziger Säugerzellen Droplet-basierte, hohen Durchsatz Einzelzelle RNA-Seq Vorbereitungen notwendig. Sequenzierung Parameter, lesen Sie Ausrichtung und nachgelagerten einzellige bioinformatische Analyse sind ebenfalls vorhanden.
Zusammenfassung
Die Analyse der einzelnen Zelle Genexpression über Tausende von einzelnen Zellen innerhalb eines Gewebes oder einer Mikroumgebung ist ein wertvolles Instrument zur Identifizierung Zelle Komposition, Diskriminierung der Funktionszustände und molekulare Signalwege, die zugrunde liegenden beobachteten Gewebe Funktionen und tierische Verhaltensweisen. Die Isolation der intakten, gesunden einzelne Zellen von Erwachsenen Säugetier-Gewebe für spätere nachgelagerte Einzelzelle molekulare Analyse kann jedoch schwierig sein. Dieses Protokoll beschreibt die allgemeine Prozesse und Qualitätskontrollen, dass unbedingt erhalten Erwachsene Einzelzelle qualitativ hochwertige Präparate aus dem Nervensystem oder Haut, die anschließende unvoreingenommene Einzelzelle RNA Sequenzierung und Analyse aktiviert. Richtlinien für die nachgelagerten bioinformatische Analyse sind ebenfalls vorhanden.
Einleitung
Mit der Entwicklung der hohen Durchsatz Einzelzelle Technologie1,2 und Fortschritte in der benutzerfreundlichen Bioinformatik über das letzte Jahrzehnt3entstand ein neues Feld der hochauflösenden Ausdruck Genanalyse – einzelne Zelle RNA Sequenzierung (ScRNA-Seq). Das Studium der einzelnen Zelle Genexpression wurde ursprünglich entwickelt, um Heterogenität innerhalb definierter Zellpopulationen, wie z. B. in Stammzellen und Krebszellen, zu identifizieren oder um seltene Populationen von Zellen4,5, zu identifizieren, die unerreichbar waren traditionelle Masse RNA Sequenzierung Techniken. Bioinformatische Werkzeuge ermöglichten die Identifizierung von neuen Sub-Populationen (Seurat)2, Visualisierung des Ordens Zellen entlang einer Psuedotime Raum (Monokel)6, Definition der aktive Signalisierung Netzwerke innerhalb oder zwischen Populationen ( MALERISCHE)7, Vorhersage der Versammlung der einzelnen Zellen in eine künstliche 3D-Raum (Seurat und vieles mehr)8. Diese neue und spannende Analysen der wissenschaftlichen Gemeinschaft zur Verfügung ScRNA-Seq schnell-die neue Standardtherapie für Genanalyse Ausdruck gewinnt.
Trotz des großen Potenzials des ScRNA-Seq kann die technischen Fachkenntnisse erforderlich, um eine saubere Dataset zu produzieren und präzise Ergebnisse interpretieren die Neuankömmlinge herausfordernd sein. Hier ist eine einfache, aber umfassende Protokoll, ausgehend von der Isolation einzelner Zellen aus dem gesamten primären Gewebe zur Visualisierung und Präsentation von Daten für die Veröffentlichung (Abbildung 1) präsentiert. Erstens kann die Isolierung der einzelnen Krebszellen anspruchsvoll, als verschiedene Gewebe in ihrem Grad der Empfindlichkeit gegenüber enzymatischen Verdauung und anschließende mechanische Dissoziation variieren. Dieses Protokoll enthält Anleitungen in den folgenden Schritten isoliert und identifiziert wichtige Qualitätskontrolle Checkpoints während des Prozesses. Zweitens kann es verwirrend sein, Verständnis für die Kompatibilität und Anforderungen zwischen einzelnen Zellentechnologie und Sequenzierung der nächsten Generation. Dieses Protokoll enthält Richtlinien zur Umsetzung einer benutzerfreundliche, Tropfen-basierte einzellige Barcoding-Plattform und Sequenzierung durchführen. Computer-Programmierung ist schließlich eine wichtige Voraussetzung für die Analyse von einzelligen transkriptomischen Datensätze. Dieses Protokoll stellt Ressourcen für erste Schritte mit der Programmiersprache R und Anleitungen für die Umsetzung der beiden beliebten ScRNA-Seq-spezifische R-Pakete. Dieses Protokoll kann zusammen, Newcomer im ScRNA-Seq-Analyse für klare, interpretierbare Ergebnisse führen. Dieses Protokoll kann auf den meisten Geweben in der Maus eingestellt werden und vor allem für die Verwendung mit anderen Organismen, einschließlich menschliches Gewebe geändert werden könnte. Anpassungen je nach Gewebe und Benutzer werden benötigt.
Es gibt mehrere Überlegungen im Auge zu behalten, während im Anschluss an dieses Protokolls; darunter 1) im Anschluss an alle Richtlinien der Qualitätskontrolle in der Schritte 1 und 2 dieses Protokolls wird empfohlen, eine tragfähige Einzelzelle Aussetzung aller Zellen in der Probe von Interesse zu gewährleisten und gleichzeitig präzise Ergebniszelle zählt (zusammengefasst in Abbildung 2 ). Sobald dies erreicht ist, und wenn die optimierte Bedingungen befolgt werden, die Qualitätskontrolle Schritte fallen gelassen werden können (zu sparen Zeit - Erhaltung RNA Qualität und Reduzierung der Zell-Verlust). Bestätigung erfolgreich isoliert hohe Rentabilität einzelner Zellen aus dem Gewebe von Interesse ist hoch empfehlen vor allen nachgelagerten Verarbeitung. (2) da einige Zelltypen empfindlicher als andere sind zu betonen, können übermäßige Dissoziation Techniken versehentlich die Bevölkerung daher Störfaktoren nachgelagerte Analyse beeinflussen. Sanfte Dissoziation ohne unnötige zellulären Scheren und Verdauung ist entscheidend für die zelluläre ertragreich und eine genaue Darstellung der Zusammensetzung des Gewebes zu erreichen. Scherkräfte auftreten, während der Verreibung, FACS und Wiederfreisetzung Schritte. (3) als mit RNA arbeiten, ist es am besten als kleine zusätzliche RNase in die Probe möglichst während der Zubereitung einzuführen. Dies wird helfen, qualitativ hochwertige RNA zu pflegen. Ribonuklease Inhibitor Lösungen mit Spülung, saubere Werkzeuge und Geräte, die nicht verwenden RNase-freie, aber vermeiden Sie DEPC-behandeltem Produkte. (4) durchführen Sie die Vorbereitungen so schnell wie möglich. Damit erhalten Sie qualitativ hochwertige RNA und Zelltod zu reduzieren. Je nach Gewebe Dissektion Länge und Tier eine solche Gruppe Gründen mehrere Sektionen/Vorbereitungen zur gleichen Zeit. (5) bereiten Sie Zellen auf Eis vor, wenn möglich zu pflegen hochwertige RNA, Zelltod reduzieren und langsam Zelle Signalisierung und transkriptionelle Aktivität. Wenn auch eiskalte Verarbeitung ideal für die meisten Zelltypen ist, Leistung einige Zelltypen (z.B. Neutrophile) bessere bei der Verarbeitung bei Raumtemperatur. (6) vermeiden Sie Calcium, Magnesium, EDTA und DEPC-behandeltem Produkte während der Vorbereitung der Zelle.
Protokoll
Alle Protokolle, die hier beschrieben sind im Einklang mit und von der University of Calgary Animal Care Committee genehmigt.
(1) trennendem Gewebe (Tag 1)
- Mäuse mit einer Überdosis von Natrium-Pentobarbital (i.p., 50 mg/kg) oder gegebenenfalls nach Tierethik Protokoll einschläfern. Dann entfernen Sie unerwünschte Haare aus den Rücken und die Beine der Maus und sterilisieren Sie die Region der Dissektion Ethanol zu.
- Zerlegen Sie das Gewebe oder die Mikroumgebung von Interesse. Für dieses Protokoll verwenden wir Haut und Nervengewebe Generalizability der Tröpfchen Barcode-basierte Einzelzelle Transkriptom nach Erwachsenen Gewebe Dissoziation zu demonstrieren.
- Verwenden Sie für den Ischiasnerv das ausführliche Protokoll finden Sie unter Stratton Et al. 9. kurz, schneiden Sie die Haut von der hinteren Region der Rücken/Beine der Maus. Machen Sie einen Schnitt entlang der Länge des Oberschenkels mit einem sterilen Skalpellklinge. Verwenden Sie feine Pinzetten und Scheren zu entlarven und entfernen den Ischiasnerv.
- Verwenden Sie für die hinteren Haut das ausführliche Protokoll finden Sie unter Biernaskie Et al. 10. kurz, sezieren Rückenhaut zurück, indem Sie Einschnitte von Schulter zu Schulter, über den Hintern und auf der Rückseite mit feinen Zangen und Scheren. Schneiden Sie die Haut in dünne Scheiben schneiden (0,5 cm Dicke) mit einer sterilen Skalpellklinge.
- Waschen Sie Gewebe 2 Mal mit eiskalten HBSS, und entfernen Sie unerwünschte Bindegewebe, Fettablagerungen oder Schmutz unter dem sezierenden Mikroskop.
- Schweben Sie für die Haut Dermis nur die Scheiben in Dispase (5 mg/mL, 5 U/mL) in HBSS für 30-40 min bei 37 ° C. Chirurgisch trennen Sie die Epidermis von der Dermis. Verwerfen Sie die Epidermis zu oder weiter zu distanzieren Sie, mit Trypsin wenn von Interesse.
- Probe in 1-2 mm Stücke mit einem sterilen Skalpellklingen Hackfleisch und frisch aufgetaut 2 mg/mL kalte Kollagenase-IV Enzym (2 mg/mL, 125 CDU/mg, F12 Medien) umgesetzt.
- Verwenden Sie für die Nerven ~ 500 µL pro 2 X Ischias Nerven. Verwenden Sie für die Haut ~ 8 mL pro 1 X Maus wieder Haut.
Hinweis: Das Gewebe sollte vollständig in die Kollagenase-IV-Lösung getaucht werden. Es ist wichtig, dass Verdauung Enzyme verarbeitet, gespeichert und entsprechend vorbereitet sind. Wenn Enzyme bei Raumtemperatur für längere Zeit bleiben, werden einzellige Isolierung erfordern übermäßige mechanische Zerreibung und Zellviabilität reduziert. Kollagenase-IV kann auch in Zellkulturmedien bestehen wo Zellviabilität optimal ist. Jedoch könnte dies Enzym-Aktivität verändern oder transkriptionellen Signatur sollte so optimiert werden, durch den Benutzer.
- Verwenden Sie für die Nerven ~ 500 µL pro 2 X Ischias Nerven. Verwenden Sie für die Haut ~ 8 mL pro 1 X Maus wieder Haut.
- Inkubieren Sie die Probe in das Enzym in einem Bad von 37 ° C für 30 min mit sanft schütteln alle 10 min. Ein Shaker platziert bei 37 ° C ist auch eine geeignete Alternative.
- Genannte mit einem P1000 Pipette 20 - 30 Mal auf 30 min nach dem zurückgekühlt.
- Wiederholen Sie Verreibung alle 30 min. bis Lösung trübe erscheint und Teile des Gewebes sind weitgehend getrennt.
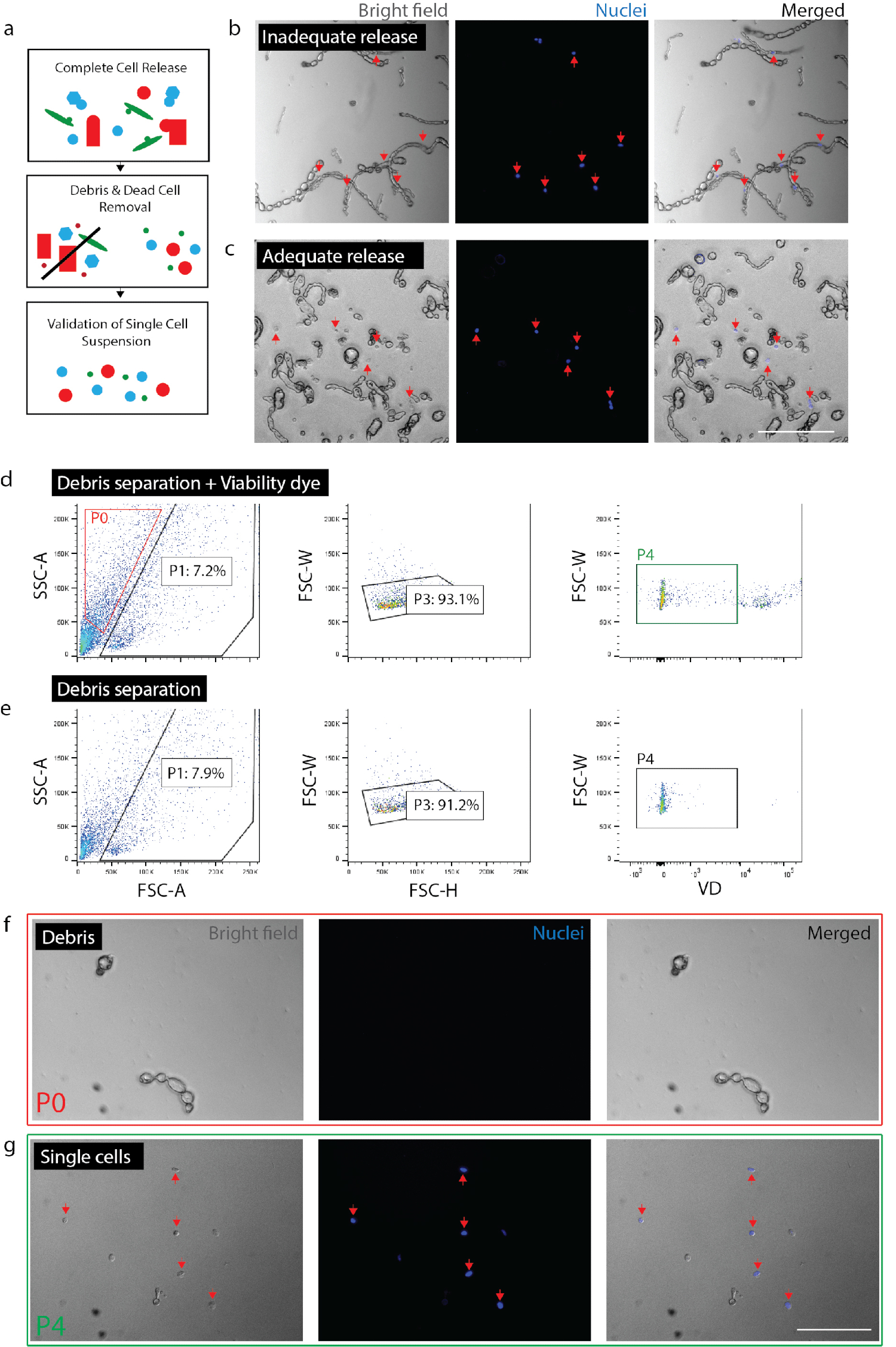
Hinweis: Stellen Sie sicher volle Freigabe von Zellen (Abb. 2 b, 2 c). Um die Vollversion zu bestätigen, Platte Zellen mit Nuc blau (2 Tropfen pro 1 mL) und nach 20 min, überprüfen Sie unter die Lupe genommen, um sicherzustellen, dass alle Kerne mit Einzelzellen anstatt Schmutz verbunden. Es ist wichtig, den Grad der Zelle Release innerhalb einer gegebenen Experiment für jeden Gewebetyp oder Zustand zu überprüfen. In fibrotische Gewebe (z.B. chronische Schädigung) oder unverletzt Erwachsenen Gewebe variieren die Freisetzung von Zellen drastisch von akuten Verletzungen oder embryonalem Gewebe. Dies ist besonders wichtig, da bestimmte Zelltypen weniger wahrscheinlich Entlassung aus dem Gewebe als andere, sind somit bevorzugt ohne diese Zellen aus nachgelagerten Analyse.- Für den Nerv distanzieren Gewebe für 0,5-1,5 Stunden insgesamt. Für die Haut, Gewebe zu trennen, für 2 Stunden insgesamt (in der letzten Stunde Inkubation DNase (1 mg/mL) zu der Hautprobe hinzufügen).
- Zweimal mit einem 40 µm-Filter filtern. Spülen Sie den Filter mit eiskalten 1 % BSA/HBSS.
- 260 X g für 8 min zentrifugieren. Dann entfernen Sie überstand.
- Aufschwemmen der Zelle Pellet in HBSS mit 1 % BSA mit einer weiten-Bohrung-Spitze, und auf Eis. Die Wiederfreisetzung Volumen basiert auf Gewebevolumens (800 mg Frischgewicht für Haut = 800 µL Volumen; 10 mg Gewicht für Nerven nass = 100 µL Volumen).
- Optional, beginnen Sie mit einem niedrigen Wiederfreisetzung Volumen und passen Sie dann nach Bedarf anhand der Durchflussmenge (Ereignisse pro Sekunde) auf FACS-Sortierer. Die effizienteste Art Dichte (Anzahl der Zellen gesammelt während abzielend Zeit zu maximieren) für Sammlungen ist 3.000-7.000 Ereignisse pro Sekunde.
- Wenn Lebensfähigkeit Farbstoff verwenden, nehmen Sie eine Subaliquot für eine ungefärbte Steuerung. Dann fügen Sie 1: 15.000 Lebensfähigkeit Farbstoff (Lager: 20.000 nM/µL) zu probieren (1,3 nM/µL Endkonzentration) mit einer weiten Bohrung Spitze Scheren reduzieren.
Hinweis: Es ist entscheidend für den Grad des Zelltods innerhalb einer gegebenen Experiment für jeden Gewebetyp oder Zustand zu überprüfen. Einige Zelltypen innerhalb einer Probe sind eher zu sterben als andere, so wird bevorzugt von nachgelagerten Analyse ausgeschlossen.- Inkubieren Sie Probe mit Lebensfähigkeit Farbstoff für 5-10 min auf Eis im Dunkeln. Fügen Sie 4 mL eiskaltes 1 % BSA/HBSS zu probieren. Zentrifugieren Sie bei 260 X g für 8 min, überschüssige Lebensfähigkeit Farbstoff zu entfernen. Behandlung der Subaliquot mit keine Lebensfähigkeit färben ungefärbten Kontrolle auf die gleiche Weise.
2. isolierende lebensfähige und gesunde Zellen (Tag 1)
- Sicherstellen Sie, dass die FACS-Anlage geeignete Fluoreszenz aktiviert Zelle sortieren (FACS) Parameter folgt.
- Vorbereiten die FACS-Maschine im Voraus, um sicherzustellen, dass sie bereit ist, sobald die endgültige Zentrifuge in Schritt 1 abgeschlossen ist, und sicherstellen, dass die Sammelfach kühl gelagert ist mit Eisblöcken.
- Verwenden Sie die folgenden Parameter: Volumenstrom: 1.0 (entspricht etwa 10 µL/min); Filter: 1,5 ND; Düsengröße: 100 µm; Forward Scatter: 80-180 V (Veränderung notwendig, um die Größe von Ereignissen zu unterscheiden); Side Scatter: 150-220 V (Veränderung notwendig, um die Granularität/Form von Ereignissen zu unterscheiden); Laser: 100-400 V (Veränderung notwendig, um die Lebensfähigkeit Farbstoff positive Vs negative Ereignisse zu unterscheiden und vergleichen dies keine Lebensfähigkeit Farbstoff Kontrolle); Tore: Ändern Sie nach Bedarf um sicherzustellen, dass alle Zellen gesammelt werden. Siehe Abb. 2d-2 g.
Hinweis: FACS-Parameter sind stark abhängig von der Zelltypen und der Sortierer eingesetzt und muss daher vom Anwender optimiert werden.
- Vorbereitung 15 mL schmalen unteren Rohre mit 8 mL eiskaltes 1 % BSA/HBSS für Musterkollektionen. Statisch im inneren Rohr und Oberflächenspannung beeinflussen Abscheideleistung. Invertieren die Rohre vor Sammlungen an der Schnittstelle zwischen der Oberfläche der Flüssigkeit zu gewährleisten und die Innenseite der Röhre ist feucht.
Hinweis: Arbeiten mit sehr niedrigen Zellzahlen an kleinen Sammelbehälter je nach Bedarf anpassen. - Sobald alle Zellen gesammelt sind, Zentrifugieren Sie Probe bei 260 X g für 8 min.
Hinweis: Vor dem Zentrifugieren, fügen Sie 1 % BSA/HBSS zu waschen/Zellen drücken nach unten von der Seitenfläche und invertieren/Mix die Röhre unmittelbar nach FACS. - Aufschwemmen Sie Zelle Pellet in 1 % BSA/HBSS und halten Sie auf dem Eis. Das maximale Volumen pro Probe, die kompatibel mit Schritt 3 Verarbeitung ist 33,8 µL, also sicherstellen, dass endgültige Zellvolumen Verdünnung/Wiederfreisetzung geeignet ideale Zellzahl in 33,8 µL zu erhalten. Weitere Verdünnung Medienoptionen für diesen Schritt (und alle früheren Verdünnungen in 1 % BSA/HBSS) DMEM, und bis zu 40 % Serum unter anderem vermeiden, Kalzium, Magnesium oder EDTA, die Reagenzien enthalten.
- Lassen Sie Zellen auf Eis für eine minimale Menge an Zeit. Im Idealfall sollten Mitarbeiter alle Geräte und Reagenzien für den folgenden Schritt (Schritt 3) während der letzten Schritte von Schritt2 vorbereiten.
- Zelle Vorbereitung kritischen Überprüfung
- Schätzungen der Zellzahlen von FACS erhalten zu bestätigen. Je nach Gewebetyp und Dissoziation können Längen, Schutt und Zellen in Größe und Form sehr ähnlich sein. So, wenn ein fluoreszierender Reporter verwendet wird, kann FACS alle Trümmer nicht ausschließen. Es wird empfohlen, dass eine endgültige Zellzahl nach FACS Sammlung durchgeführt wird, um zu verstehen, wie viel Prozent der Ereignisse (nach FACS) sind in der Tat Zellen für ein bestimmtes Präparat (Abbildung 2 g). Führen Sie Zellzahl unter Verwendung eines Hemocytometer oder automatisierte Zelle Zähler (zweimal wiederholen) und Berechnung des Prozentsatzes der lebensfähigen Zellen, die durch die Gesamtereignisse nach FACS Maschine gesammelt dargestellt ist.
- Handy-Vorbereitung zu validieren. Überprüfen, dass keine großen Partikeln (> 100 µm) vorhanden sind, wie sie Geräte in nachgelagerten Stufen verstopfen können. Unzureichende Beseitigung der Trümmer riskieren verstopfen die einzelligen Mikrofluidik-Chip. Platte verbleibende Zellen mit Nuc blau (siehe oben) um sicherzustellen, dass keine großen Trümmer Fragmente vorhanden sind. Dies ermöglicht auch für die Bestätigung, dass einzelne Zellen (d. h.nicht zusammenkleben) geben Sie Gewissheit, dass nachgeschaltete Einzelzelle Genanalyse Einzelzellen, anstatt mehrere Zellen darstellt.
- Entscheiden Sie sich für Zellzahlen Sequenz: gibt es eine große Auswahl an Erwachsenen Gewebe abgeleitet Zellzahlen pro Probe, die mit bis zu 8 Proben in das System geladen werden kann, die gleichzeitig ausgeführt werden können. Autoren haben überall von 500 – 50.000 Zellen pro Probe geladen und erhalten gute Qualität ScRNA-Seq Datasets. Weitere Diskussion über die am besten geeignete Zellzahlen Laden finden Sie im Abschnitt Diskussion. Die endgültige Ausgabe der sequenzierten Zellzahlen hängt stark von der Qualität der Einzel-Zellen isoliert. Ladezellen 10.000 Erwachsenen Gewebe abgeleitet kann überall von 1.000 bis 4.000 sequenzierten Zellen (10-40 % Rendite) zurück. Wenn Sie interessieren sich für Sequenzierung hohe Zellzahlen (~ 10.000 Zellen, die maximale Anzahl für dieses System empfohlen), werden dann 25.000-100.000 Zellen geladen.
(3) GEM (Gel Perle in Emulsion) Generation und Barcoding (Tag 1)
Hinweis: Schritte 3 bis 6 dieses Protokolls sollen in Verbindung mit die häufigste Microdroplet-einzellige Plattform, hergestellt von 10 X Genomics verwendet werden. Ausführliche Richtlinien für die Schritte 3 und 4 in des Herstellers aufgeführt sind Protokoll (siehe die einzelne Zelle Chrom 3' Protokoll)11,12 und in Verbindung mit diesem Protokoll einzuhalten. Um beste Ergebnisse zu erzielen muss Schritt 3 sofort nach der Dissoziation (Schritt 1) und Zelle isoliert (Schritt2) Schritte am Tag 1 dieses Protokolls abgeschlossen sein.
- Chip nach dem Hersteller Protokoll 11,12vorzubereiten. Dieses Microdroplet-basierte einzellige Plattform nutzt Technologie dieser Proben ~ 750.000 Barcodes, um jede Zelle Transkriptom separat zu indizieren. Dies wird erreicht durch die Partitionierung Zellen in Gel Perle in dem generierten cDNA teilen einen gemeinsame Barcode-Emulsionen (GEMs). Bei der GEM-Generierung Zellen werden geliefert, so dass die meisten (90-99 %) der generierten Edelsteine enthalten keine Zellen, während der Rest zum größten Teil, eine einzelne Zelle enthalten.
- Platzieren Sie den Chip in der Chip-Halterung.
- Bereiten Sie Zelle master-Mix auf dem Eis.
- Nicht genutzte Brunnen 50 % Glycerin hinzufügen und gut 1, 90 µL der Zelle-master-Mix hinzufügen 90 µL Gel Perlen um gut 2 und 270 µL der Partitionierung Öl gut 3.
- Decken Sie den Chip mit der Dichtung.
- Laden Sie den Chip und führen Sie in eine einzelne Zelle-Controller.
- Werfen Sie das Fach, platzieren Sie den Chip im Fach zurückziehen Sie das Fach und drücken Sie Play. Eine einzelne Zelle 3' Gel Perle in ein Juwel beinhaltet Grundierungen, enthält eine Teilsequenz Illumina R1 (gelesen 1 Sequenzierung Grundierung), ein 16 Nukleotiden (nt) 10 X Barcode, 10 nt einzigartige molekulare Bezeichner (UMI) und eine Poly-dT-Grundierung-Sequenz. Während des Laufs sind Gel-Perlen in der Steuerung freigegeben und gemischt mit Zelle lysate und master Mix.
- Sammeln Sie 100 µL der Probe und Ort in einem PCR-Röhrchen.
- Ort PCR-Röhrchen in voreingestellten PCR Maschine und führen Sie die PCR nach dem Kit. Nach Inkubation, Edelsteine umfasst Full-length, Barcode cDNA aus Poly-Adenylated-mRNA.
- Stellen Sie nach der Flucht bei-20 ° C über Nacht bis zu 1 Woche vor dem nächsten Schritt vor.
4. Sanierung, Verstärkung, Bibliotheksbau und Bibliothek Quantifizierung (Tag 2)
Hinweis: Detaillierte Richtlinien für die Schritte 4 in des Herstellers Protokoll 11,12, beschrieben werden und müssen in Verbindung mit diesem Protokoll befolgt werden.
- Verwenden Sie Silan magnetische Beads, um übrig gebliebene biochemische Reagenzien/Primer aus GEM Reaktionsgemisch zu entfernen.
- Verstärken in voller Länge, Barcode cDNA, ausreichende Masse für Bibliotheksbau zu generieren.
- Bewerten Sie DNA-Ertrag. Beurteilen Sie vor Bibliotheksbau DNA-Ertrag der Probe. Damit wird festgelegt, wie viele Zyklen im nachgeschalteten PCR Schritt (Probe Index PCR während Bibliotheksbau) verwenden. Je nach RNA-Gehalt einer bestimmten Probe, die je nach Aktivierung Staaten (z. B.Kontrolle Vs verletzt, etc.), Zelltyp und Zellausbeute variieren können, kann die empfohlene Zykluszahl ändern.
- Für die Sequenzierung ~ 3.000 Gewebe-abgeleitete Zellen (irrelevant für Aktivierungsstatus), die Autoren haben festgestellt, dass 14 Zyklen (Proben: ~ 10-100 ng DNA) ist standard.
- Verwenden Sie eine Bioanalyzer für DNA-Analysen. Finden Sie für den Benutzer führen13.
- Sample-Fragment und wählen Sie die Größe der DNA. Verwenden Sie vor Bibliotheksbau enzymatische Fragmentierung und Größe Auswahl Protokolle, um entsprechende cDNA Amplikons Größe zu erhalten.
- Probe für Bibliotheksbau vorbereiten. Während R1 (gelesen 1 Primer Sequenz) der Moleküle während der Inkubation GEM hinzugefügt wird; P5, P7 (eine Probe Index) und R2 (lesen 2 Primer Sequenz) während der Bibliotheksbau hinzugefügt werden.
- Bewerten Sie DNA-Ertrag. Die meisten Sequenzierung Einrichtungen erfordern Einreichung der endgültigen Bibliotheken, die DNA-Ausbeute und Qualität Informationen enthalten. So laufen Sie die Bioanalyzer nach Abschluss des gesamten Protokolls und vor dem Transport zur Sequenzierung Anlage.
- Proben bei-80 ° C bis zu 2 Monate lang zu speichern.
- Vor der Sequenzierung, Proben mit den DNA-Quantifizierung Kit zu quantifizieren. Dies kann bei der Sequenzierung Einrichtung erfolgen.
5. Bibliothek Sequenzierung (Tag 3)
Hinweis: Die einzelligen Transkriptom Barcoding-Plattform in diesem Protokoll verwendeten generiert Illumina-kompatiblen gepaart-End Bibliotheken beginnend und endend mit P5 und P7-Sequenzen. Obwohl Mindesttiefe erforderlich, um zu beheben, dass Zelltyp Identität so wenig wie 10.000-50.000 mal gelesen/Zelle15,16sein kann, empfiehlt sich ~ 100.000 mal gelesen/Zelle als eine optimale Kostendeckung Kompromiss für Erwachsene in-vivo-Zellen (unter Berücksichtigung einer Zelle Arten oder minimal aktivierten Zelle Staaten erreichen Sättigung bei 30.000-50.000 mal gelesen/Zelle).
- Transport von cDNA-Bibliotheken auf Trockeneis, eine Sequenzierung-Anlage mit einem entsprechenden Illumina-Sequenzer ausgestattet.
- Geben Sie die folgenden Informationen zur Sequenzierung Anlage:
- Einzelheiten der Probe: Probieren Sie Index-IDs für jede Bibliothek; Spezies; genomische Datenbank für primären Assembly (d. h.GRCm38 für Maus); Electropherogram zeigt Fragment Größen von Bioanalyzer (zwischen 200 und 9.000 bp); cDNA-Konzentration (ng/µL) und die gesamte Bibliothek Konzentration (Gesamt Erträge reichen von 200 – 1400 ng); Volumen (µL) der Probe.
- Sequenzierung Anfragen bieten: quantifizieren Proben mit den DNA-Quantifizierung Kit; Adapter/Indextyp (TruSeq DNA); Plattentyp (Eppendorf twin.tec, voller Rock - empfohlen für DNA); Technologie/Bibliothek Typ (10 X, vollständige Sequenzierung Anweisungen und Empfehlungen der Zyklus)17-Sequenzierung.
- Führen Sie flache Sequenzierung (optional): Studien analysieren mehrere biologische Proben profitieren vom pooling Proben (Aggregation) um ein einzelnes Gen-Barcode-Matrix enthält Daten aus allen Proben zu generieren. Um Batch-Effekte zwischen Proben bei der Bündelung zu minimieren, sollte die lesen Sie Tiefe zwischen verschiedenen Bibliotheken standardisiert werden. Um dies zu tun, ist eine präzise Annäherung an einzelne Zelle Zahlen notwendig. Der MiSeq-Sequencer ermöglicht flache Sequenzierung und ist eine kostengünstige und praktische Möglichkeit, genaue Zelle Schätzungen zu erhalten.
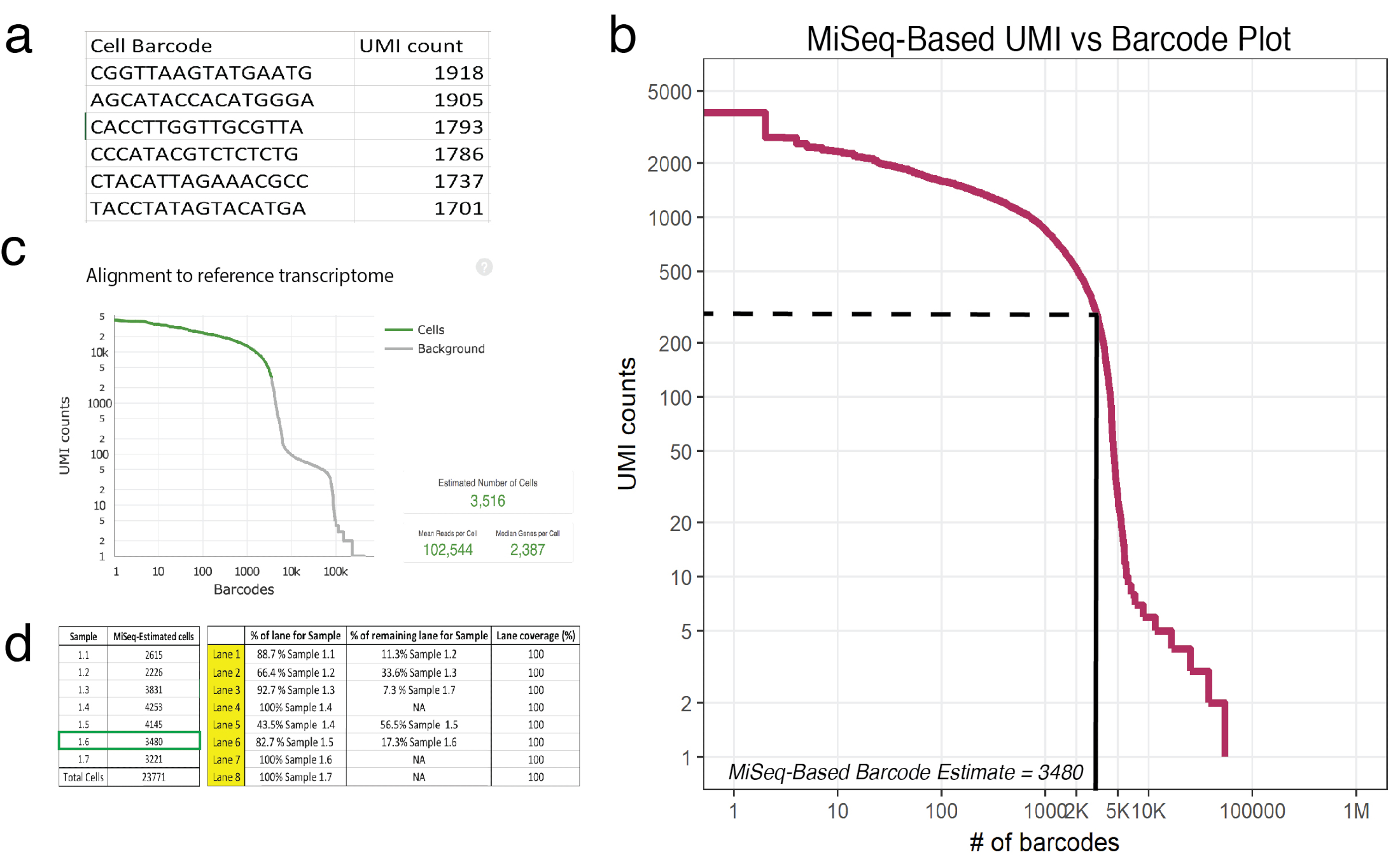
Hinweis: Ein Lauf mit einem MiSeq SR50 Sequencer bietet ausreichende Deckung um ca. 20.000 Zellen genau zu schätzen. Diese Ausführung wird die Anzahl der UMI erholt für jeden eindeutigen Barcode annähern. In Abbildung 3ader Header eine Beispielausgabe (Probe 1.6) (CSV) wird angezeigt, Inserat Barcodes und seine entsprechenden UMI gilt als selbstbewusst zugeordneten liest bestimmt.- Wenden Sie sich an ein Bioinformatiker mit der Programmiersprache R vertraut. Beziehen sich auf DataCamp-Tutorials für mehr Informationen18.
- Bewerten Sie Rohdaten aus der Sequencer mit dem mitgelieferten R-Skript als eine Vorlage19gewonnen. RAW-Daten bezieht sich auf die Anzahl von UMIs jedes einzigartige Zelle Barcode zugeordnet. Das Skript liest eine CSV-Datei, wo die erste Spalte ist eine Liste von Barcodes und die zweite Spalte sind die entsprechenden Umi, zählt. Dieses Skript wird ein Grundstück (Abb. 3 b) sowie die geschätzte Anzahl der Barcode-Zellen in jeder Probe zur Verfügung stellen. Stellen Sie ein Skript, um sicherzustellen, dass die eingegebene Zahl von UMI zählt für eine gegebene Probe zum Zeitpunkt der ersten steilen Drop ein Drittel. In Abbildung 3 bfällt dieser Ellenbogen rund 225 UMIs 3.480 Barcode Zellen entspricht.
- Vergleichbar mit ganzer Tiefe Sequenzierung mit Leseweite (wo 3.516 Zellen erfolgreich wurden sequenziert, Abbildung 3 c), flachen Sequenzierung Schätzungen vorhergesagt 3.480 Zellen.
- Entweder Nutzung Zelle Erholung Annäherungen (von Schritt 5.3) oder verwenden Sie die Recovery-Diagramm gefunden in des Herstellers Protokoll20 , Lane-Verteilung für die tieferen Sequenzierung zu planen. Jede Probe sollte vergleichbare Abdeckung erhalten, so dass wenn flache Sequenzierung zeigt, dass es unterschiedliche Anzahlen von Zellen in jeder Probe (was oft der Fall ist) dann Lane Verteilung entsprechend berechnet werden soll. Ein Leseweite Durchflusszelle (umfasst 8 Bahnen) kann bis zu 2,4 Milliarden gekoppelten Benutzerdef liest sequenzieren. Beispiel Flow Zelle Aufbau ist in Abbildung 3dvorgestellt.
6. Verarbeitung Dateien lesen
Hinweis: Eine einzelne Zelle 3' Bibliothek unter Verwendung dieses Protokolls Sequenzierung Rohdaten in binäre Basis Call (BCL) Format generiert. Die Zelle Ranger Paket verwendet wird, um textbasierte FASTQ Dateien von BCL-Dateien zu erzeugen, führen genomische und transkriptomischen Ausrichtungen, gen zählt Entpackung und Aggregation von Proben. In diesem Abschnitt werden die wichtigsten Schritte, mit denen Benutzer BCL Rohdaten von eine Sequenzierung Anlage herunterladen und gefilterten gen-Barcode Matrizen bereit für nachgeschaltete Bioinformatik zu generieren vorgestellt.
- Verwenden Sie einen zentralen Server für die Ausführung des Programms. BCL-Dateien, FASTQ Dateien und ein Großteil der nachgeschalteten Bioinformatik Verarbeitung erfordert erhebliche Verarbeitungsleistung.
- Laden Sie alle lesen Sie raw-Dateien auf Server (oder FASTQ-Dateien, wenn diese verfügbar sind).
- Wenden Sie sich an Server-Administrator ein Konto auf einem zentralen Server oder Cluster einrichten und mit Unix-21vertraut machen.
- Verwenden Sie einen Fetch-Befehl für das Server-Betriebssystem aus der Sequenzierung Facility Server alle Dateien herunterzuladen.
- Die meisten Sequenzierung Einrichtungen bieten einen Befehl zum Herunterladen von Dateien aus einen sicheren Weg, der von der Befehlszeile aus ausgeführt werden kann (siehe Beispiel unten).
- Ersetzen Sie die "< Username >" und "< Kennworts >" Platzhalter in der Kommandozeile mit Anmeldeinformationen zur Verfügung gestellt.
Wget - O - "https://your_sequencing_facilitys_server.com/path_to_raw_read_files/--keine-Cookies--keine-Check-Zertifikat - Post-Daten" J_username = Benutzername & J_password = Passwort "| Wget--keine-Cookies--keine-Check-Zertifikat - Post-Daten "J_username = Benutzername & J_password = Passwort ' - Ci -
- Wenn nur ein absoluter Pfad zu Dateien (d.h. https://your_sequencing_facilitys_server.com/path_to_raw_read_files/) bereitgestellt wird, legen Sie diesen Pfad in eine Fetch-Befehl.
- Dateien entpacken: Wenn Ende der Dateien mit der Erweiterung ".gz" heruntergeladen haben, es hat komprimiert wurden mit dem Befehl "Gzip". Zum Entpacken, entpacken ausführen Befehl in der Befehlszeile (siehe Beispiel unten).
Gunzip raw_read_files.gz - Laden Sie die neueste Version von Zelle Ranger auf dem Server als eine eigenständige .tar-22.
-
Kritische: Vor dem Download sicherzustellen Sie, dass das Linux-System Mindestanforderungen23entspricht. Sorgen Sie für ein Minimum von 8-Core Intel-Prozessor mit 64 GB RAM und 1 TB Speicherplatz.
Hinweis: Cell Ranger bietet vorgefertigte menschliche und Nagetier Referenz Transkriptom. Diese können geändert werden, mit Cellranger Mkref Befehl, um Gene wie GFP24zu erkennen.
-
Kritische: Vor dem Download sicherzustellen Sie, dass das Linux-System Mindestanforderungen23entspricht. Sorgen Sie für ein Minimum von 8-Core Intel-Prozessor mit 64 GB RAM und 1 TB Speicherplatz.
- Generieren Sie FASTQ Dateien vom Sequencer Basis Anruf Dateien (BCL) mit dem Cellranger-Mkfastq-Befehl.
Hinweis: Das Programm richten Sie roh liest (aus FASTQ-Dateien) zu einem Referenz-Genom und gen-Zelle Matrizen für nachgelagerte Analyse zu generieren. Es nutzt STAR-Aligner, das Spleißen-aware Ausrichtung der Lesezugriffe auf eine Referenz-Genom ausführt. Nur getrost zugeordneten liest (d.h.liest kompatibel mit einem einzigen Gen Annotation) dienen zum zählen von UMI.- Z. B. dem Befehl Cellranger Mkfastq:
Cellranger Mkfastq - Id = Sample_name \
--= / Pfad/zu/Probe laufen \
--csv=csv_file_containing_lane_sample_index.csv
- Z. B. dem Befehl Cellranger Mkfastq:
- Führen Sie Cellranger Graf auf FASTQ Dateien generiert, mit Mkfastq, einzellige gen zählt zu generieren.
- Z. B. dem Befehl Cellranger zählen:
Cellranger Graf - Id = Sample_name \
--Transkriptom Refdata-Cellranger-mm10-1.2.0 = \
--Fastqs = / absolutes/Pfad/zu/Fastq/Dateien \
--Probe = Same_sample_name_supplied_to_cellranger_mkfastq \
--Localcores = 30
- Z. B. dem Befehl Cellranger zählen:
- Multi-Bibliothek Aggregation (optional): Proben zu kombinieren, Cellranger Anzahl Ausgänge mit Cellranger Aggr zu bündeln. Dies führt zu einer einzigen Gen-Barcode-Matrix mit Daten aus mehreren Bibliotheken gebündelt. Cellranger Aggr Beispielbefehl:
Cellranger Aggr--Id = Sample_name \
Csv - Csv_with_libraryID_ & _path_to_molecule_h5.csv = \
--normalisieren = zugeordnet
: Anm. die Bibliotheken können werden mit drei Modi der Normalisierung (zugeordnete, roh, keiner). Abgebildet wird empfohlen, da es höher Tiefe Bibliotheken fügt, bis alle Bibliotheken gleich Sequenzierung Tiefe25haben. - Importieren Sie für sofortige Visualisierung/Analyse der Daten die .cloupe-Ausgabe-Datei (generiert mit Cellranger Graf oder Cellranger Aggr) in 10 x Lupe Zelle Browser26.
7. erweiterte Analyse der ScRNA-Seq-Datasets
Hinweis: Eine vollständige ScRNA-Seq-Tools-Datenbank finden Sie bei ScRNA-Werkzeuge3,27. Unten ist ein Rahmen für unbeaufsichtigtes Zelle clustering mit Seurat2 und pseudotemporal mit Monokel6bestellen. Obwohl ein Großteil dieser Arbeit auf einem lokalen Computer erfolgen kann, die folgenden Schritte angenommen, Berechnung über einen institutionellen Server abgeschlossen sein wird.
- Laden Sie die neueste Version von Miniconda auf Server-Konto mit der Linux-Plattform-28.
- Installieren Sie die neueste Version von R mit Conda29.
- Plot-Daten mit Hilfe der mitgelieferten Seurat R-Skripts als eine Vorlage30.
Hinweis: Seurat ist eine R-basierte Toolkit, die Qualitätskontrollen, clustering, differenzielle gen Expressionsanalyse, Marker-gen Identifizierung, Reduzierung der Dimensionalität und Visualisierung von ScRNA-Seq-Daten ermöglicht. Eine umfassende Beschreibung von Seurat Codierung und Tutorials finden Sie auf der Satija Lab Website31. - Plot-Daten mit Hilfe der mitgelieferten Monocle R-Skripts als eine Vorlage32.
Hinweis: Monokel ist ein anderes R-basierte Toolkit, das ermöglicht die Visualisierung der Ausdruck Änderungen über Pseudotime und Zelle Schicksal Entscheidungen zugrunde liegenden Gene identifiziert. Eine umfassende Beschreibung von Monocle Codierung und Tutorials finden Sie auf der Monocle Website33. - R-Pakete wie kBET können eingesetzt werden, um zu testen und korrigieren Batch Effekte durch Bündelung von Datasets34.
8. NCBI GEO und SRA Einreichungen
Hinweis: Da einfach Zugriff auf raw Sequenzierung Dateien gewährleisten Reproduzierbarkeit und Reanalyse, trat Einreichungen auf öffentlich zugänglichen Online-Repositories sind empfohlen oder erforderlich vor dem Manuskript einreichen. National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) und Reihenfolge lesen Archiv (SRA) sind öffentlich zugängliche Datenrepositories für Hochdurchsatz-Sequenzierung Daten35,36.
- Registrieren Sie sich für NCBI GEO Submitter Konto37.
- Komplette GEO-Vorlage umfasst drei Komponenten in ein Verzeichnis/Ordner (mit dem Titel als die GEO Einreicher Benutzername) kompiliert: 1) Metadaten Datensatz (eine Tabelle pro Projekteinreichung); (2) raw-Daten-Dateien; (3) verarbeiteten Daten-Dateien.
- Herunterladen Sie und füllen Sie der Metadaten Tabelle38 aus. Die folgende öffentlichen GEO-Vorlage kann als ein Führer (GSE100320)39verwendet werden. Platzieren Sie die Tabelle in das Verzeichnis.
- Ort Raw Daten-Dateien aus Cellranger Graf -Skript für alle Bibliotheken in das Verzeichnis generiert.
- Ort verarbeitet Datendateien (gefilterte barcodes.tsv, genes.tsv und matrix.mtx) aus Cellranger Graf -Skript für alle Bibliotheken in das Verzeichnis generiert.
- Verwenden Sie GEO Einreicher FTP-Serveranmeldeinformationen zum Verzeichnis aller drei Komponenten übertragen. Für Linux/Unix-Benutzer: Ncftp, Lftp, ftp, Sftp und Ncftpput verwendet werden.
- GEO für alle Überweisungen38zu benachrichtigen.
Ergebnisse
Das Repertoire des open-Source-Pakete entwickelt, um ScRNA-Seq-Datasets analysieren40 mit der Mehrheit der diese Pakete verwenden R-basierten Sprachen3sprunghaft angestiegen. Hier werden repräsentative Ergebnisse mit zwei dieser Pakete präsentiert: Bewertung unbeaufsichtigt Gruppierung von einzelne Zellen basierend auf Genexpression und Zell-Heterogenität und dekonstruieren biologische Bestellung einzelne Zellen entlang einer Flugbahn um zu lösen Prozesse.
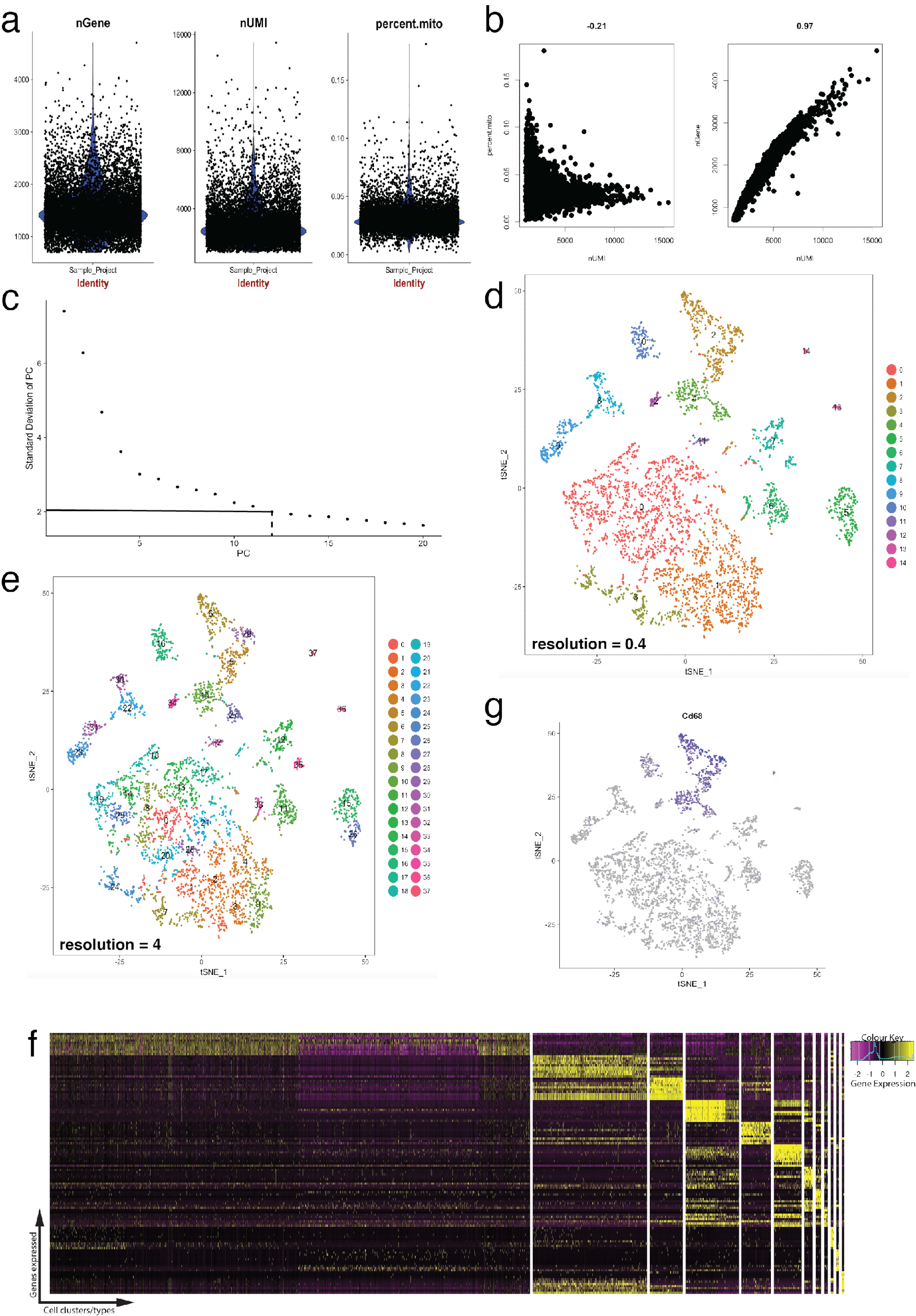
Abbildung 4 zeigt die Verwendung von Seurat für Vorverarbeitung Qualitäts-Checks und nachgelagerten Bioinformatik-Analyse. Erstens ist Filtration und Entfernung von abweichenden Zellen aus Analyse essentiell für die Qualitätsprüfung. Dies geschah mit Violine (Abb. 4a) und streuen Grundstücke (Abbildung 4 b), um den Prozentsatz der mitochondrialen Genen, Anzahl der Gene (nGene) und Anzahl der UMI (nUMI), Zelle Dubletten und Ausreißer zu identifizieren zu visualisieren. Jede Zelle mit einer klaren Ausreißer Anzahl von Genen, UMI oder Prozentsatz der mitochondriale Gene war mit Seurat FilterCells Funktion entfernt. Da Seurat Hauptkomponente (PC) nutzt Analyse Scores zu Clustern Zellen, Bestimmung der statistisch signifikanten PCs enthalten ist ein entscheidender Schritt. Ellenbogen Grundstücke (Abb. 4 c) dienten für die PC-Auswahl, welche PCs über das Plateau der "Standardabweichung des PC" Achse wurden ausgeschlossen. Die Auflösung der Cluster wurde auch manipuliert, zeigen, dass die Anzahl der Cluster geändert werden kann, angefangen bei 0,4 (niedrige Auflösung führt zu weniger Zellcluster, Abbildung 4 d) bis 4 (hohe Auflösung führt zu höheren Zellcluster, Abbildung 4e ). Mit niedriger Auflösung ist es wahrscheinlich, dass jedem Cluster einen definierte Zelltyp darstellt, während bei hoher Auflösung dies auch Subtypen oder vorübergehende Zustände einer Zellpopulation darstellen kann. In diesem Fall wurden mit niedriger Auflösung Clustereinstellungen für die weitere Analyse Ausdruck Heatmaps (mit Seurat DoHeatmap Funktion) verwendet, um die höchst exprimierten Gene in einem bestimmten Cluster (Abb. 4f) identifizieren. In diesem Fall wurden die höchst exprimierten Gene identifiziert, durch die Beurteilung differentielle Expression in einem bestimmten Cluster im Vergleich zu allen anderen Cluster kombiniert, zeigt, dass jeder Cluster eindeutig durch definierte Gene vertreten war. Darüber hinaus können einzelne Kandidaten-Gene auf tSNE Grundstücke mit Seurat FeaturePlot Funktion (Abb. 4 g) visualisiert werden. Dies ermöglichte Entzifferung ob gab es Cluster, die Makrophagen zu vertreten. Mit FeaturePlot, fanden wir, dass beide-2 Cluster und 4 wurden Cd68 - Pfanne-Makrophagen Marker zum Ausdruck bringt.
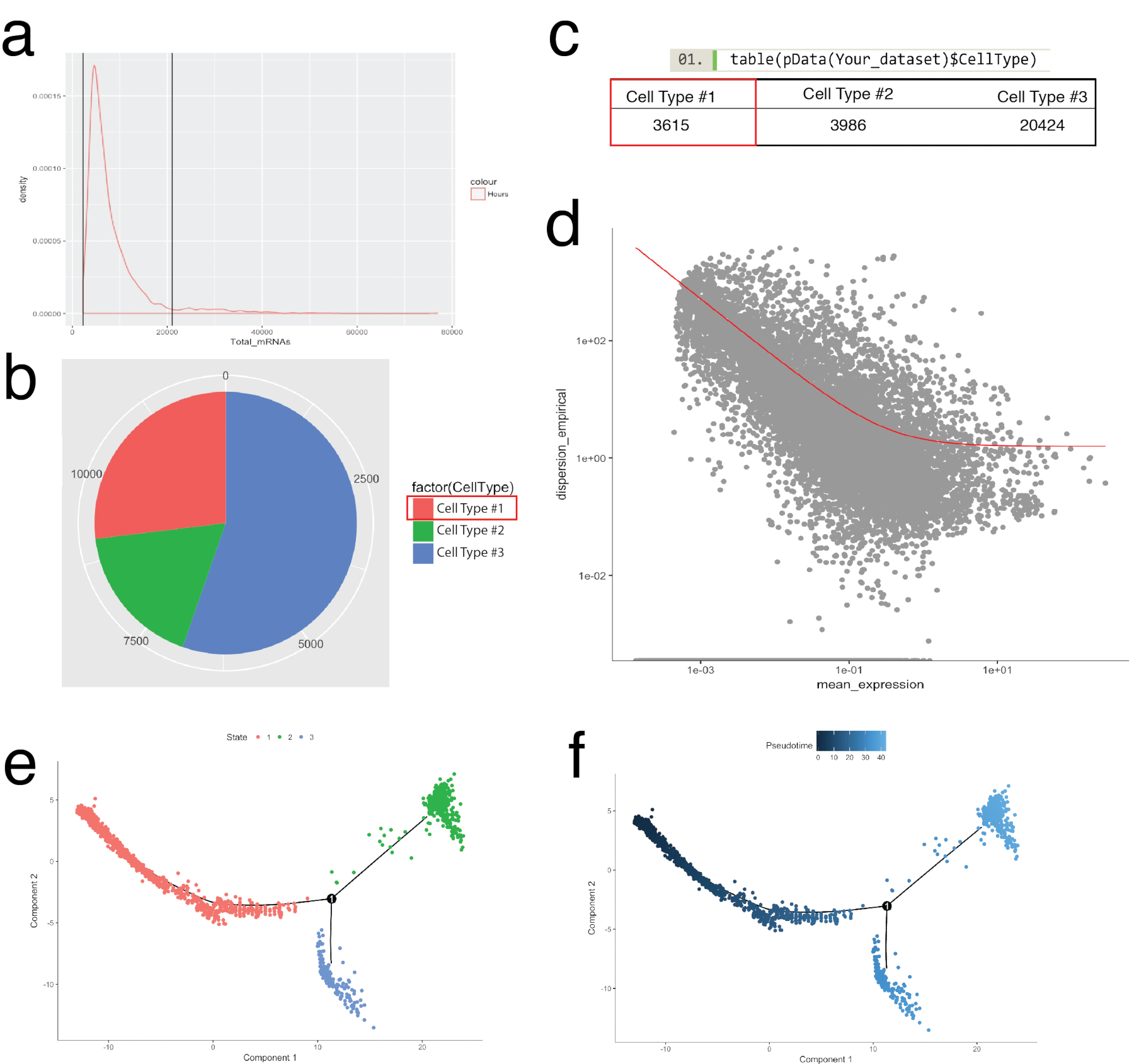
Das Monokel-Paket wurde für erhärten Zellcluster in Seurat identifiziert und für den Aufbau der Zelle Trajektorien oder pseudotemporal bestellen, zur biologische Prozesse (Abbildung 5) zu rekapitulieren. Pseudotemporal Bestellung kann für Proben verwendet werden, wo sollen einzellige Expressionsprofile einen biologischen zeitlichen Verlauf folgen. Zellen pseudotemporal Kontinuum aufzulösenden Zwischenzustände, Gabelung Punkte von zwei alternative Zelle Schicksale bestellbar und gen Unterschriften zugrunde liegenden Erwerb von jedem Schicksal zu identifizieren. Erstens, ähnlich wie Seurat Filtration, minderwertige Zellen wurden entfernt, so dass die Verteilung der mRNA über alle Zellen Log normal und fiel zwischen oberen und unteren Grenzen wie in Abbildung 5aidentifiziert wurde. Dann mit Monokel NewCellTypeHierarchy Funktion Einzelzellen eingestuft wurden und mit bekannter Abstammung Markergene (Abbildung 5 b, 5 c) gezählt. Beispielsweise wurden Zellen PDGF-Rezeptor Alpha oder Fibroblasten spezifische Protein 1 auszudrücken Zelle Typ #1 zugewiesen, erstelle ich ein Kriterium für die Definition von Fibroblasten. Als nächstes wurde dieser Population (Zelle Typ #1) bewertet, um Fibroblasten Trajektorien zu entschlüsseln. Um dies zu tun, war Monokel Differenzial-GeneTest-Funktion genutzt, die im Vergleich der Zellen, die extreme Zustände innerhalb der Bevölkerung darstellt und differenzielle Gene für die Bestellung der übrigen Zellen in der Bevölkerung (Abb. 5D). Durch die Anwendung vielfältige Lernmethoden (eine Art von nicht-linearen Dimensionalität Reduktion) über alle Zellen, wurde eine Koordinate auf einem pseudotemporal Weg zugewiesen. Diese Bahn wurde dann durch Zellstatus (Abb. 5e) und Pseudotime (Abb. 5f) visualisiert.

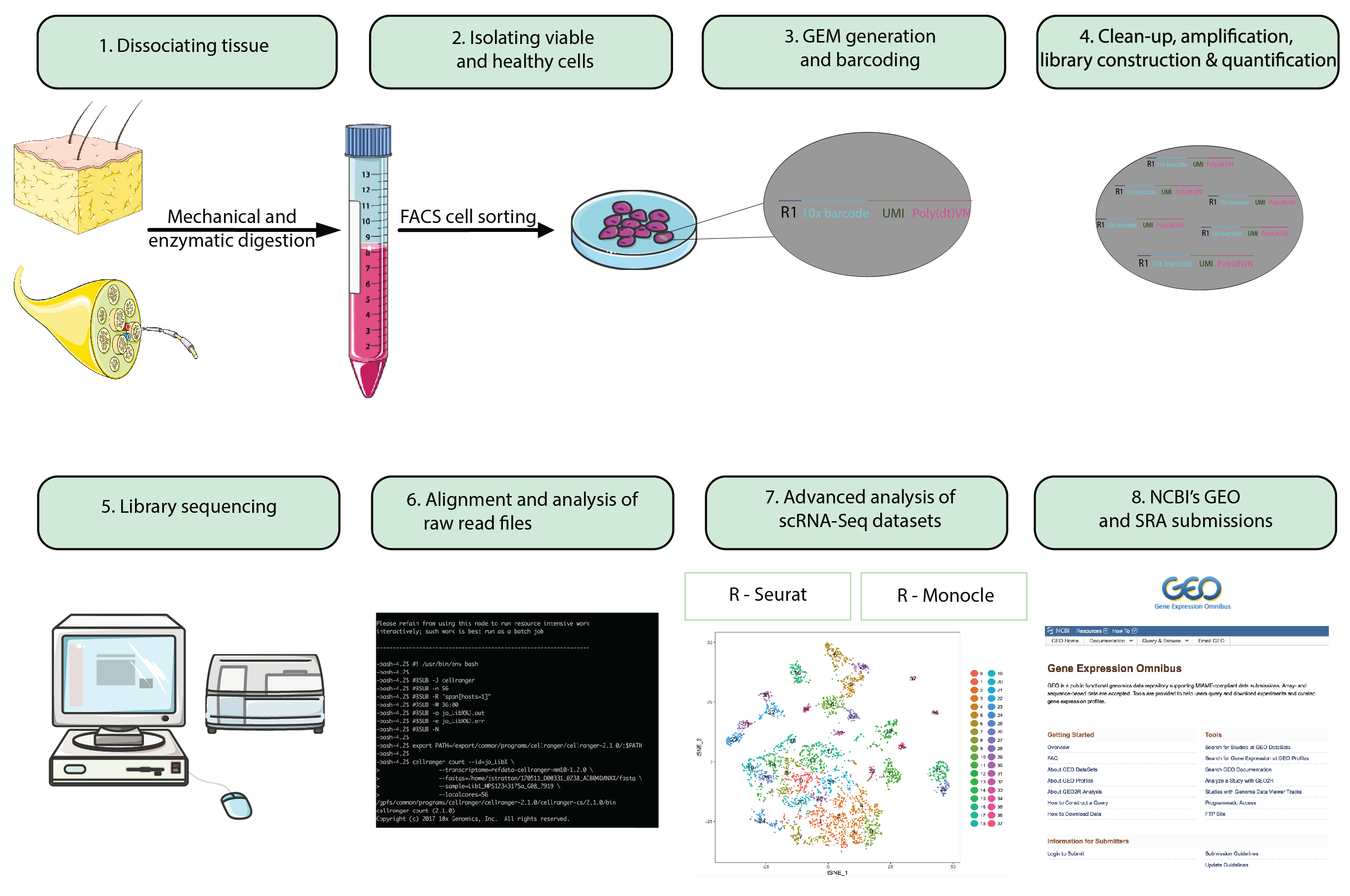
Abbildung 1: Flussdiagramm. Wenige Schritte vom ganze Tier Vorbereitung zu analysieren einzelne Zelle RNA-Seq-Datensätze für die Übermittlung der endgültigen Datasets zu einem öffentlich zugänglichen Repository. Gel-Perlen in Emulsion (GEMs) beziehen sich auf Perlen mit Barcode-Oligonukleotide, die Tausende von Einzelzellen zu Kapseln. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 2: tragfähige einheitliche Zellsuspension aus Nervengewebe erstellen. (a) Cartoon Überblick über Qualitätskontrollen. (b) Zellen und Trümmer mit Zellen aufgenommen noch in Trümmern (rote Pfeile). (c) Zellen aus dem Schutt (rote Pfeile) entlassen. (d) Zellen isoliert durch FACS. P0: Schutt Bruchteil; P1: zellähnliche Bruchteil; P3: Ausschluß von Duplets; P4: Lebensfähigkeit Farbstoff (Sytox Orange) negative Bruchteil. (e) keine Lebensfähigkeit Farbstoff Kontrolle. (f) Bild P0 Bruchteil vertreten isoliert Schutt. (g) Bild der P4-Fraktion vertritt isolierende lebensfähige Zellen (rote Pfeile). (b) (c) (f) und (g) hatte nuklearen Farbstoff hinzugefügt 20 Minuten vor Bildgebung. Maßstabsleisten: 80 µm. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: flache Sequenzierung prognostiziert die Anzahl der wiederhergestellten Zellen in 10 X Proben. (a) ein Beispiel (Beispiel 1.6) für MiSeq generiert Csv Inserat Zelle Barcodes und seine entsprechenden UMI gilt als selbstbewusst zugeordneten liest bestimmt. (b) Barcode Rang Plot für Probe 1.6 zeigt einen deutlichen Rückgang im UMI Graf als Funktion der Zelle Barcodes. Die solide und gestrichelte Linien repräsentieren die Cutoff zwischen Zellen und Hintergrund durch Sichtkontrolle ermittelt. (c) Zelle Barcodes beobachtet, mit der Zelle Ranger Pipeline Post-Leseweite offenbart, dass flache Sequenzierung genau die Anzahl der Zellen für Probe 1.6 angenähert. (d) ein Beispiel für eine-Durchflusszelle Setup basierend auf flachen Sequenzierung abgeleitet Zelle Schätzungen. Für Probe 1.6, da flache Sequenzierung 3480 Zellen vorausgesagt 1,17 Bahnen wurden zugewiesen, um sicherzustellen > 100.000 Mal pro Zelle Sequenzierung Berichterstattung in Leseweite gelesen. Hinweis: Alle Bahnen müssen 100 % hinzufügen. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: Qualitätskontrolle und Bioinformatik der einzelligen RNA-Seq Dataset mithilfe Seurat R Paket. (a) Grundstücke Qualitätskontrolle Metriken, die Anzahl der Gene, Anzahl der eindeutigen molekularen Kennungen (UMIs) und der Prozentsatz der Transkripte, die Zuordnung zu den mitochondrialen Genom enthalten. (b) Probe gen Grundstücke Nachweis von Zellen mit abweichenden mitochondrialer Transkripte und UMIs. (c) Probe Ellenbogen Grundstück für ad-hoc-Bestimmung der statistisch signifikanten PCs verwendet. Die gestrichelte und Dot-gestrichelte Linien repräsentieren die Cutoff, wo eine klare "Ellenbogen" in der Grafik ersichtlich. PC Abmessungen vor diesem Bogen werden in nachgeschalteten Analyse einbezogen. (d, e) Graph-basierte Zellcluster visualisiert in zwei verschiedenen Auflösungen in einem niedrig-dimensionalen Raum mit einem tSNE Grundstück. (f) top Markergene (gelb) für jeden Cluster auf einem Ausdruck Heatmap mit Seurat DoHeatmap Funktion visualisiert. (g) Visualisierung Marker Ausdruck, z. B. Cd68 gen, Makrophagen (lila) mit Seurat FeaturePlot Funktion darstellt. Dies deutet darauf hin, dass diese Cluster 2 und 4 (im Panel d) dieses Datensatzes Makrophagen darstellt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: Zell-Kategorisierung und entlang Peudotemporal Flugbahn mit Monokel Toolkit bestellen. (a) Prüfung der Verteilung der mRNA (abgeleitet von UMI zählt) auf alle Zellen in einer Probe. Nur Zellen mit mRNA zwischen 0 - ~ 20.000 für nachgelagerte Analyse verwendet wurden. (b, C) Zuweisen und Zelltypen basierend auf bekannten Linie Zelle Marken zählen. Z. B. Zellen PDGF-Rezeptor Alpha oder Fibroblasten spezifische Protein 1 zum Ausdruck bringen waren Zelle Typ #1 zugewiesen, Pan-Fibroblasten darstellt mit Monokel NewCellTypeHierarchy Funktion. Anzahl der verschiedenen Zelltypen kann als Kreisdiagramm (b) und als Tabelle (c) visualisiert werden. (d) Verwendung von Zelle Art #1 (Fibroblasten) als Beispiel, die Gene, die für die Bestellung von Zellen können mit einem Streudiagramm, das Gen Streuung vs. mittlere Ausdruck zeigt visualisiert werden verwendet. Die rote Kurve zeigt die Cutoff für Gene, die für die Bestellung berechnet den Mittelwert-Varianz-Modell mit Monokel EstimateDispersions-Funktion verwendet. Gene, die diese Cut-off Treffen dienten für die nachgeschaltete pseudotime Bestellung. (e, f) Visualisierung der Zelle Trajektorien in einem reduzierten zweidimensionalen Raum farbige Zelle "Staat" (e) und von Monocle zugewiesen "Pseudotime" (f). Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
Diskussion
Dieses Protokoll zeigt, wie die angemessene Vorbereitung einzelner Zellen entdecken die transkriptionelle Heterogenität aus Tausenden von einzelnen Zellen und Funktionszustände oder einzigartige zelluläre Identitäten innerhalb eines Gewebes zu unterscheiden kann. Das Protokoll erfordert keine Reporter fluoreszierende Proteine oder transgenen Werkzeuge und kann auch auf die Isolierung einzelner Zellen aus verschiedenen Geweben von Interesse, die auch von Menschen; unter Berücksichtigung jedes Gewebe ist einzigartig und dieses Protokoll erfordert ein gewisses Maß an Anpassung/Änderung.
Die vielfältigen und höchst dynamischen transkriptionelle Programme innerhalb von Zellen betonten den Wert der einzelligen Genomics. Neben qualitativ hochwertigen RNA zu isolieren, ist eine kritische Probe Vorbereitungsschritt für qualitativ hochwertige Datensätze notwendig sicherzustellen, dass Zellen vollständig aus Gewebe freigesetzt werden und Zellen gesund und intakt sind. Dies ist relativ geradlinig für Sammeln von Zellen, die leicht sind veröffentlichte z. B. zirkulierenden Zellen oder Geweben, wo Lose Zellen beibehalten werden, z. B. in lymphatischen Geweben. Dies kann jedoch eine Herausforderung für andere adulten Geweben aufgrund der hoch entwickelten zellulären Architektur über große Entfernungen, umgebenden extrazellulären Matrix und die oft starren Zellskelett Proteine, die bei der Aufrechterhaltung der Zellstruktur. Auch mit entsprechenden Dissoziation Techniken für die Vollversion von Zellen gibt es Potenzial, dass die strenge und oft langwierige Verarbeitung erforderlich mRNA Qualität und Zelle Integrität verändern würde. Darüber hinaus wirken sich die hohen Temperaturen verwendet für Enzym-gestützte Dissoziation auch transkriptionelle Signaturen29,30. Die Absicht des Protokolls ist vorzulegen Qualitätskontrolle prüft mit Geweben wie Markhaltige Erwachsenen Nerven und der extrazellulären Matrix-reiche Erwachsene Haut, um zu demonstrieren wie Optimierung kann helfen, um diese Hindernisse zu überwinden.
Ein wichtiger Aspekt bei der Gestaltung jedes ScRNA-Seq-Experiment ist die Wahl der Sequenzierung Tiefe. Sequenzierung hoch gemultiplext und gelesen werden kann Tiefe kann variieren von sehr niedrigen mit Drop-Seq2 auf bis zu 5 Millionen Mal gelesen/Zelle14 mit einer Full-length RNA-Seq-Methode wie Smart-seq. Die meisten ScRNA-Seq Experimente erkennt mäßigen bis hohen Ausdruck Protokolle mit Sequenzierung so niedrig wie 10.000 mal gelesen/Zelle, was für Zelle Art Klassifikation41,42in der Regel ausreichend ist. Flache Sequenzierung Tiefe ist der Wert bei Sequenzierung Kosten sparen, wenn Sie versuchen, seltene Zellpopulationen über komplexe Gewebe erkennen, wo Tausende von Zellen erforderlich ist, um selbstbewusst seltenere Populationen zuschreiben. Aber geringer Tiefe Sequenzierung ist nicht ausreichend, wenn detaillierte Informationen über Genexpression und Prozesse im Zusammenhang mit subtilen transkriptionelle Unterschriften notwendig ist. Derzeit wird geschätzt, dass die große Mehrheit der Gene in einer Zelle mit 500.000 mal gelesen/Zelle, erkannt werden, aber dies je nach Protokoll variieren kann und Gewebe Typ43,44. Während in voller Länge Abschrift Sequenzierung die Notwendigkeit für die Montage umgeht und daher, Roman oder seltene Splice-Varianten erkennen kann, begrenzt Sequenzierung Kosten oft Skalierung solche Ansätze um Tausende von Zellen bestehend aus einem komplexe Gewebe-System zu untersuchen. Im Gegensatz dazu getaggt 3' einzellige Bibliotheken wie beschrieben in diesem Protokoll in der Regel geringeren Komplexität haben und flacheren Sequenzierung erforderlich. Es ist wichtig zu beachten, dass Bibliotheken generiert mit dem beschriebenen Protokoll auf einem der fünf unterstützten Sequenzer sequenziert werden können: (1) NovaSeq, (2) Leseweite 3000/4000 (3) Leseweite 2500 schnellen Lauf und hohe Leistung, (4) NextSeq 500/550 und (5) MiSeq.
Ein alternativer Ansatz zur einzelnen Zelle RNA-Seq, der reduziert die Notwendigkeit für empfindliche Gewebe und Zellen, die Handhabung noch einige der Vorteile der einzelnen Zelle RNA-Seq unterhält, ist die Analyse der RNA aus einzelnen Kernen45. Dieser Ansatz ermöglicht eine schnellere Verarbeitung Verringerung der RNA-Abbau und mehr extreme Maßnahmen zur Gewährleistung der angemessenen Veröffentlichung von Kernen und somit wahrscheinlich ein selbstbewusster Erfassung der transcriptional Profile repräsentieren alle Zellen in einem bestimmten Gewebe ermöglicht. Dies würde, natürlich, bieten nur einen Teil der transkriptionelle Aktivität in einer bestimmten Zelle, also je nach was experimentelle Ziele von Interesse sind, dieser Ansatz möglicherweise oder möglicherweise nicht geeignet.
Neben der vollständigen Charakterisierung der zellulären Identitäten innerhalb eines bestimmten Gewebes ist eines der wertvollsten Analysen für ScRNA-Seq-Datasets die Beurteilung der transkriptionellen Zwischenzustände in "definierten" Zell-Populationen. Diese vermittelnden Staaten können vermitteln Einblicke in die Linie Beziehungen zwischen Zellen in bestimmten Bevölkerungsgruppen, die nicht mit traditionellen Masse möglich war, die RNA-Seq nähert. Mehrere ScRNA-Seq bioinformatische Werkzeuge wurden jetzt entwickelt, um dies aufzuklären. Solche Tools können die Prozesse beteiligt, zum Beispiel Krebszellen Übergang zu einem onkogenen/metastasierten Zustand, Stammzellen, die Reifen zu vielfältigen terminal Schicksale oder Immunzellen pendelt zwischen aktiven und ruhenden Zustand beurteilen. Subtile Transkriptom Unterschiede in den Zellen möglicherweise auch bezeichnend für Linie Vorurteile, dass neu entwickelte Bioinformatic Hilfsmittel wie FateID,47ableiten können. Da die Unterschiede zwischen den Zellen den Übergang schwierig sein können angesichts der transkriptionellen Unterschiede feststellen kann subtil sein, tiefer Sequenzierung möglicherweise notwendigen46. Glücklicherweise werden Abdeckung einer flach sequenzierten Bibliothek erhöht, wenn das Dataset weiter durch erneutes Ausführen der Bibliothek auf einem anderen Durchflusszelle Sondieren Sie interessieren.
Zusammen genommen, bietet dieses Protokoll einen leicht anzupassen Workflow, der Benutzern ermöglicht, Hunderte bis Tausende von Einzel-Zellen innerhalb eines Experiments transcriptionally Profil. Die endgültige Qualität eines ScRNA-Seq-Datasets stützt sich auf optimierte Zelle Isolation, Durchflusszytometrie, cDNA Bibliothek Generierung und Interpretation der Roh-gen-Barcode-Matrizen. Zu diesem Zweck bietet dieses Protokoll einen umfassenden Überblick über alle wichtigen Schritte, die leicht geändert werden können, um Studien von verschiedenen Gewebearten zu ermöglichen.
Offenlegungen
Keine Angaben
Danksagungen
Wir anerkennen die Support-Mitarbeiter in der UCDNA-Dienstleistungen-Anlage sowie die Animal Care Mitarbeiter der Einrichtung an der University of Calgary. Wir danken Matt Workentine für seine Unterstützung der Bioinformatik und Jens Durruthy für seine technische Unterstützung. Diese Arbeit wurde finanziert durch einen CIHR Zuschuss (r.m. und j.b.), ein CIHR neue Investigator Award, j.b. und eine Alberta Kinder Health Research Institute Fellowship (j.s.).
Materialien
| Name | Company | Catalog Number | Comments |
| Products | |||
| RNAse out | Biosciences | 786-70 | |
| Pentobarbital sodium | Euthanyl | 50mg/kg | |
| HBSS | Gibco | 14175-095 | |
| Dispase 5U/ml | StemCell Technologies | 7913 | 5 mg/ml |
| Collagenase-4 125 CDU/mg | Sigma-Aldrich | C5138 | 2 mg/ml |
| DNAse | Sigma-Aldrich | DN25 | 10mg/ml |
| BSA | Sigma-Aldrich | A7906 | |
| 15 ml Narrow bottom tube VWR® High-Performance Centrifuge Tubes | VWR | 89039-666 | |
| Sytox Orange Viability Dye | Molecular Probes | 11320972 | 1.3 nM/µl |
| Nuc Blue Live ReadyProbes | Invitrogen | R37605 | |
| Agilent 2100 Bioanalyzer High senitivity DNA Reagents | Agilent | 5067-4626 | |
| Kapa DNA Quantification Kit | Kapa Biosystems | KK4844 | |
| Chromium Single Cell 3' reagents | 10x Genomics | ||
| Equipment | |||
| BD FACSAria III | BD Biosciences | ||
| Agilent 2100 Bioanalyzer Platform | Agilent | ||
| Illumina® HiSeq 4000 | Illumina | ||
| Illumina® MiSeq SR50 | Illumina | ||
| 10X Controller + accessories | 10x Genomics | ||
| Software | |||
| The Cell Ranger | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome | |
| Loupe Cell Browser | 10x GENOMICS | support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| R | https://anaconda.org/r/r |
Referenzen
- Shalek, A. K., et al. Single-cell RNA-seq reveals dynamic paracrine control for cellular variation. Nature. 510, 363-369 (2014).
- Macosko, E. Z., et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 161, 1202-1214 (2015).
- Zappia, L., Phipson, B., Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. bioRxiv:206573. , (2018).
- Dulken, B. W., Leeman, D. S., Boutet, S. C., Hebestreit, K., Brunet, A. Single cell transcriptomic analysis defines heterogeneity and transcriptional dynamics in the adult neural stem cell lineage. Cell Reports. 18 (3), 777-790 (2017).
- Llorens-Bobadilla, E., et al. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell. 17 (3), 329-340 (2015).
- Trapnell, C., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 32, 381-386 (2014).
- Aibar, S., et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 14, 1083-1086 (2017).
- Mayer, C., et al. Developmental diversification of cortical inhibitory interneurons. Nature. 555 (7697), 457-462 (2018).
- Stratton, J. A., et al. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro. 4 (3), (2017).
- Biernaskie, J. A., McKenzie, I. A., Toma, J. G., Miller, F. D. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nature Protocols. 1 (6), 2803-2812 (2007).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- 10X Genomics. Chromium Single Cell 3' Training Module. , Available from: http://go.10xgenomics.com/training-modules/single-cell-gene-expression (2018).
- Agilent. , Available from: https://www.agilent.com/en-us/library/usermanuals?N=135 (2018).
- Kolodziejczyk, A. A. Single Cell RNA-Sequencing of Pluripotent States Unlocks Modular Transcriptional Variation. Cell Stem Cell. 17, 471-485 (2015).
- Jaitin, D. A., et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 343, 776-779 (2014).
- Pollen, A. A., et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nature Biotechnology. 32, 1053-1058 (2014).
- 10X Genomics. Sequencing Requirements for Single Cell 3'. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/sequencing/doc/specifications-sequencing-requirements-for-single-cell-3 (2018).
- Datacamp. Introduction to R. , Available from: https://www.datacamp.com/courses/free-introduction-to-r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics#39; Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- 10X Genomics. User Guides. , Available from: https://www.10xgenomics.com/resources/user-guides/ (2018).
- UNIX Tutorial for Beginners. , Available from: http://www.ee.surrey.ac.uk/Teaching/Unix/ (2018).
- 10X Genomics. Creating a Reference Package with cellranger mkref. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/advanced/references (2018).
- 10X Genomics. System Requirements. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/overview/system-requirements (2018).
- 10X Genomics. Software Downloads. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest (2018).
- 10X Genomics. Aggregating Multiple Libraries with cellranger aggr. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/aggregate#depth_normalization (2018).
- 10X Genomics. Loupe Cell Browser Gene Expression Tutorial. , Available from: https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/tutorial (2018).
- scRNA-tools. A table of tools for the analysis of single-cell RNA-seq data. , Available from: https://www.scrna-tools.org/ (2018).
- Conda. Downloading conda. , Available from: https://conda.io/docs/user-guide/install/download.html (2018).
- Anaconda. r / packages / r 3.5.1. , Available from: https://anaconda.org/r/r (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Satija Lab. Seurat - Guided Clustering Tutorial. , https://satijalab.org/seurat/pbmc3k_tutorial.html (2018).
- Droplet-based, high-throughput single cell transcriptional analysis of adult mouse tissue using 10X Genomics' Chromium Single Cell 3' (v2) system: From tissue preparation to bioinformatic analysis. , Available from: https://figshare.com/s/97b83e649e5eefd01357 (2018).
- Monocle. , Available from: http://cole-trapnell-lab.github.io/monocle-release/docs/#constructing-single-cell-trajectories (2018).
- Github. An R package to test for batch effects in high-dimensional single-cell RNA sequencing data. , (2018).
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 30, 207-210 (2002).
- Leinonen, R., Sugawara, H., Shumway, M. The sequence read archive. Nucleic Acids Research. 39, D19-D21 (2011).
- NIH. GenBank Submission Portal Wizards. , Available from: https://www.ncbi.nlm.nih.gov/account/register/?back_url=/geo/submitter/ (2018).
- NIH. Submitting data. , Available from: https://submit.ncbi.nlm.nih.gov/geo/submission/ (2018).
- Shah, P. T., et al. Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell. 173, 1045-1057 (2018).
- Anon, Method of the Year 2013. Nature Methods. 11, 1(2013).
- Adam, M., Potter, A. S., Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: a molecular atlas of kidney development. Development. 144, 3625-3632 (2017).
- Wu, Y. E., Pan, L., Zuo, Y., Li, X., Hong, W. Detecting activated cell populations using single-cell RNA-seq. Neuron. 96, 313-329 (2017).
- Zeigenhain, C., et al. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 65 (4), 631-643 (2017).
- Wu, A. R., et al. Quantitative assessment of single-cell RNA-sequencing methods. Nature Methods. 11 (1), 41-46 (2014).
- Habib, N., et al. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science. 353 (6302), 925-928 (2016).
- Janes, K. A. Single-cell states versus single-cell atlases - two classes of heterogeneity that differ in meaning and method. Current Opinions in Biotechnology. 39, 120-125 (2016).
- Herman, J. S., Sagar,, Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods. 15 (5), 379-386 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten